


Abstract
The objective of the present study was to examine the cassette dosing method in determination of brain-to-plasma concentration ratio (area under the concentration-time profiles for plasma/area under the concentration-time profiles for brain, Kp). Eleven model compounds, amprenavir, citalopram, digoxin, elacridar, imatinib, (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester (Ko143), loperamide, prazosin, quinidine, sulfasalazine, and verapamil, were selected to compare their Kp determined from discrete dosing in wild-type mice and their Kp from cassette dosing in wild-type, Mdr1a/1b(−/−), Bcrp1(−/−), and Mdr1a/1b(−/−)/Bcrp1(−/−) mice at 1 to 3 mg/kg. The mice brain and plasma were collected at 0.25, 1, and 3 h and were analyzed using high-performance liquid chromatography-tandem mass spectrometry methods. The Kp determined from discrete dosing versus cassette dosing in the wild-type mice were within 2-fold for all the compounds except sulfasalazine and Ko143. The brain concentrations of sulfasalazine and Ko143 and the plasma concentrations of Ko143 were below the lower limit of quantitation. In addition, the Kp values estimated by mass spectrometry responses, namely the ratio of compound peak area to internal standard peak area, were within 2-fold of the Kp observed from the actual concentrations. Furthermore, the ratios of Kp in Mdr1a/1b(−/−), Bcrp1(−/−), and Mdr1a/1b(−/−)/Bcrp1(−/−) mice versus the Kp in the wild-type mice from cassette dosing were consistent with the ones reported in the literature where the compounds were dosed discretely. These results demonstrate that drug-drug interactions at the blood-brain barrier are unlikely at a subcutaneous dose of 1 to 3 mg/kg and support the use of the cassette dosing approach to assess brain penetration in drug discovery.
Introduction
The blood-brain barrier (BBB) consists of a continuous layer of endothelial cells joined by tight junctions at the cerebral vasculature. It represents a physical, enzymatic, and transporter barrier to restrict and regulate the penetration of compounds into and out of the brain (Davson and Segal, 1995). The main mechanisms limiting the delivery of drugs from blood into the brain are that the BBB exhibits very low paracellular permeability and expresses multiple drug transporters. Two efflux drug transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (Bcrp) are the main efflux transporters expressed at the luminal side of the BBB, and their functional importance in limiting brain penetration of various compounds has been demonstrated (Schinkel et al., 1994; Chen et al., 2003; Breedveld et al., 2005; Enokizono et al., 2007; Polli et al., 2009; Zhou et al., 2009; Agarwal et al., 2011). In mice, P-gp is the product of Mdr1a (Abcb1a) and Mdr1b (Abcb1b) genes, and Bcrp is the product of Bcrp1 (Abcg2) gene (Schinkel, 1999; Scherrmann, 2005).
A useful parameter to assess the efficiency of a drug to cross the BBB is the ratio of unbound brain concentration to unbound plasma concentration (Kp,uu) (Liu et al., 2008; Hammarlund-Udenaes et al., 2009). The common method to estimate Kp,uu is to determine in vivo plasma and brain concentrations (Kp) and in vitro unbound fraction in plasma and brain tissue (Maurer et al., 2005). Kp can be determined at steady state after intravenous infusion or from the area under the curve (AUC) of brain and plasma concentrations after a single dose. These experimental approaches are low throughput and resource-intensive. In the present study, we evaluated the cassette dosing approach to increase the throughput and reduce resource consumption in determination of Kp.
Traditionally pharmacokinetic parameters were generated by dosing compounds discretely. To increase the throughput of pharmacokinetic studies, cassette dosing (also called N-in-1 dosing) has been used in animal pharmacokinetic studies in drug discovery screening (Manitpisitkul and White, 2004). Extensive research work has been published to assess the cassette dosing approach for screening systemic pharmacokinetics in drug discovery. However, much less original research work has been published to evaluate cassette dosing to study brain penetration. Frick et al. (1998) briefly described in a review for the cassette dosing method to study a series of compounds in mice and observed good agreement between the cassette dosing and discrete dosing. However, neither the compounds nor the methods were disclosed in the review. Zhang et al. (2004) reported use of cassette dosing with three to four compounds at 3 mg/kg for each compound in each cassette to determine Kp, but the authors did not compare the Kp between discrete and cassette dosing and did not disclose the compound structures or their transport characteristics.
