


Abstract
Nicotinic acetylcholine receptors (nAChR) are diverse members of the neurotransmitter-gated ion channel superfamily and play critical roles in chemical signaling throughout the nervous system. The present study establishes the acute functional effects of bupropion, phencyclidine, and ibogaine on two human nAChR subtypes. Function of muscle-type nAChR (α1βγδ) in TE671/RD cells or of ganglionic nAChR (α3β4α5±β2) in SH-SY5Y neuroblastoma cells was measured with86Rb+ efflux assays. Functional blockade of human muscle-type and ganglionic nAChR is produced by each of the drugs in the low to intermediate micromolar range. Functional blockade is insurmountable by increasing agonist concentrations in TE671/RD and SH-SY5Y cells for each of these drugs, suggesting noncompetitive inhibition of nAChR function. Based on these findings, we hypothesize that nAChR are targets of diverse substances of abuse and agents used in antiaddiction/smoking cessation strategies. We also hypothesize that nAChR play heretofore underappreciated roles in depression and as targets for clinically useful antidepressants.
Nicotinic acetylcholine receptors (nAChR) are diverse members of the neurotransmitter-gated ion channel superfamily (see reviews, Lukas and Bencherif, 1992; Galzi and Changeux, 1994; Lindstrom, 1996; Lukas, 1998). nAChR are found throughout the nervous system, where they play critical and novel roles in physiology. nAChR are composed of multiple and diverse subtypes encoded by at least 16 distinct genes (α1-α9, β1-β4, γ, δ, and ε). Muscle-type nAChR are composed as pentamers of two α1 and one each of β1, δ, and either γ (fetal) or ε (adult) subunits. One form of ganglionic nAChR contains α3, β4, and α5 with or without β2 subunits (Lukas et al., 1993;Conroy and Berg, 1995).
Bupropion (Wellbutrin; Zyban; Glaxo Wellcome, Research Triangle Park, NC) has been well established as an antidepressant and has recently been shown to act as an aid to smoking cessation (Hurt et al., 1997). However, mechanisms of these actions of bupropion are still unclear. The current, presumed mechanism involves modulation of noradrenergic and dopaminergic systems implicated in addiction (Ascher et al., 1995).
Ibogaine (Endabuse; NDA International, New York, NY) is a putative antiaddiction drug that attenuates self-administration of cocaine and morphine in animal models and has been shown to be a competitive inhibitor of N-methyld-aspartate receptors (Popik et al., 1995). Ibogaine has been previously shown to inhibit nicotinic receptor-mediated catecholamine release from cultured chromaffin cells (Schneider et al., 1996). Ibogaine has also recently been shown to noncompetitively block function measured with22NaCl influx of ganglionic nAChR in rat pheochromocytoma PC-12 cells (concentration that inhibits response by 50% (IC50) ∼20 nM) and muscle-type nAChR in human TE671/RD cells (IC50 ∼ 2 μM;Badio et al., 1997).
Numerous studies have been done on the psychopharmacology of phencyclidine (PCP) (Marwah and Pitts, 1986). PCP-N-methyld-aspartate receptor interaction has been indicated in drug-induced models of schizophrenia (see reviews,Halberstadt, 1995; Thornberg and Saklad, 1996). PCP has also been reported as early as 1979 to interact with nAChR (Kloog et al., 1979;Albuquerque et al., 1980).
These drugs have biological and/or clinical relevance. The current studies were undertaken to examine roles that nicotinic receptors may play as targets for these drugs, in models of drug-induced psychosis, and in strategies for the development of future antiaddiction drugs.
Materials and Methods
Materials.
Bupropion HCl was purchased from Research Biochemicals International (Natick, MA). Ibogaine HCl was kindly provided by Dr. Henry Sershen (Nathan S. Kline Institute, Orangeburg, NY). PCP, carbamylcholine (carb), and common salts were purchased from Sigma (St. Louis, MO). All drugs were prepared fresh from powder the day of the assay. 86Rb+ was obtained from New England Nuclear (Boston, MA). Dulbecco’s modified Eagle’s medium, trypsin, penicillin/streptomycin solution, amphotericin B, and horse sera were obtained from Irvine Scientific (Santa Ana, CA), and fetal calf sera was obtained from Hyclone (Logan, UT).
