


Abstract
Calcific aortic valve stenosis (CAS) is the most frequent heart valve disease in the elderly, accompanied by valve calcification. Tumor necrosis factor-α (TNF-α), a pleiotropic cytokine secreted mainly from macrophages, has been detected in human calcified valves. However, the role of TNF-α in valve calcification remains unclear. To clarify whether TNF-α accelerates the calcification of aortic valves, we investigated the effect of TNF-α on human aortic valve interstitial cells (HAVICs) obtained from patients with CAS (CAS group) and with aortic regurgitation or aortic dissection having a noncalcified aortic valve (control group). HAVICs (2 × 104) were cultured in a 12-well dish in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. The medium containing TNF-α (30 ng/ml) was replenished every 3 days after the cells reached confluence. TNF-α significantly accelerated the calcification and alkaline phosphatase (ALP) activity of HAVICs from CAS but not the control group after 12 days of culture. Furthermore, gene expression of calcigenic markers, ALP, bone morphogenetic protein 2 (BMP2), and distal-less homeobox 5 (Dlx5) were significantly increased after 6 days of TNF-α treatment in the CAS group but not the control group. Dorsomorphin, an inhibitor of mothers against decapentaplegic homologs (Smads) 1/5/8 phosphorylation, significantly inhibited the enhancement of TNF-α-induced calcification, ALP activity, Smad phosphorylation, and Dlx5 gene expression of HAVICs from the CAS group. These results suggest that HAVICs from the CAS group have greater sensitivity to TNF-α, which accelerates the calcification of aortic valves via the BMP2-Dlx5 pathway.
Introduction
Calcific aortic valve stenosis (CAS) is the most frequent heart valve disease in the elderly (Chikwe et al., 2003). Aortic valves from patients with CAS are characterized by massive fibrosis thickening of valve leaflets and extensive focal ectopic calcification (Mohler, 2004). Because the irreversible aortic valve calcification in CAS causes the restriction of internal treatment, the most viable treatment is surgical aortic valve replacement, which is extremely invasive for patients (Iunga et al., 2003). To develop a noninvasive treatment using medical drugs, it is necessary to determine the detailed mechanism of aortic valve calcification. Although several groups have focused on the mechanism of aortic valve calcification in CAS (Rajamannan et al., 2003; Clark-Greuel et al., 2007), no effective noninvasive treatment has been established.
CAS has been recognized as an active process that may be related with several inflammatory factors (Mohler et al., 1999). Inflammation is a prominent feature of aortic valve calcification and may develop because of endothelial dysfunction fueled by atherosclerotic risk factors (Tanaka et al., 2005; Goldbarg et al., 2007). Macrophages and T lymphocytes were identified in calcified aortic valve lesions (Wallby et al., 2002; Freeman and Otto, 2005). These cells release cytokines, such as tumor necrosis factor-α (TNF-α), transforming growth factor-β1 (Jian et al., 2003), and interleukin-1 (IL-1) (Kaden et al., 2003), all of which contribute to extracellular matrix formation, remodeling, and local calcification. Transforming growth factor-β1 and IL-1 accelerate the calcification of aortic valves via the bone morphogenetic protein 2 (BMP2) pathway. In addition, intracellular signal transduction of TNF-α elicits a wide spectrum of other cellular responses, including the modulation of the differentiation and proliferation of a variety of cell types and induction of apoptosis via several signaling pathways; such as the extracellular signal-regulated kinase (ERK), the p38 mitogen-activated protein (MAP) kinase, and the nuclear factor-κB (NF-κB) pathways (Bradley, 2008).
TNF-α, a pleiotropic cytokine, is secreted mainly by activated macrophages and T lymphocytes in response to many factors such as oxidized low-density lipoprotein (Jovinge et al., 1996), damaged extracellular matrix (Alexandraki et al., 2006), or bacterial infection (Guzik et al., 2006). TNF-α influences many aspects of atherogenesis (Brett et al., 1989), including increasing permeability of endothelial cells (Tintut et al., 2000) and promotion of monocyte adhesion and foam cell formation (Riches et al., 1996; Parhami et al., 1997). All of those factors have been proven to be associated with the progression of CAS (Libby et al., 1995). TNF-α has been detected in both human and mouse atherosclerotic lesions and calcified aortic valve, and it plays an important role in the calcification of the aortic valve (Moe and Chen, 2004; Al-Aly et al., 2007). Data have been reported that TNF-α promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-κB signaling pathway (Hess et al., 2009), and NF-κB is responsible for TNF-α-induced BMP2 expression and activity (Wu et al., 2007). Runt-related gene 2 (Runx2) and muscle segment homeobox 2 (Msx2) act downstream of BMP2 and closely associate to the region of physiological calcification. In osteoblasts, Msx2, and Distal-less homeodomain 5 (Dlx5) have been shown to be coregulated genes that are essential in bone development, including the Runx2 and the osteocalcin genes (Hassan et al., 2006). In myogenic C2C12 cells, Dlx5 is a direct gene target of BMP2 signaling, and it regulates Runx2 expression (Lee et al., 2005). However, the detailed molecular mechanism of TNF-α-induced calcification of aortic valve remains unclear.