Although it is plausible that drug transporter substrates and inhibitors may incidentally exist in one cassette and the brain penetration for the drug transporter substrates could be modified by the inhibitors, we hypothesized that if cassette dosing is conducted at a low dose such as at 1 to 3 mg/kg, the possibility of drug-drug interactions at the BBB may be low. To test this hypothesis, we selected a set of 11 compounds, namely amprenavir, citalopram, digoxin, elacridar, imatinib, (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester (Ko143), loperamide, prazosin, quinidine, sulfasalazine, and verapamil, and examined whether there is any difference for their Kp by dosing and analyzing the 11 compounds individually and by dosing and analyzing them as a cassette in mice. This set of compounds contains known potent P-gp and Bcrp inhibitors and typical P-gp and Bcrp substrates to create the “worst” scenario of potential drug-drug interactions at the BBB. We also examined whether we can estimate the Kp directly from mass spectrometer responses without using any standard curves to quantitate the actual plasma and brain concentrations. Furthermore, we assessed brain penetration of the 11 compounds in P-gp knockout mice [Mdr1a/1b(−/−)], Bcrp knockout mice [Bcrp1(−/−)], and P-gp and Bcrp knockout mice [Mdr1a/1b(−/−)/Bcrp1(−/−)] using the cassette dosing approach.
Materials and Methods
Chemicals.
Citalopram, digoxin, loperamide, prazosin, quinidine, sulfasalazine, and verapamil were obtained from Sigma-Aldrich (St. Louis, MO). Amprenavir, amprenavir-D4, elacridar, and imatinib were obtained from Toronto Research Chemicals, Inc. (North York, ON, Canada). Ko143 was obtained from Enzo Life Sciences, Inc. (Farmingdale, NY). All chemicals used in the experiments were of the highest available grade.
Animal Experiments.
Male wild-type (FVB), P-gp knockout [Mdr1a/1b(−/−)], Bcrp1 knockout [Bcrp1(−/−)], and P-gp/Bcrp knockout mice [Mdr1a/1b(−/−)/Bcrp1(−/−)] of approximately 9 weeks of age, weighing 25 to 30 g, were obtained from Taconic Farms (Germantown, NY). Upon arrival, the mice were maintained for at least 5 days on a 12-h light/dark cycle in a temperature- and humidity-controlled environment with free access to food and water. In the discrete dosing study, FVB mice were administered a single subcutaneous dose (n = 3/time point) of a single compound at 1 mg/kg for citalopram, elacridar, imatinib, loperamide, prazosin, and verapamil and at 3 mg/kg for amprenavir, quinidine, Ko143, digoxin, and sulfasalazine. In the cassette dosing study, FVB, P-gp, Bcrp, and P-gp/Bcrp knockout mice were administered a single subcutaneous dose (n = 3/time point) of a mixture of 11 compounds at 1 mg/kg for citalopram, elacridar, imatinib, loperamide, prazosin, and verapamil and at 3 mg/kg for amprenavir, quinidine, Ko143, digoxin, and sulfasalazine. Dosing solutions of each drug were prepared in 100% N-methyl-2-pyrrolidone and were dosed at 1 ml/kg. Mice were euthanized in a CO2 chamber at 0.25, 1, and 3 h postdose. Whole blood was collected by cardiac puncture into Costar tubes (Corning, Inc., Corning, NY) containing heparin and was stored on ice until centrifuged for the preparation of plasma. Whole brains were collected by decapitation, rinsed in phosphate-buffered saline, weighed, and immediately frozen on dry ice. All studies were conducted in accordance with approved Genentech Animal Care and Use Procedures.
Sample Analysis.