Model Cell Lines and Cell Culture.
The human clonal cell line TE671/RD expresses muscle-type nAChR containing α1, β1,γ, and δ subunits. TE671/RD nAChR function is detectable with86Rb+ efflux assays (Lukas, 1986, 1989). The human neuroblastoma cell line SH-SY5Y expresses ganglionic nAChR containing α3, β4, and α5 with or without β2 subunits. SH-SY5Y cell nAChR function is also detectable with86Rb+ efflux assays (Lukas et al., 1993). SH-SY5Y cells also express nAChR containing α7 subunits that have high-affinity binding sites for α-bungarotoxin, but these α7-nAChR do not contribute to 86Rb+ functional responses under the conditions used here (Lukas et al., 1993; Puchacz et al., 1994).
Assays of nAChR Function.
86Rb+efflux assays with intact SH-SY5Y or TE671/RD cultured on 24-well plates were performed according to Bencherif et al. (1995). Levels of nonspecific ion flux were comparable (and unaffected by ibogaine, PCP, or bupropion) whether defined with samples containing agonist (carb) plus 100 μM d-tubocurarine or with blank samples that contained no agonist. Specific nAChR function was defined as total, experimentally determined ion flux in the presence of agonist with or without test drugs, minus nonspecific ion flux. Typical values for specific and nonspecific 86Rb+ efflux are 40,000 and 5000 cpm, respectively, for TE671/RD cell samples (∼50 μg of protein) loaded with ∼100,000 of 350,000 cpm of86Rb+ applied (quantified by Cerenkov counting at 40% efficiency). Typical values for specific and nonspecific86Rb+ efflux are 6000 and 2000 cpm, respectively, for SH-SY5Y cell samples (∼50 μg of protein) loaded with ∼60,000 of 350,000 cpm of 86Rb+ applied (quantified by Cerenkov counting at 40% efficiency).
Data Analysis.
Dose-response curves were fit to data points by the general equation Y = b + [(a−b)/(1+[c/X] n)] where Y is the observed specific86Rb+ efflux response (% of control),X is the experimental concentration of the drug,b is the lowest value of observed ion flux (typically equal to nonspecific flux), a is the maximum value of observed ion flux, c is the EC50 value for agonist dose-response profiles or the IC50 value for antagonist dose-response profiles at fixed agonist concentration, andn is the Hill coefficient (<0 for antagonist dose-response profiles; >0 for agonist dose-response profiles). Best fit, nonlinear regression least-squares curves were determined by an iterative process, and values of a, b, c, andn were derived for each experiment, normalizinga and b values to percentage of control flux. Results from replicate experiments were plotted as averages (±S.E.M.) and fit again to the logistic equation to derive the parameters reported below.
Results
Acute Effects of Drugs on nAChR Function.