This study was designed to verify whether TNF-α accelerates the calcification of human aortic valve interstitial cells (HAVICs) from CAS compared with a control group. Results indicate that TNF-α accelerated calcification of HAVICs by elevating alkaline phosphatase (ALP) activity and ALP gene expression in the CAS group. We further investigated the molecular mechanism of TNF-α induced calcification of HAVICs in the CAS group and found critical roles for BMP2 and Dlx5 in aortic valve calcification.
Materials and Methods
HAVICs Isolation and Culture.
Human aortic valves were obtained from patients with CAS (CAS group, n = 6; age 66.5 ± 25.6 years; male/female 3:3) and patients with aortic dissection or aortic regurgitation (control group, n = 4; age 66 ± 10 years; male/female 3:1) without any signs of calcification followed by an aortic valve replacement. There was no statistically significant difference in clinical factors associated with CAS between these two groups (data not shown). All patients gave written informed consent, and this study was approved by the institutional review boards of the Hospital of Hirosaki University.
Human aortic valve specimens were gently cut into 2 ± 1-mm pieces and cultured in DMEM containing 10% fetal bovine serum (FBS). The fourth passage of cells was used in all experiments. After HAVICs reached 100% confluence, they were further cultured in the presence or absence of TNF-α for 6 or 12 days. The medium was changed every 3 days. All chemicals were of analytical grade and obtained from Wako Pure Chemicals (Osaka, Japan).
Identification of Calcification.
Cells were seeded into a 12-well plate and grown for 3 days until confluent and then they were further cultured with or without TNF-α (3–100 ng/ml) for 12 days. The degree of calcification was measured by Alizarin Red S (Puchtler et al., 1969) and Von Kossa stains (Von Kossa, 1901) at 0 and 12 days. The former method stains calcium in calcium phosphate precipitations by reacting with Alizarin Red S dye, and the latter stains phosphate in calcium phosphate precipitations by reacting with Von Kossa solution including silver nitrate. The stained cells were examined under a digital camera (Nikon, Tokyo, Japan). The amount of released Alizarin Res S dye from extracellular matrix by incubation in 100 mM aqueous cetyl-pyridinium chloride solution was quantified by spectrophotometry at 550 nm (Stanford et al., 1995).
ALP Activity Assay.
Cells were seeded into a 12-well plate and grown for 3 days until confluent, and then they were further cultured in the presence or absence of TNF-α (30 ng/ml) for 12 days. The proteins were collected from the cells at 0 and 12 days using a cell lysis buffer containing 500 μl of 0.05% Triton X-100. We used the LabAssay ALP Kit from Wako Pure Chemicals to measure ALP activity.
Measurement of Gene Expression.
Total RNA was isolated from cells using a QuickGene RNA cultured cell kit S (Fujifilm, Tokyo, Japan). An aliquot of total RNA was reverse-transcribed to obtain cDNA by using random primers. For real-time polymerase chain reaction (PCR), cDNA was amplified (ABI PRISM 7000; Invitrogen, Carlsbad, CA) under the reaction conditions: 40 cycles of PCR (95°C for 15 s, and 60°C for 1 min) after an initial denaturation (95°C for 1 min). The reaction volume was adjusted to 20 μl containing 3 μl of a 1:4 dilution of the first-strand reaction product, 0.6 μl of 10 μM specific forward and reverse primers, 0.4 μl of 50× ROX reference dye, 5.4 μl of pure water, and 10 μl of SYBR qPCR. The primers used for ALP, BMP2, tumor necrosis factor receptor superfamily member 1A (TNFRSF1A), Dlx5, Runx2, Msx2, NF-κB, and glyceraldehyde 3-phosphate dehydrogenase (G3PDH) were designed by Primer Express version 2.0 (Invitrogen), and their sequences are shown in Table 1. Amplification of the housekeeping gene G3PDH served as a normalization standard. Real-time PCR data were represented as cycle threshold levels and normalized by the individual G3PDH control cycle threshold values. Relative gene expression was calculated using the 2(−ΔΔC(T)) method (Livak and Schmittgen, 2001).
Primers used for quantitative real-time PCR
The Effects of Various Drugs on TNF-α-Induced Calcification of HAVICs.