Standard curves and quality control samples were prepared by spiking a known amount of a mixture of the 11 compounds into a blank mixed matrix of mouse plasma and brain homogenate (1:1 v/v). The brain tissue of each mouse was homogenized in 4 volumes (w/v) of water. Twenty-five microliters of plasma sample was mixed with 25 μl of blank brain homogenate, and 25 μl of brain homogenate sample was mixed with 25 μl of blank plasma. A total 50 μl of samples, 50 μl of calibration standards, or 50 μl of quality controls was mixed with 15 μl of internal standard (amprenavir-D4) and 150 μl of acetonitrile. After vortexing and centrifugation at 1500g for 10 to 15 min, 150 μl of supernatant was transferred to a 96-well plate and was diluted with 50 μl of water before analysis by high-performance liquid chromatography combined with tandem mass spectrometry.
Samples were analyzed using two sets of standard curves and two sets of quality controls in each analytical run. The system consisted of an Accela pump (Thermo Fisher Scientific, Waltham, MA), an HTS-PAL autosampler (Leap Technologies, Chapel Hill, NC), and an AB Sciex API 5000 (AB Sciex, Foster City, CA) mass spectrometer with a turbo ion spray interface. A 20-μl aliquot of each sample was injected onto a reverse-phase HALO C18 column. The lower limit of quantitation (LLOQ) for the compounds in the plasma and brain homogenate ranged from 0.122 to 7.80 ng/ml. The assay accuracy was between 75 and 125%.
Data Analysis.
The total brain drug concentration was corrected for the residual blood in the brain vasculature by subtracting 1.03% of the plasma concentration determined in the corresponding samples (Fridén et al., 2010). The AUC values were calculated using the trapezoid rule from 0 to 3 h.
Results
Comparison of Kp from Discrete Dosing and Cassette Dosing in Wild-Type Mice.
Plasma and brain concentrations of the 11 model compounds after a single subcutaneous administration at 1 or 3 mg/kg in either discrete or cassette dosing were above the detection limits except Ko143 and sulfasalazine. The plasma and brain concentrations of Ko143 were below its LLOQ (0.244 ng/ml for the plasma and 1.22 ng/g or ng/ml for the brain), although the chromatographic peaks were able to be identified in the brain samples. The plasma concentrations of sulfasalazine were above its LLOQ (7.8 ng/ml), but the brain concentrations were below its LLOQ (39 ng/g). The shape of the plasma and brain concentration-time profiles appeared similar for the discrete and cassette dosing (Fig. 1). The AUC values for plasma and brain from the discrete and cassette dosing are listed in Table 1. The Kp values from the discrete and cassette dosing are listed in Table 2 and are presented in Fig. 2. Although the plasma or brain AUC values were different between the discrete and cassette dosing groups, their Kp values were similar between the two groups (Tables 1 and 2). A good correlation was observed for the Kp from the discrete dosing and the Kp from cassette dosing with R2 = 0.99 on a logarithmic scale. The Kp values from the cassette dosing were within 2-fold of the Kp from the discrete dosing for 9 of the 11 compounds. For sulfasalazine, its brain concentrations were below the LLOQ, so both cassette and discrete dosing demonstrated essentially no or low brain penetration, and Kp for this compound can be considered as similar in cassette and discrete dosing. The correlation for Ko143 Kp from cassette and discrete dosing cannot be assessed because its plasma and brain concentrations were below the LLOQ. The above results demonstrate that cassette dosing and discrete dosing generate similar Kp values.

Mouse plasma and brain concentration-time profiles of 10 compounds after a discrete or cassette dose of 1 to 3 mg/kg. The plasma and brain concentrations of Ko143 were below the LLOQ values. Circles and squares represent plasma and brain concentrations, respectively; closed and open symbols represent discrete and cassette dosing, respectively. Data points represent mean and S.D. from triplicate experiments.