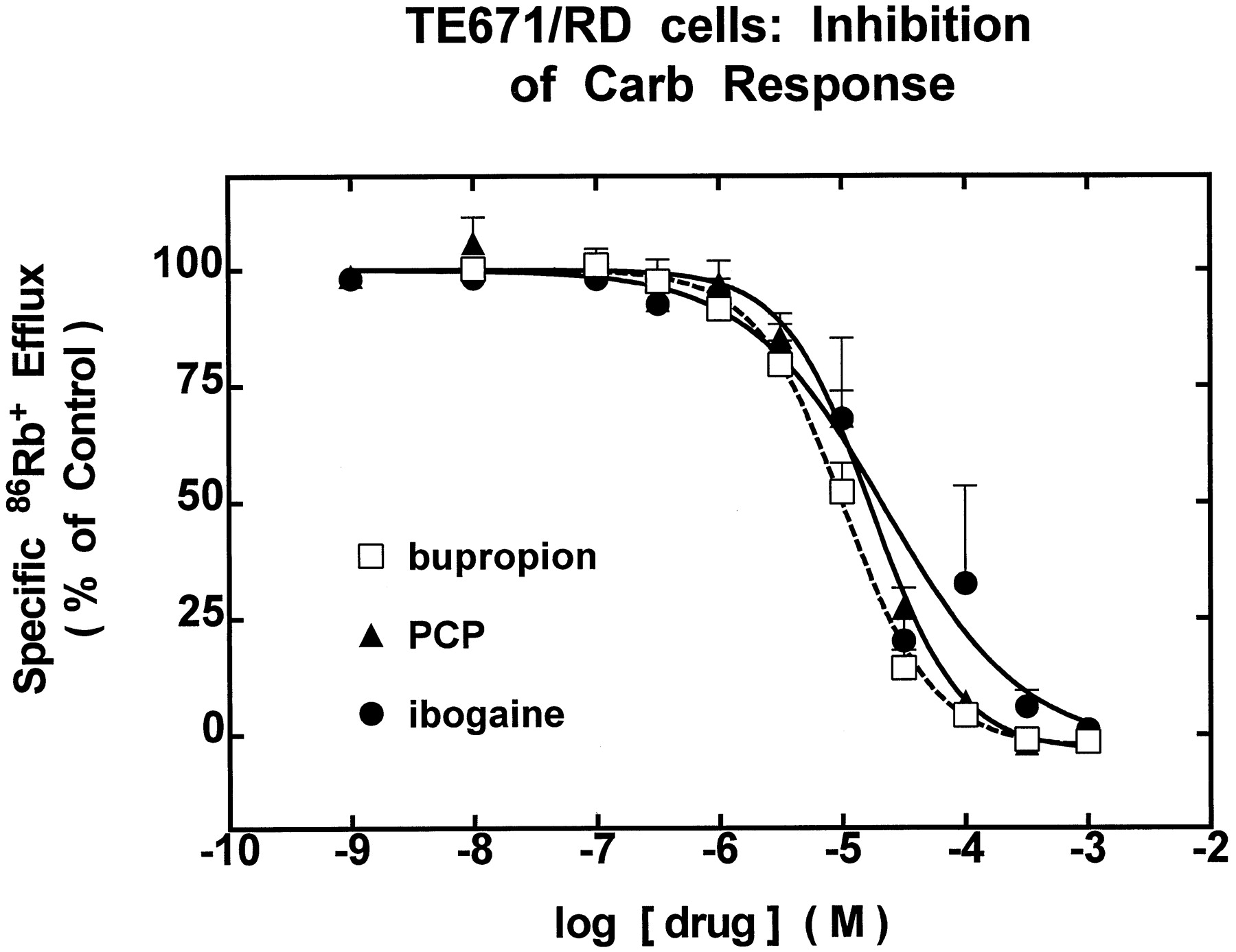
86Rb+ efflux assays with cell lines TE671/RD (muscle-type α1-nAChR) or SH-SY5Y (ganglionic α3β4-nAChR) were used to evaluate acute effects of bupropion, ibogaine, or PCP on nAChR function. In these studies, cells were exposed simultaneously to test concentrations of drug and 1 mM carb. All drugs tested produced similar dose-dependent inhibition of muscle-type nAChR function in TE671/RD cells (Fig.1), indicating half-maximal block at 10.5 μM bupropion, 17.6 μM PCP, and 22.3 μM ibogaine (see Table1). Each of the drugs also produced similar dose-dependent inhibition of ganglionic nAChR function in SH-SY5Y cells in the low micromolar range (Fig.2). Ibogaine was the most potent of these drugs, producing half-maximal inhibition at 1.1 μM, followed by bupropion (1.4 μM) and PCP (5.9 μM; see Table 1).