Primary cultured cells were seeded into a 12-well plate and cultured for 3 days until confluent, and then the cells were pretreated with each inhibitor of Smads1/5/8 phosphorylation (dorsomorphin; 3 μM; Sigma-Aldrich, St. Louis, MO), NF-κB p65 subunit translocation into nucleus (SN-50, 10 μM; Merck, Darmstadt, Germany), inhibitor of nuclear factor κ-B kinase subunit β activity (4-(2′-aminoethyl)amino-1,8-dimethylimidazo[1,2-a]quinoxaline (BMS-345541, 10 μM; Merck), and MAP kinase signaling [1,4-diamino-2, 3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U-0126, 10 μM; Cayman Chemical, Ann Arbor, MI) and trans-1-(4-hydroxycyclohexyl)-4-(fluorophenyl)-5-(2-methoxypyrimidin-4-yl)imidazole (SB239063, 3 μM; Sigma-Aldrich)] for 2 h and were subsequently treated with TNF-α (30 ng/ml) for 6 and 12 days. We examined alterations in calcification and ALP activity on day 12. The gene expression was examined by real-time PCR on day 6.
Western Blot Analysis of Smad and pSmad.
Primary cultured cells were seeded into a six-well plate and cultured for 3 days until confluent, and then these cells were pretreated with dorsomorphin (3 μM), a potent inhibitor of Smads1/5/8 phosphorylation (Anderson and Darshan, 2008) for 2 h and subsequently treated with TNF-α (30 ng/ml) for 1 h. Cytoplasmic extracts were obtained by lysing the cells in 20 mM Tris-HCl, pH 7.4. After a Bradford protein assay, proteins (each 3 μg) were resolved by SDS/polyacrylamide gel electrophoresis, separated on 8% SDS-polyacrylamide gels, and transferred to polyvinylidene difluoride membranes, Immobilon-FL (Millipore Corporation, Billerica, MA). Membranes were incubated with primary antibody diluted in 5% bovine serum albumin-Tween-Tris-buffered saline (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween 20) (TTBS) for 1 h (Smad 1:200 or pSmad 1:500). Membranes were then washed three times with TTBS buffer (10 min each) and incubated with Alexa Fluor 680 goat anti-rabbit secondary antibody for 1 h at room temperature. After three washes with TTBS buffer, proteins were detected using the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE).
Statistical Analysis.
All statistical analyses were carried out using KyPlot 5.0 software (Kyenslab, Tokyo, Japan). Group comparisons were performed by analysis of variance with the Student–Newman–Keuls post hoc correction procedure. Values are presented as means ± S.E.M. P < 0.05 was considered to indicate statistical significance.
Results
Effects of TNF-α on the Calcification of HAVICs.
To determine whether TNF-α effects the calcification of HAVICs, HAVICs obtained from patients with CAS (CAS group; age 66.5 ± 25.6 years; n = 6) and patients with aortic dissection or aortic regurgitation (control group; age 66.0 ± 10.0 years; n = 4) were cultured in the presence or absence of TNF-α for 12 days after reaching confluence. Calcification of aortic valve obtained from the control group was not detected by a computed tomography scanning. We confirmed TNF-α-induced calcification of HAVICs from the CAS group was dose-dependent, and the most suitable concentration of TNF-α was 30 ng/ml (Supplemental Fig. 1). HAVICs in the CAS group were positively stained by both Alizarin Red S (Fig. 1A) and Von Kossa staining (Fig. 1B), indicating the acceleration of calcification by TNF-α (30 ng/ml) treatment. The spectrophotometric quantification using the Alizarin Red S dye showed that the calcification was accelerated by TNF-α in the CAS group, but those significant changes were not observed in the control group (Fig. 1C). We further examined whether TNF-α-induced calcification correlated to changes in its ALP activity. After HAVICs were cultured with TNF-α for 12 days, ALP activity was significantly increased in the CAS group, but not in the control group (Fig. 1D). These data suggest that TNF-α accelerated calcification and ALP activity of HAVICs from patients with CAS and these cells had greater sensitivity to TNF-α than those in the control group.

TNF-α accelerates calcification of HAVICs obtained from patients with CAS. HAVICs were cultured in DMEM containing 10% FBS. After reaching confluence (day 0), HAVICs were further cultured for 12 days (day 12). A and B, typical images of Alizarin Red S staining (A) and Von Kossa staining (B) of HAVICs from a patient with CAS (67-year-old woman) and a control patient (70-year-old man; aortic dissection) in the presence or absence of TNF-α (30 ng/ml). C, quantification of Alizarin Red S staining at day 12 via extraction with cetyl-pyridinium chloride. The amount of released dye was quantified by spectrophotometry at 550 nm. All ratios were calculated versus the control group at day 0 as 100%. White bars, day 0; gray bars, TNF-α (−) at day 12; black bars, TNF-α (+) at day 12. Bars represent the mean ± S.E.M. (control group: n = 4; CAS group: n = 6). Significant differences: *, P < 0.05 versus CAS group in the absence of TNF-α at day 12; #, P < 0.01 versus control group in the presence of TNF-α at day 12. D, ALP activity at day 12. The cells were solubilized with 1% Triton X-100 in 0.9% NaCl, and the supernatants were assayed for ALP activity using a commercially available kit (Laboassay ALP). One unit was defined as the activity producing 1 nmol of p-nitrophenol for 30 min. All ratios were calculated versus the control group at day 0. White bars, day 0; gray bars, TNF-α (−) at day 12; black bars, TNF-α (+) at day 12. Bars represent the mean ± S.E.M. (control group: n = 4; CAS group: n = 6). Significant differences: **, P < 0.001 versus CAS group in the absence of TNF-α at day 12; #, P < 0.01 versus control group in the presence of TNF-α at day 12.