Mouse brain and plasma AUC(0–3 h) quantified as concentration or as mass spectrometer response following a discrete or cassette dose of the 11 compounds
Brain and plasma AUC(0–3 h) ratio observed in wild-type, P-gp, Bcrp, or P-gp/Bcrp knockout mice
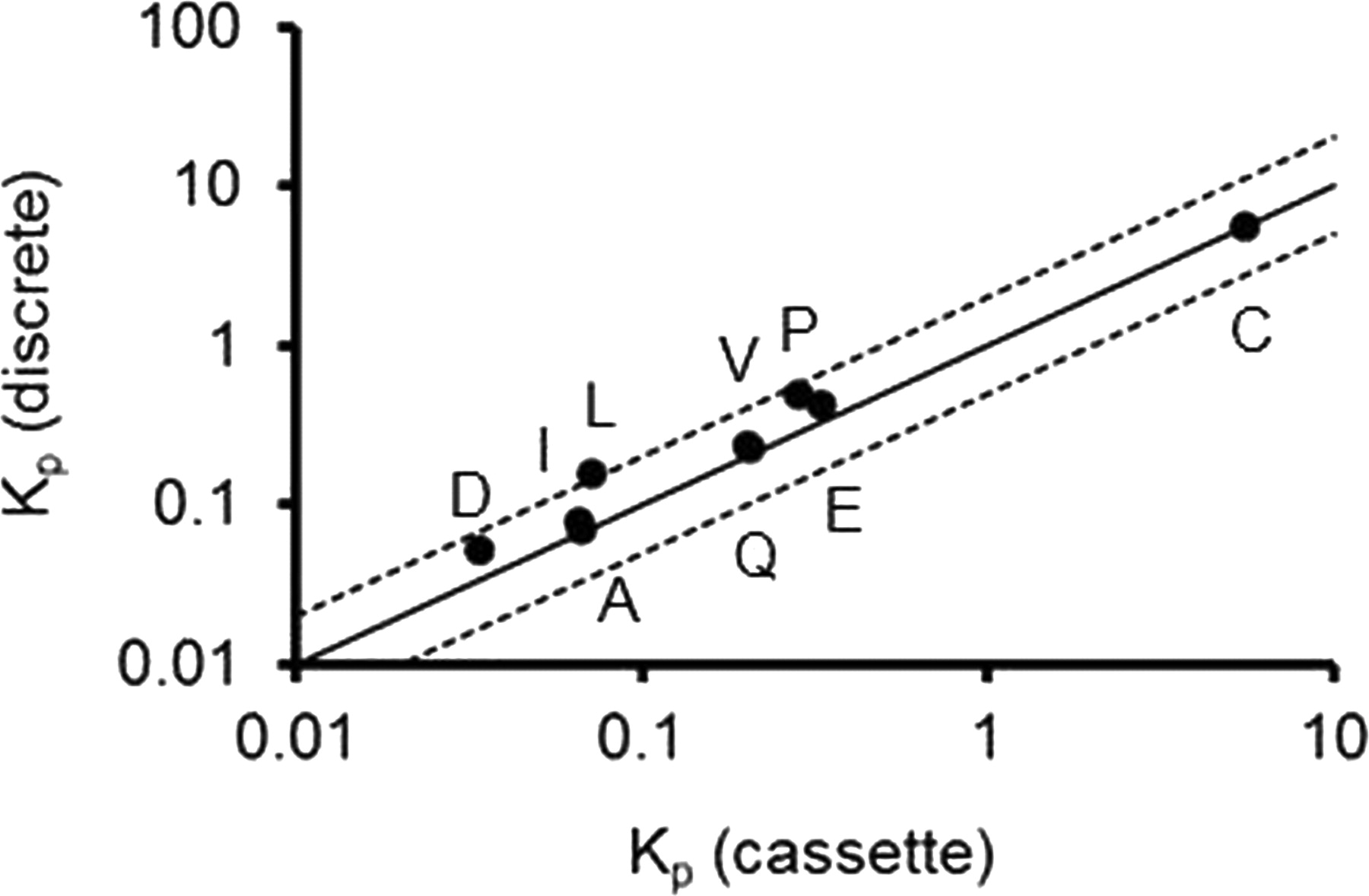
Relationship between Kp determined from discrete dosing and cassette dosing of nine compounds. The solid and dotted lines represent unity and 2-fold of error. A, amprenavir; C, citalopram; D, digoxin; E, elacridar; I, imatinib; L, loperamide; P, prazosin; Q, quinidine; V, verapamil.