TE671/RD Cells: inhibition of carb response. Drug dose dependence for functional blockade of muscle-type nAChR (α1β1γδ) in TE671/RD cells. Specific86Rb+ efflux (ordinate, percentage of control) measured as described in Materials and Methods was determined in the presence of the indicated concentrations (abscissa, molar log scale) of bupropion (■), PCP (▴), or ibogaine (•). Smooth curves drawn through data points (means ± S.E.M. from at least three separate experiments) are from iterative fits to the logistic function setting a = 100% (see Materials and Methods). Results yield IC50 values indicated in the text and log IC50 values provided in Table 1. Hill coefficients and r2 values (in parentheses) are −1.19 ± 0.11 for bupropion (0.98), −1.23 ± 0.16 for PCP (0.97), and −0.77 ± 0.24 for ibogaine (0.80), respectively. Values for the parameter, b, for bupropion, PCP, and ibogaine are −2.71 ± 2.24%, −3.44 ± 3.61%, and −2.79 ± 13.6% of control86Rb+ efflux, respectively.
Inhibition constants for nAChR antagonists

SH-SY5Y cells: inhibition of carb response. Drug dose dependence for functional blockade of ganglionic α3β4-nAChR in SH-SY5Y cells. Specific 86Rb+ efflux (ordinate, percentage of control) measured as described in Materials and Methods was determined in the presence of the indicated concentrations (abscissa, molar log scale) of bupropion (■), PCP (▴), or ibogaine (•). Smooth curves drawn through data points (means ± S.E.M. from at least three separate experiments) are from iterative fits to the logistic function setting a = 100% (see Materials and Methods). Results yield IC50 values indicated in the text and log IC50values provided in Table 1. Hill coefficients andr2 values (in parentheses) are −1.45 ± 0.32 for bupropion (0.92), −0.87 ± 0.19 for PCP (0.87), and −1.00 ± 0.19 for ibogaine (0.90), respectively. Values for the parameter, b, for bupropion, PCP, and ibogaine are 3.22 ± 3.57%, −10.5 ± 6.93%, and 0.71 ± 4.02% of control86Rb+ efflux, respectively.