Gene Expression of Calcigenic Makers on TNF-α-Induced Calcification of HAVICs.
We investigated gene expression of various calcigenic markers to confirm the intracellular signaling pathway of TNF-α-induced calcification in HAVICs. Although no significant time-dependent changes in gene expression at day 12 were noted, we confirmed that the gene expression of ALP and BMP2 was significantly increased by TNF-α in the CAS group at day 6 in comparison with those in the control group (Fig. 2, A and B). Gene expression of Runx2 did not change, but a higher basal level was maintained than that of the control group (Fig. 2C). On the other hand, Dlx5, a transcription factor of the Smad pathway that is closed to Runx2 activity, had significantly increased gene expression in the presence of TNF-α in the CAS group at day 6 (Fig. 2D). These data indicate the possibility that TNF-α-induced calcification of HAVIC from the CAS group proceeds through the Smad pathway and Dlx5 plays an important role in the acceleration of aortic valve calcification by TNF-α.
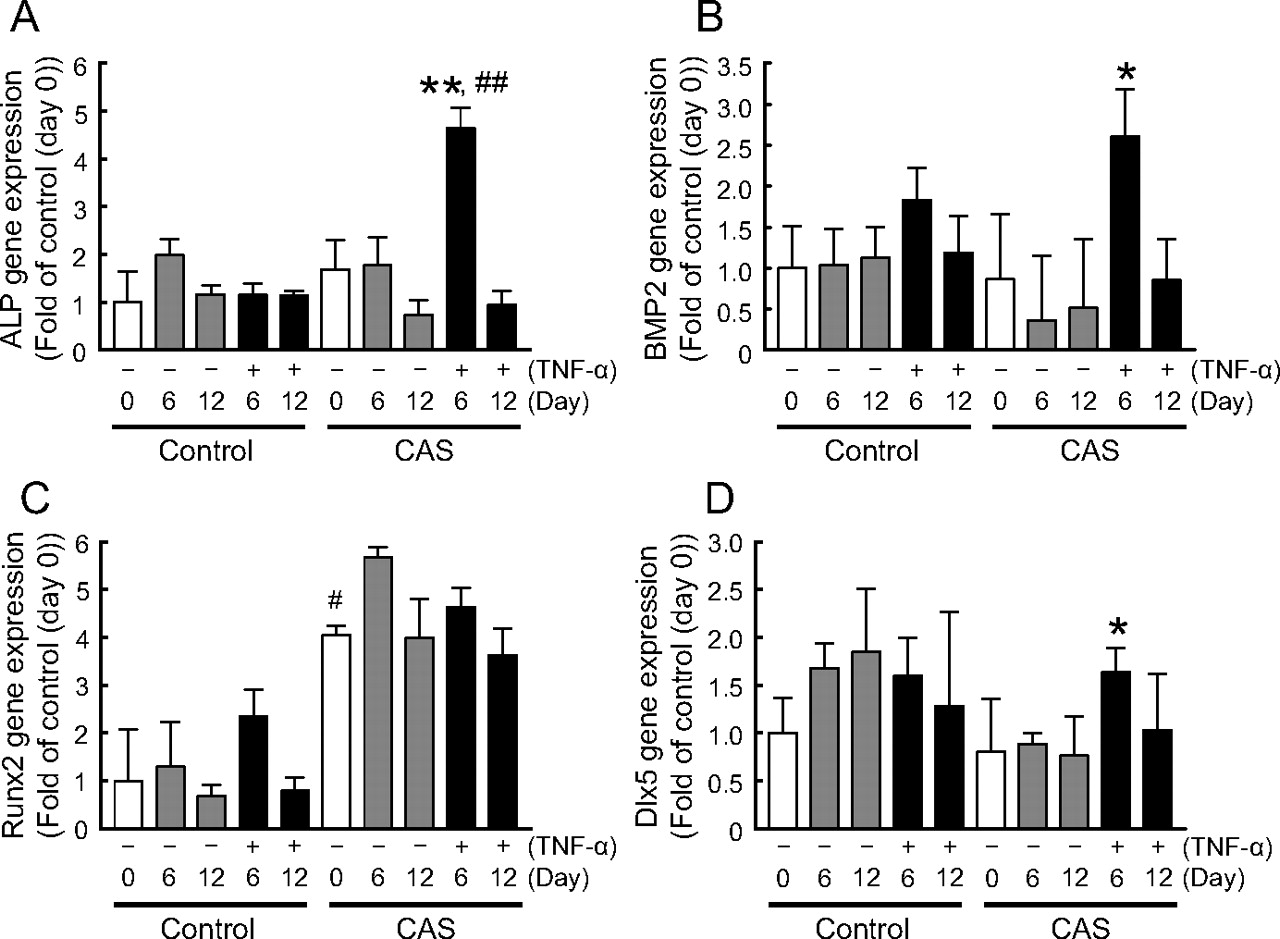
Real-time PCR analysis of calcigenic mRNA expression in HAVICs from CAS and control groups. After reaching confluence (day 0), HAVICs were further cultured in DMEM containing 10% FBS for 6 (day 6) or 12 (day 12) days. Gray and black bars represent the mRNA levels in HAVICs in the absence and presence of TNF-α, respectively. ALP (A), BMP2 (B), Runx2 (C), and Dlx5 (D) gene expression in HAVICs from the CAS and control groups was measured at days 6 and 12. All ratios were calculated versus the control group at day 0. Gene expression levels were normalized by the G3PDH gene. Bars represent the mean ± S.E.M. (control group: n = 4; CAS group: n = 6). Significant differences: *, P < 0.05 and **, P < 0.001 versus CAS group in the absence of TNF-α at day 6; #, P < 0.05 versus control group at day 0; ##, P < 0.01 versus control group in the presence of TNF-α at day 6.
TNF-α-Induced BMP2 Gene Expression in HAVICs via the NF-κB Signaling Pathway.
To determine the mechanism of TNF-α-induced BMP2 gene expression in HAVICs, we first investigated the gene expression of TNFRSF1A, a receptor of TNF-α (Fig. 3C). However, this was not enhanced by TNF-α in the CAS group. We further investigated the effects of inhibitor of NF-κB p65 subunit translocation into the nucleus, SN-50 (10 μM), and inhibitor of nuclear factor κ-B kinase subunit β inhibitor, BMS-345541 (10 μM) on