Comparison of Kp Determined on the Basis of Concentration Versus Mass Spectrometer Response after Discrete or Cassette Dosing in Wild-Type Mice.
We examined whether we can estimate the Kp without using standard curves to quantitate the actual brain and plasma concentrations and instead using the mass spectrometer response, namely peak area ratio between the compounds and the internal standard in the plasma and brain mixed matrix. The AUC values calculated by using the mass spectrometer response for plasma (AUCp,R) and brain (AUCb,R) in unit of response · hour (R · h) from the discrete and cassette dosing are listed in Table 1, and the corresponding Kp values are presented in Table 2. The Kp calculated from the concentrations versus the Kp calculated from the response were very similar, with correlation coefficient R2 = 0.98 for discrete dosing (Fig. 3A) and R2 = 0.97 for cassette dosing (Fig. 3B) on a logarithmic scale. The Kp values obtained from the response of mass spectrometry were within 2-fold of the Kp from the measured concentration. These results demonstrate that the Kp values estimated from the mass spectrometer response without using a standard curve are consistent with the Kp values determined from the actual concentrations.
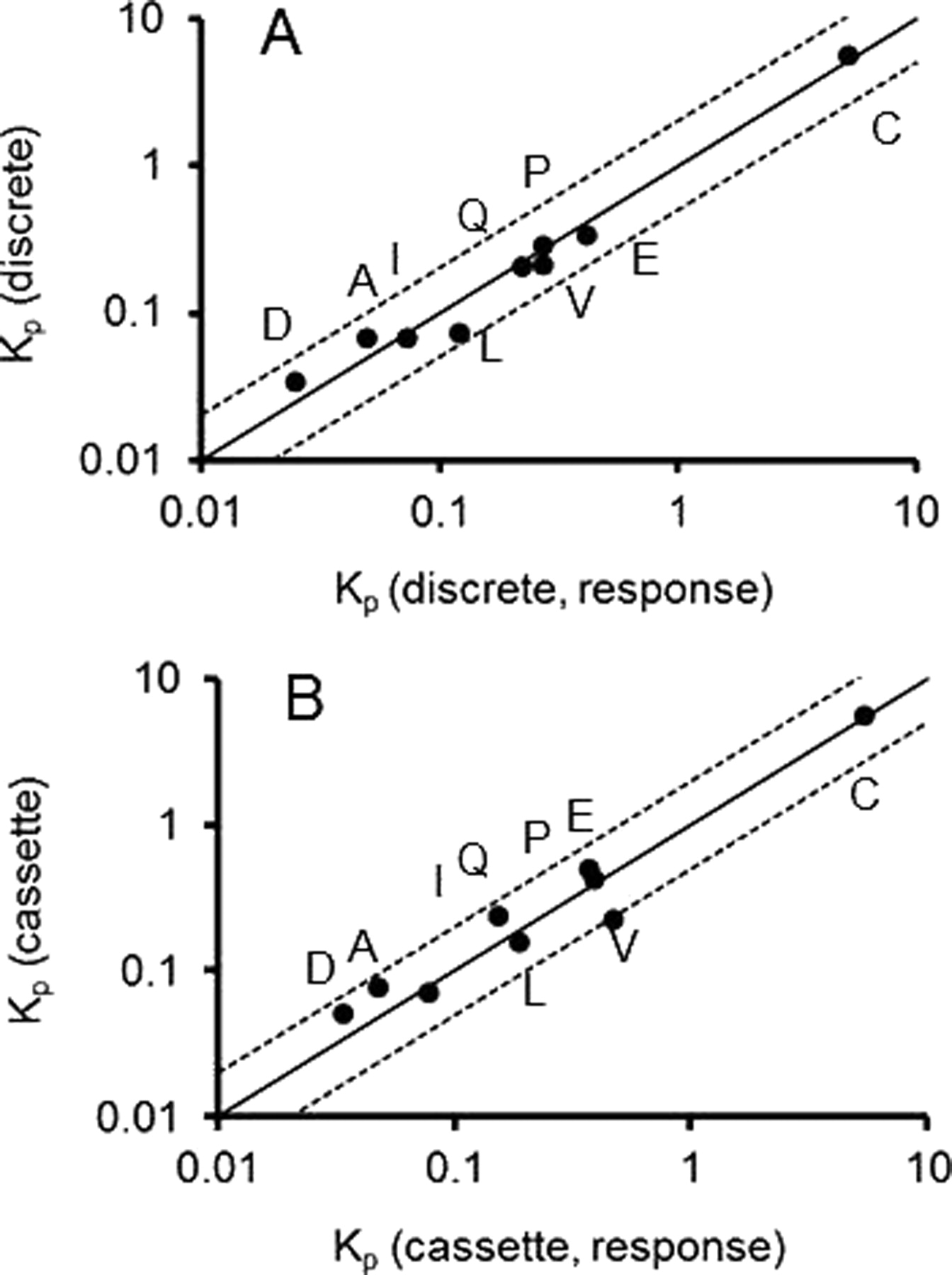
Relationship between the Kp determined from concentration, and the Kp determined from mass spectrometer response [Kp(response)] of nine compounds from discrete dosing (A) and cassette dosing (B). The solid and dotted lines represent unity and 2-fold of error. A, amprenavir; C, citalopram; D, digoxin; E, elacridar; I, imatinib; L, loperamide; P, prazosin; Q, quinidine; V, verapamil.
Brain Penetration in P-gp, Bcrp, and P-gp/Bcrp Knockout Mice after Cassette Dosing.
We examined brain penetration for the 11 compounds using the cassette dosing approach in P-gp, Bcrp, and P-gp/Bcrp knockout mice. The plasma and brain AUC values are shown in Table 1. The Kp values are listed in Table 2, and the correlation with the Kp from wild-type mice is presented in Fig. 4. The ratios of Kp in knockout mice versus the Kp from wild-type mice (KO/WT ratio) are presented in Table 3. For citalopram, the KO/WT ratios in all knockout