Mechanisms of nAChR Functional Block.
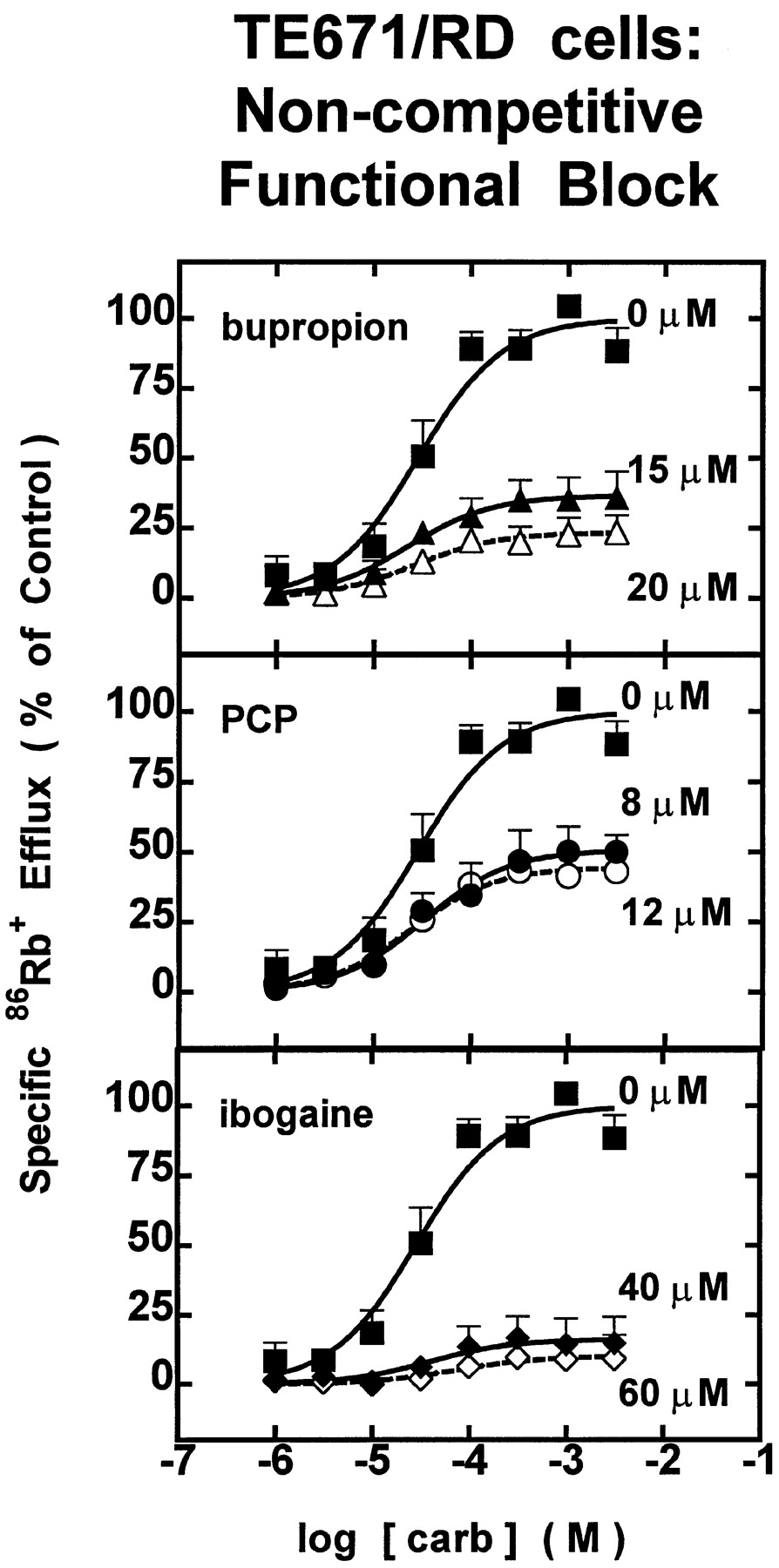
Carb dose-response profiles were obtained either alone or in the presence of bupropion, PCP, or ibogaine at concentrations near their respective IC50 values for each nAChR subtype to illuminate mechanisms of inhibition. For each of the drugs tested, the functional block produced near the IC50 value was insurmountable by increasing concentration of carb in both TE671/RD (Fig.3) and SH-SY5Y (Fig.4) cells, suggesting that these drugs act noncompetitively to inhibit nAChR function.

TE671/RD cells: noncompetitive functional block. Dose-response profiles for carb-stimulated86Rb+ efflux through TE671/RD cells expressing muscle-type nAChR (α1β1γδ) at different concentrations of test drug. Measurements of specific 86Rb+ efflux (ordinate, percentage of 1 mM carb control) were made with TE671/RD cells assayed in the presence of carb alone (▪) at the indicated doses (abscissa, molar log scale) or in the presence of carb with 15 μM (▴) or 20 μM (▵) bupropion (top panel), with 8 μM (•) or 12 μM (○) PCP (middle panel), or with 40 μM (♦) or 60 μM (⋄) ibogaine (bottom panel). Data points are from two experiments (mean ± S.E.M.). Smooth curves drawn through the data points are from iterative fits to the logistic function setting b = 0% (seeMaterials and Methods). Values for the parameter, a (as a percentage of specific 86Rb+ efflux), and r2 values (in parentheses) are 100.0 ± 5.4% (0.92) for untreated control, 36.5 ± 3.1% (0.82) for 15 μM bupropion, 23.4 ± 2.8% (0.70) for 20 μM bupropion, 50.8 ± 4.3% (0.84) for 8 μM PCP, 44.6 ± 2.4% (0.92) for 12 μM PCP, 16.2 ± 3.8% (0.43) for 40 μM ibogaine, and 10.0 ± 3.0% (0.38) for 60 μM ibogaine. None of the carb dose- response profiles produced any significant shift in EC50 value.