TNF-α-induced calcification of HAVICs using Alizarin Red S staining. Both SN-50 and BMS-345541 significantly inhibited TNF-α-induced calcification of HAVICs from the CAS group at day 12 (Fig. 3, A and B). These inhibitors strongly inhibited TNF-α-induced gene expression of NF-κB and BMP2 (Fig. 3, D and E). These results suggested that TNF-α-induced BMP2 gene expression in HAVICs proceeds via the NF-κB signaling pathway. Although the MAP kinase pathway is another pathway possibly involved in aortic valve calcification, two inhibitors of this pathway, U-0126 (10 μM, ERK inhibitor) and SB239063 (3 μM, p38 kinase inhibitor), did not inhibit the TNF-α-induced calcification of HAVICs from the CAS group (Supplemental Fig. 2).

TNF-α-induced calcification was inhibited by NF-κB inhibitors. A and B, HAVICs were cultured in DMEM containing 10% FBS in the presence or absence of SN-50 (A) or BMS-345541 (BMS; B). After reaching confluence (day 0), HAVICs were further cultured for 12 days (day 12). Calcification of HAVICs from the CAS group was measured and quantified using Alizarin Red S staining. All ratios were calculated versus the CAS group at day 0 as 100%. White bars, day 0; gray bars, TNF-α (−) at day 12; black bars, TNF-α (+) at day 12; horizontal bars, TNF-α (+) + SN-50 (+) at day 12; hatched bars, TNF-α (+) + BMS-345541 (+) at day 12. Bars represent the mean ± S.E.M. (CAS group: n = 6). Significant differences: #, P < 0.05 versus TNF-α (−) at day 12; *, P < 0.05 versus TNF-α alone at day 12. C, real-time PCR analysis of TNFRSF1A mRNA expression in HAVICs from control and CAS groups at day 6. All ratios were calculated versus the control group at day 0. Gene expression levels were normalized by the G3PDH gene. White bars, day 0; gray bars, TNF-α (−) at day 6; black bars, TNF-α (+) at day 6. Bars represent the mean ± S.E.M. (control group: n = 4; CAS group: n = 6). D and E, real-time PCR analysis of NF-κB (D) and BMP2 (E) mRNA expression in HAVICs from CAS groups at day 6. All ratios were calculated versus the CAS group at day 0. Gene expression levels were normalized by the G3PDH gene. White bars, day 0; gray bars, TNF-α (−) at day 6; black bars, TNF-α (+) at day 6; horizontal bars, TNF-α (+) + SN-50 (+) at day 6; hatched bars, TNF-α (+) + BMS-345541 (+) at day 6. Bars represent the mean ± S.E.M. (CAS group: n = 6). Significant differences: #, P < 0.05 and ##, P < 0.01 versus TNF-α (−) at day 6; *, P < 0.05 versus TNF-α alone at day 6.