mice were less than 2 (Table 3; Fig. 4). For Bcrp substrate, sulfasalazine, its plasma AUC increased 12- to 8-fold from 351 to 4050 ng · h/ml in Bcrp knockout mice and to 2640 ng · h/ml in P-gp/Bcrp knockout mice. However, its brain concentration was below the LLOQ in all strains of mice (Table 1). For P-gp substrates, amprenavir, digoxin, loperamide, quinidine, and verapamil, their KO/WT ratios in P-gp and P-gp/Bcrp knockout mice were similar and greater than 2 (Table 3; Fig. 4). For P-gp and Bcrp dual substrates elacridar, imatinib, and prazosin, their KO/WT ratios in P-gp/Bcrp knockout mice were greater than their KO/WT ratios in P-gp or Bcrp knockout mice (Table 3; Fig. 4). The plasma concentrations of Ko143 were below the LLOQ in all the samples. Very low amount in the wild-type mice brains was detected, but the concentrations were below the LLOQ (Table 1). However, its brain concentrations in knockout mice were above the LLOQ, and its brain concentrations in P-gp and P-gp/Bcrp mice were approximately 3-fold higher than that in Bcrp mice (Table 1). The KO/WT ratios in Bcrp knockout mice were lower or near 2 for all the compounds (Table 3; Fig. 4B).

A, relationship between the Kp in P-gp knockout mice (Kp,P-gp) versus Kp in wild-type mice (Kp). B, the Kp in Bcrp knockout mice (Kp,Bcrp) versus Kp. C, the Kp in P-gp and Bcrp knockout mice (Kp, P-gp/Bcrp) versus Kp. The solid and dotted lines represent unity and 2-fold of error, respectively. A, amprenavir; C, citalopram; D, digoxin; E, elacridar; I, imatinib; L, loperamide; P, prazosin; Q, quinidine; V, verapamil.
Ratio of Kp in P-gp, Bcrp, and P-gp/Bcrp knockout mice versus Kp in wild-type mice
Discussion
The results from the present study indicate that drug-drug interactions mediated by inhibition of efflux drug transporter P-gp or Bcrp at the BBB appear unlikely in cassette dosing at 1 to 3 mg/kg. In addition, the Kp may directly be estimated from the mass spectrometer response without using standard curves to quantitate the actual plasma and brain drug concentrations. Furthermore, the brain penetration in P-gp, Bcrp, and P-gp/Bcrp knockout mice observed in cassette dosing is similar to the values reported in the literature.