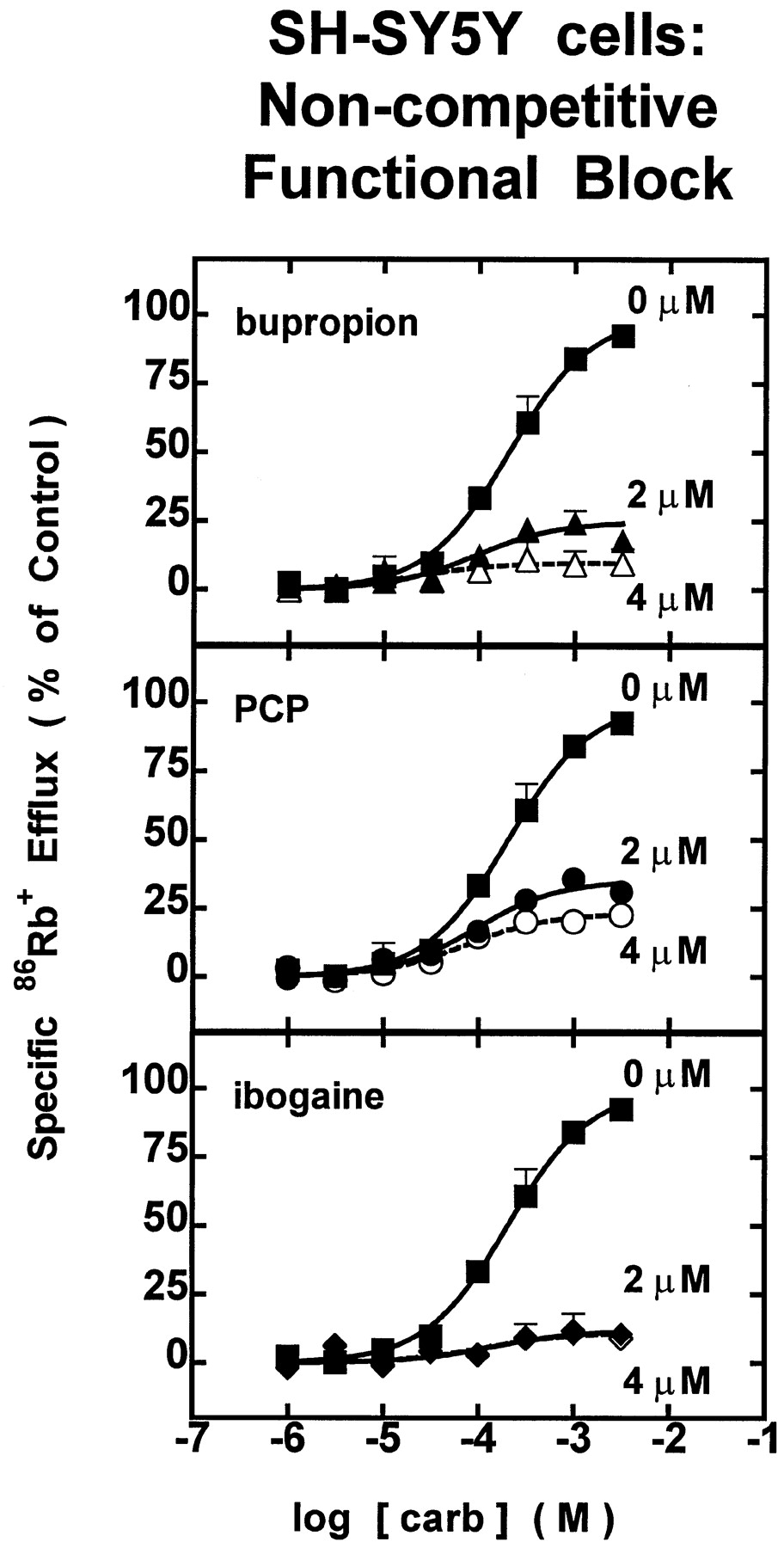
SH-SY5Y cells: noncompetitive functional block. Dose-response profiles for carb-stimulated86Rb+ efflux through SH-SY5Y cells expressing ganglionic nAChR (α3β4) at different concentrations of test drug. Measurements of specific 86Rb+ efflux (ordinate, percentage of 1 mM carb control) were made with SH-SY5Y cells assayed in the presence of carb alone (▪) at the indicated doses (abscissa, molar log scale) or in the presence of carb with 2 μM (▴) or 4 μM (▵) bupropion (upper panel), with 2 μM (•) or 4 μM (○) PCP (middle panel), or with 2 μM (♦) or 4 μM (⋄) ibogaine (lower panel). Data points are from two experiments (mean ± S.E.M.). Smooth curves drawn through the data points are from iterative fits to the logistic function setting b = 0% (seeMaterials and Methods). Values for the parameter, a (as a percentage of specific 86Rb+ efflux), andr2 values (in parentheses) are 100.0 ± 4.8% (0.97) for untreated control, 24.7 ± 2.4% (0.82) for 2 μM bupropion, 9.7 ± 2.3% (0.70) for 4 μM bupropion, 35.2 ± 2.4% (0.84) for 2 μM PCP, 23.2 ± 1.6% (0.92) for 4 μM PCP, 11.7 ± 4.2% (0.43) for 2 μM ibogaine, and 11.0 ± 2.6% (0.38) for 4 μM ibogaine. None of the carb dose-response profiles produced any significant shift in EC50 value.