Effect of Dorsomorphin on TNF-α-Induced Calcification of HAVICs from the CAS Group.
We used dorsomorphin, an inhibitor of Smads1/5/8 phosphorylation, to confirm whether TNF-α-induced calcification of HAVICs from the CAS group occurs via the Smad pathway. In the presence of dorsomorphin, TNF-α-induced calcification (Fig. 4A; Supplemental Fig. 3), ALP activation (Fig. 4B), and gene expression of Dlx5 (Fig. 5B) were significantly inhibited. Gene expression of ALP (Fig. 4C) and BMP2 (Fig. 5C) induced by TNF-α was decreased in the presence of dorsomorphin in spite of the lack of significance. Furthermore, in the Western blot analysis, we confirmed that TNF-α (30 ng/ml) significantly increased the level of pSmad1/5/8 (active form of Smads) in HAVICs from the CAS group within 1 h of treatment, which was significantly inhibited in the presence of dorsomorphin (3 μM) (Fig. 5A). On the other hand, dorsomorphin did not affect the gene expression of Runx2 (Fig. 5D) in HAVICs from the CAS group. In addition, two NF-κB inhibitors, SN-50 and BMS-345541, acted the same as dorsomorphin on the TNF-α-induced ALP activation, expression of ALP and Dlx5 genes, and Smad phosphorylation (Supplemental Fig. 4). These data suggest that TNF-α induces calcification of HAVICs from the CAS group via the BMP2-Dlx5 pathway together with the ALP activation.
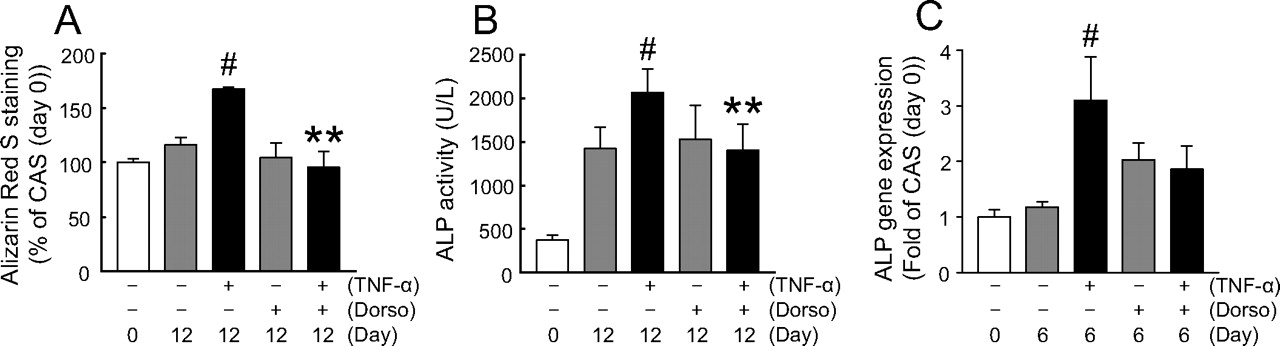
Dorsomorphin inhibited calcification and ALP activity of HAVICs from the CAS group induced by TNF-α. A, quantification of Alizarin Red S staining of HAVICs from the CAS group at days 0 or 12 in the presence or absence of TNF-α (30 ng/ml) or dorsomorphin (3 μM). The amount of released dye was quantified by spectrophotometry at 550 nm. All ratios were calculated versus the control group at day 0 as 100%. White bars, day 0; gray bars, TNF-α (−) at day 12; black bars, TNF-α (+) at day 12. Bars represent the mean ± S.E.M. (n = 6). Significant differences: **, P < 0.001 versus TNF-α alone at day 12; #, P < 0.05 versus TNF-α (−) and dorsomorphin (−) at day 12. B, ALP activity at day12. All ratios were calculated versus the control group at day 0. White bars, day 0; gray bars, TNF-α (−) at day 12; black bars, TNF-α (+) at day 12. Bars represent the mean ± S.E.M. (n = 6). Significant differences: **, P < 0.001 versus TNF-α alone at day 12; #, P < 0.05 versus TNF-α (−) and dorsomorphin (−) at day 12. C, real-time PCR analysis of ALP mRNA expression in HAVICs from the CAS groups at day 6 in the presence or absence of dorsomorphin (dorso, 3 μM). All ratios were calculated versus the control group at day 0. Gene expression levels were normalized by the G3PDH gene. White bars, day 0; gray bars, TNF-α (−) at day 6; black bars, TNF-α (+) at day 6. Bars represent the mean ± S.E.M. (n = 6). Significant differences: #, P < 0.05 versus TNF-α (−) and dorsomorphin (−) at day 6.