Similar Kp from Discrete Dosing and from Cassette Dosing.
We hypothesized that for cassette dosing at low doses such as subcutaneous injection of 1 to 3 mg/kg, the possibility of drug-drug interactions at the BBB is low even with compounds that are specifically designed to inhibit P-gp or Bcrp. To test this hypothesis, we selected 11 compounds including potent P-gp and Bcrp inhibitors and typical P-gp and Bcrp substrates to create the “worst” scenario of potential drug-drug interactions at the BBB. To classify the efflux effects of P-gp and Bcrp at the BBB, we used an arbitrary value 2-fold as the cutoff value (Table 4). A compound is considered a non-P-gp or Bcrp substrate if its KO/WT ratio in P-gp and Bcrp knockout mice is less than 2, a P-gp substrate if its KO/WT ratio is greater than 2 in P-gp knockout mice, a Bcrp substrate if its KO/WT ratio is greater than 2 in Bcrp knockout mice, or a P-gp and Bcrp substrate if its KO/WT ratio is greater than 2 in P-gp/Bcrp knockout mice. The 11 compounds in this study can be classified into five groups: 1) non-P-gp substrate: citalopram; 2) P-gp substrates: amprenavir, digoxin, loperamide, quinidine, and verapamil; 3) Bcrp substrate: sulfasalazine; 4) P-gp/Bcrp dual substrates: elacridar, imatinib, and prazosin; and 5) P-gp inhibitors: quinidine and verapamil; Bcrp inhibitor: Ko143; and P-gp/Bcrp dual inhibitor: elacridar.
The ratio of Kp in in vivo transport activities of 11 compounds
The Kp values obtained from the discrete and cassette dosing are within 2-fold and showed a good correlation. These data demonstrate that inhibition of the drug efflux transporters such as P-gp and Bcrp is negligible at 1 to 3 mg/kg after a cassette dose of compounds containing potent P-gp and Bcrp inhibitors and specific substrates. Our results are consistent with the results reported in the literature for two potent P-gp and Bcrp inhibitors: elacridar and Ko143. Elacridar is a third-generation P-gp inhibitor with Ki of 1.6 nM for P-gp inhibition and is also a potent Bcrp inhibitor with in vitro EC90 of 51 to 61 nM (Allen et al., 2002; Sugimoto et al., 2011). Its in vivo mouse and rat plasma IC50 for inhibition of P-gp at the BBB ranged from 114 to 282 ng/ml, and its IC50 for inhibition of Bcrp was 790 ng/ml determined in a brain perfusion study (Cutler et al., 2006; Bihorel et al., 2007; Oostendorp et al., 2009; Kuntner et al., 2010; Sugimoto et al., 2011). Thus, with 2-fold as a cutoff value for the effects of elacridar on Kp, no change for the Kp values of P-gp and/or Bcrp substrates occurs when elacridar plasma concentrations are below 114 ng/ml. In the present study, the mouse plasma concentrations for elacridar were less than 15 ng/ml from time 0 to 3 h, so it is expected no much increase for the Kp values for P-gp substrates. Ko143 is a potent and selective Bcrp inhibitor. Its cell EC90 is 23 to 26 nM (Allen et al., 2002), and its effects on the oral absorption of topotecan have been demonstrated after a 10 mg/kg oral dose. However, no studies have been reported to examine the in vivo effects of Ko143 in enhancement of the brain penetration for Bcrp substrates.
Estimation of Kp Directly from Mass Spectrometer Response.