Discussion
The primary findings of this study are that bupropion, PCP, and ibogaine are potent functional inhibitors of human muscle-type and ganglionic nAChR subtypes, and that this functional block is noncompetitive. We favor the hypothesis that these drugs act as open channel blockers rather than allosteric modifiers (Albuquerque et al., 1980), but the precise mechanism at the human nAChR subtypes studied is subject to clarification through electrophysiological studies. The rank order potency for muscle-type nAChR is bupropion > PCP > ibogaine, and for ganglionic nAChR is ibogaine > bupropion > PCP.
Our findings show that PCP inhibits human muscle-type and ganglionic nAChR with IC50 values of 17.6 and 5.86 μM, respectively. Yamamoto et al. (1992) report that 10 μM PCP fully blocks a slowly evolving nicotine-stimulated K+flux from nerve growth factor-differentiated rat PC12 cells expressing ganglionic nAChR. Although this value for PCP IC50 is in close agreement with our finding, the pharmacological profile for the K+ efflux response reported by Yamamoto et al. (1992) does not match that of ganglionic nAChR in PC12 cells (Lukas, 1989; compared-tubocurarine and hexamethonium sensitivities). Perhaps there are two PCP-sensitive receptors/channels in PC12 cells; nAChR and a receptor/channel with comparable PCP sensitivity mediating a slower K+ efflux response. Blood serum levels of 1.6 μM PCP have been reported with corresponding cerebrospinal fluid concentrations as high as 6 μM after high-dose intoxication (Donaldson and Baselt, 1979). Thus, inhibition of nAChR function in vivo may contribute to effects of PCP exposure and psychosis induced by this highly addictive drug.
The results of this study show that ibogaine inhibits human muscle-type and ganglionic nAChR with IC50 values of 22.3 and 1.06 μM, respectively. Previous reports have shown that low micromolar concentrations of ibogaine inhibit nicotinic receptor-mediated catecholamine release (Schneider et al., 1996). Recent reports have also shown that ibogaine inhibits22Na+ influx through human muscle-type nAChR in TE671/RD cells and rat ganglionic nAChR in PC12 cells with IC50 values of 2.0 μM and 20 nM, respectively (Badio et al., 1997). By comparison, our findings indicate substantially lower affinities of nAChR for ibogaine. It is possible that human α3β4-nAChR investigated in our study have lower affinity for ibogaine than do rat α3β4-nAChR from PC12 cells (Badio et al., 1997). Nevertheless, tissue distribution of ibogaine after i.p. or s.c. administration in rats is 109 ng/ml (314 nM) in plasma and up to 11 μg/g (∼32 μM) in fat (Hough et al., 1996). Thus, actions of ibogaine at nAChR would be predicted to occur in vivo, even if human nAChR have comparably lower affinities for the compound like those shown in our study.