Dorsomorphin inhibited Smad phosphorylation and gene expression of Dlx5 of HAVICs from the CAS group induced by TNF-α. A, inhibition of TNF-α-induced phosphorylation of Smads1/5/8 by dorsomorphin. HAVICs were cultured in DMEM containing 10% FBS. After reaching confluence (day 0), HAVICs were further cultured in DMEM containing 10% FBS and TNF-α (30 ng/ml) for 1 h in the presence or absence of dorsomorphin (3 μM). Smads1/5/8 phosphorylation was determined by Western blot. Band intensity was normalized to the mean value obtained from the nontreatment group. Lane 1, nontreatment; lane 2, 30 ng/ml TNF-α; lane 3, 30 ng/ml TNF-α + 3 μM dorsomorphin. Values are the mean ± S.E.M. of three experiments. *, P < 0.05 versus TNF-α (+), #, P ≤ 0.05 versus nontreatment. B to D, real-time PCR analysis of Dlx5 (B), BMP2 (C), and Runx2 (D) mRNA expression in HAVICs from CAS groups at day 6 in the presence or absence of dorsomorphin (dorso, 3 μM). All ratios were calculated versus the CAS group at day 0. Gene expression levels were normalized by the G3PDH gene. White bars, day 0; gray bars, TNF-α (−) at day 6; black bars, TNF-α (+) at day 6. Bars represent the mean ± S.E.M. (n = 6). Significant differences: *, P < 0.05 versus TNF-α alone at day 6; #, P < 0.05 versus TNF-α (−) and dorsomorphin (−) at day 6.
Discussion
It is well known that calcification occurs frequently in the aortic valves of CAS. Previous studies have demonstrated the presence of TNF-α in the extracellular matrix of calcified valve tissue. Because aortic valve calcification occurs via ongoing endothelial injury associated with inflammatory cell infiltration (Moe et al., 2004; Hess et al., 2009), TNF-α may contribute to the calcification process of the aortic valve. In our study, we demonstrated that TNF-α-induced calcification and ALP activation of HAVICs obtained from the CAS group was significantly increased. We also demonstrated that gene expression of calcigenic makers ALP and BMP2 was significantly increased by TNF-α in the CAS group. These findings suggest that HAVICs from the CAS group are more sensitive to TNF-α than those from the control group.
Generally, TNF regulates many physical process including apoptosis, cell proliferation, increasing permeability of endothelial cells, and promoting monocyte adhesion and foam cell formation by binding to TNFRSF1A (Jovinge et al., 1996). TNFRSF1A was expressed in HAVICs and may regulate the activation of NF-κB (Bouwmeester et al., 2004). A study has reported that TNF-α elevated BMP2 expression via the NF-κB pathway (Mohler et al., 2001). BMP2 was shown to be expressed by myofibroblasts and preosteoblasts in the valves where calcification was identified (Mohler et al., 2001; Yetkin and Waltenberger, 2009). Although we could not get up-regulation of the TNFRSF1A gene by TNF-α, TNF-α-induced calcification, ALP activation, and NF-κB and BMP2 gene expression were significantly inhibited in the presence of SN-50 and BMS-345541, inhibitors of NF-κB signaling. These data show that TNF-α activates the NF-κB signaling pathway and translocates NF-κB p65 subunit into the nucleus for up-regulation of the BMP2 and NF-κB genes and supports the previous findings. Another pathway for aortic calcification is via MAP kinase, which accelerates IL-6 expression (Nishimura et al., 1998). It is well known that IL-6 activates ALP, resulting in aortic valve calcification. However, two MAP kinase inhibitors, U-0126 (ERK inhibitor) and SB239063 (p38 kinase inhibitor), did not inhibit TNF-α-induced calcification, which eliminated the participation of the MAP kinase pathway on the TNF-α-induced calcification.