Here we present the first evidence that bupropion inhibits function of nAChR. Bupropion blocks function of human muscle-type and ganglionic nAChR with IC50 values of 10.5μM and 1.44 μM, respectively. Studies have found that peak plasma levels of bupropion in humans can reach a maximum of 0.52 μM (Hsyu et al., 1997). Other studies have found that plasma levels of its major metabolite, hydroxybupropion, reach doses of 4 μM (Golden et al., 1988). Given the apparent clinical activity of hydroxybupropion as well as the extremely long half-life of bupropion and its metabolites (see review,Ascher et al., 1995), we hypothesize that antidepressant effects of bupropion reflect, in part, inhibition of nAChR function in vivo. Moreover, we suggest that bupropion’s utility as an aid to smoking cessation may also relate to its effects as an antagonist at nAChR. Orally administered mecamylamine, which is a classical nAChR antagonist, also promotes smoking cessation when used in combination with nicotine provided via transdermal patches (Rose et al., 1994). Taken collectively with the current findings, these observations suggest the general principle that functional block of nAChR, perhaps as an adjunct to nicotine replacement therapy and presumably via noncompetitive mechanisms, facilitates smoking cessation.
Based on our findings, effects of ibogaine, PCP, or bupropion at human muscle-type nAChR in vivo are likely to be modest. For example, for IC50 values of ∼10 to 20 μM and plasma/tissue concentrations for these drugs of ∼1 μM, there would be ∼5 to 10% functional inhibition of muscle-type nAChR. However, effects of these drugs in vivo would be more substantial at human ganglionic nAChR. For example, for IC50 values ∼1 μM for bupropion and ibogaine and ∼6 μM for PCP, and for tissue concentrations for these drugs of ∼1 μM, there would be ∼20 to 50% functional inhibition of ganglionic nAChR. Toward elucidation of centrally-mediated psychoactive actions of these compounds, we also are establishing effects of bupropion, PCP and ibogaine at numerically predominant brain-nAChR subtypes containing α4 and β2 or α7 subunits. Also of interest due to our current studies of α3β4-nAChR are effects of bupropion, PCP, and ibogaine at brain nAChR that contain α3 subunits. Although not numerically predominant, α3 subunits and the nAChR subtypes that contain them are located in catecholamine-rich brain regions implicated in pleasure and reward (Lukas, 1998). Hence, these central nAChR containing α3 subunits might play disproportionately important roles in drug dependence and in responses to ibogaine, PCP, and/or bupropion. Pharmacokinetic issues also bear on nicotinic actions of these drugs centrally. Brain concentrations for bupropion are 7 to 9 times higher than plasma ratios in rats, mice, and guinea pigs (Suckow et al., 1986). Given that brain concentrations of bupropion in humans may thus approach 3 to 5 μM, there is good reason to expect that bupropion and perhaps the other drugs would have pronounced effects on nAChR function in the brain in vivo.
In conclusion, bupropion, PCP, and ibogaine at bioavailable doses block function of two different nAChR subtypes. These findings are of general interest in identification of receptors involved in drug dependence and abuse. The current results also have implications in the design, discovery, and characterization of agents with potential as aids to smoking cessation, as antidepressants, and in treatment of drug abuse and dependence.
Acknowledgments
We thank Dr. Henry Sershen for his generous donation of ibogaine HCl.
Footnotes
-
Send reprint requests to: Ronald J. Lukas, Division of Neurobiology, Barrow Neurological Institute, 350 W. Thomas Road, Phoenix, AZ 85013. E-mail:rlukas{at}mha.chw.edu
-
1 This work was supported by a grant from the Arizona Disease Control Research Commission (9730) and by faculty endowment and laboratory capitalization funds from the Men’s and Women’s Boards of the Barrow Neurological Foundation. The contents of this report are solely the responsibility of the authors and do not necessarily represent the views of the aforementioned rewarding agencies.
- Abbreviations:
- nAChR
- nicotinic acetylcholine receptors
- PCP
- phencyclidine
- carb
- carbamylcholine
- IC50
- concentration that inhibits response by 50%
- Received March 24, 1998.
- Accepted July 20, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}