The downstream mechanisms of BMP2 expression are complex and still unclear. The main pathways related to the aortic valve calcification are the Smad and wingless/Int (Wnt)/β-catenin pathways. Msx2, a transcription factor in the Wnt/β-catenin pathway, and Runx2, a transcription factor in the Smad pathway, have been found in calcified aortic valves (Rajamannan et al., 2005; Yang et al., 2009) and play an important role in osteoblast differentiation and bone formation. However, in our study, gene expression of Msx2 was not affected by TNF-α (data not shown). Although Runx2 gene expression was not increased by TNF-α, HAVICs from the CAS group had greater Runx2 gene expression than those in the control group. Furthermore, gene expression of Dlx5 was increased in the CAS group and was significantly inhibited by not only dorsomorphin, an inhibitor of Smads1/5/8 phosphorylation, but also two NF-κB inhibitors, SN-50 and BMS-345541. TNF-α-induced calcification, ALP activation, and Smad phosphorylation were also inhibited by them. Dlx5, a transcription factor in the Smad pathway, also regulates osteoblast differentiation and bone formation in other tissues such as jaw subdivision and the thoracic aorta (Depew et al., 2002; Doherty et al., 2004). Our data first demonstrated that TNF-α-induced calcification of aortic valves proceeds via the BMP2-Dlx5 pathway. Lee et al. (2005) suggested that Dlx5 is a direct target gene of BMP2 signaling and Dlx5 is a key upstream regulator of Runx2 by transactivating its distal promoter. It is possible that HAVICs from the CAS group highly expressed the Runx2 gene and differentiated into osteoblast-like cells by TNF-α-induced Dlx5 expression, resulting in ALP activation and then ectopic calcification. The detailed mechanism of ALP activation and calcification of HAVICs from the CAS group induced by Dlx5 awaits further investigation.
In conclusion, we demonstrated that HAVICs in the CAS group were more sensitive to TNF-α than those in the control group, and TNF-α accelerated calcification of HAVICs from patients with CAS by elevating ALP activity via the BMP2-Dlx5 pathway. As depicted in Fig. 6, TNF-α may accelerate BMP2 gene expression mainly via NF-κB signaling and translocation of the NF-κB p65 subunit into the nucleus. BMP2 stimulates the Dlx5 gene expression, resulting in ALP activation followed by ectopic calcification via Runx2 activation. It is hoped that these results will help to further clarify the mechanism of aortic valve calcification and develop new therapies for aortic valve stenosis.

A possible mechanism of TNF-α-induced calcification of HAVICs obtained from the CAS group. The filled arrows indicate the pathway supported by the data, and the open arrows indicate alternative pathway that are not supported by the data. TNF-α accelerates BMP2 gene expression via translocation of NF-κB into the nucleus, which stimulates the Dlx5 gene expression, resulting in ALP activation followed by calcification, via Runx2 activation. It is well known that TNF-α stimulates MAP kinase signaling, resulting in ALP activation. However, two MAP kinase inhibitors (U-0126 and SB239063) did not inhibit TNF-α-induced calcification (Supplemental Fig. 2). Although Msx2 produced by the Wnt/β-catenin pathway can also stimulate ALP gene expression, in HAVICs from the CAS group, BMP2 did not affect Msx2 gene expression.
Authorship Contributions
Participated in research design: Yu, Seya, Daitoku, Motomura, Fukuda, and Furukawa.
Conducted experiments: Yu, Seya, Daitoku, Motomura, Fukuda, and Furukawa.
Contributed new reagents or analytic tools: Yu, Seya, Daitoku, Motomura, Fukuda, and Furukawa.
Performed data analysis: Yu, Seya, and Furukawa.
Wrote or contributed to the writing of the manuscript: Yu, Seya, Daitoku, Fukuda, and Furukawa.
Other: Motomura, Fukuda, and Furukawa acquired funding for the research.
Acknowledgments
We thank the surgeons in the Department of Thoracic Cardiovascular Surgery, Hirosaki University Graduate School of Medicine for support and Izumi Miki (Hirosaki University Graduate School of Medicine) for technical support.
Footnotes
This work was supported by the Ministry of Education, Science, Sports, and Culture of Japan [Grant-in-Aid 20590245]; and Hirosaki University Educational Improvement Promotional Aid. K.-I.F. received supported from the Research Foundation for Pharmaceutical Science (Japan).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.177915.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- CAS
- calcific aortic valve stenosis
- TNF-α
- tumor necrosis factor-α
- HAVIC
- human aortic valve interstitial cell
- ALP
- alkaline phosphatase
- FBS
- fetal bovine serum
- BMP2
- bone morphogenetic protein 2
- Dlx5
- distal-less homeobox 5
- Smad
- mothers against decapentaplegic homologs
- IL
- interleukin
- ERK
- extracellular signal-regulated kinase
- MAP
- p38 mitogen-activated protein
- NF-κB
- nuclear factor-κB
- Msx2
- muscle segment homeobox 2
- Runx2
- runt-related gene 2
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- TNFRSF1A
- tumor necrosis factor receptor superfamily
- G3PDH
- glyceraldehyde 3-phosphate dehydrogenase
- TTBS
- Tween-Tris buffered saline
- PCR
- polymerase chain reaction
- Wnt
- wingless/Int
- BMS-345541
- 4-(2′-aminoethyl)amino-1,8-dimethylimidazo[1,2-a]quinoxaline
- U-0126
- 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene
- SB239063
- trans-1-(4-hydroxy-cyclohexyl)4-(fluorophenyl)-5-(2-methoxypyrimidin-4-yl)imidazole.

- Received December 3, 2010.
- Accepted December 28, 2010.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}