The main objective in brain penetration study is to determine Kp and not the absolute brain and plasma concentrations. In the present study, we examined a method to estimate the Kp by using the compound chromatographic peak area relative to an internal standard peak area without determining the actual plasma and brain concentrations. This method significantly reduced the bioanalytical work because it is not required to prepare and analyze the standards. The issue for this method is that plasma and brain homogenate may have different matrix effects on the mass spectrometer responses, and therefore, the Kp determined from the responses cannot represent the Kp determined from the actual concentration. To eliminate the different matrix effects, we mixed the brain homogenate with blank plasma and mixed plasma with blank brain homogenate so the matrices were identical for the brain homogenate and plasma samples. The Kp values calculated from the instrument response were almost identical to the ones from the measured concentration, supporting the use of this simplified bioanalytical method in drug discovery. A similar analytical approach has been used in the determination of unbound fractions in brain homogenate or slices where the absolute concentrations are not critical but the concentration ratios are important (Fridén et al., 2011).
Similar Brain Penetration after Discrete or Cassette Dosing in P-gp, Bcrp, and P-gp/Bcrp Knockout Mice.
Our data from the cassette dosing method are consistent with literature results in classifying whether the brain penetration of a compound is impaired by P-gp or Bcrp or both P-gp and Bcrp. The observed KO/WT ratio of citalopram in P-gp knockout mice was 1.9 (Doran et al., 2005). In the present study, the KO/WT ratio of citalopram in P-gp, Bcrp, or P-gp/Bcrp was less than 2, indicating P-gp and Bcrp have negligible effects on the brain penetration of citalopram. For P-gp substrates, amprenavir, digoxin, loperamide, quinidine, and verapamil, whose KO/WT ratio was reported in the range of 8.3 to 36 in P-gp knockout mice, showed a KO/WT ratio of 5.3 to 20 in the present study. No data are reported for their KO/WT ratios in Bcrp or P-gp/Bcrp knockout mice, except quinidine, which has a KO/WT ratio of unity in Bcrp knockout mice. Our data demonstrated similar KO/WT ratios in P-gp and P-gp/Bcrp mice for these compounds, and the KO/WT ratios in Bcrp mice were less than 2, indicating mainly P-gp at the BBB impairs the brain penetration for these compounds. For Bcrp substrate sulfasalazine, a 13-fold increase in plasma exposure in Bcrp knockout mice over the wild-type mice after an intravenous dose was observed (Zaher et al., 2006). In our study, plasma exposure increased 8- to 12-fold in Bcrp and P-gp/Bcrp knockout mice. Sulfasalazine was not detected in the brains in all strains of mice, suggesting P-gp and Bcrp may not be important in impairing Bcrp substrate sulfasalazine brain penetration. For P-gp and Bcrp dual substrates elacridar, imatinib, and prazosin, the reported KO/WT ratios in P-gp/Bcrp were 5.5 to 28 with less increase in P-gp or Bcrp knockout mice. In the present study, their KO/WT ratios increased 2.6 to 9.6 fold with less increase for their KO/WT ratios in P-gp or Bcrp knockout mice, confirming that removal of either P-gp or Bcrp from the BBB has a limited effect on the brain penetration, but removal of both P-gp and Bcrp has a profound effect on the brain penetration (Kusuhara and Sugiyama, 2009; Kodaira et al., 2010).
The available data support that P-gp is the main efflux protein at the BBB and that Bcrp at the BBB may play an important role for P-gp and Bcrp dual substrates. However, our data were not able to demonstrate the importance of Bcrp in impairing brain penetration for selective Bcrp substrate sulfasalazine. A similar observation was reported in the literature for a selective Bcrp substrate 2-(4-(2-(2-(4-methoxyphenyl)-5-methyloxazol-4-yl)ethoxy)benzyl)-tetrahydrofuran-2-carboxylic acid (PF-407288), whose KO/WT ratio was close to unity in Bcrp knockout mice (Zhou et al., 2009). The underlying mechanism for the lack of enhancement of brain penetration for selective Bcrp substrates in Bcrp knockout mice or both P-gp and Bcrp knockout mice may be due to up-regulation of other efflux.
Ko143 is a potent in vitro and in vivo Bcrp inhibitor, but no data have been reported for the in vivo disposition of Ko143. In the present study, the plasma concentration was below the LLOQ in all mouse strains after a 3 mg/kg subcutaneous dose. However, its brain concentrations were below the LLOQ in wild-type mice but were above its LLOQ in P-gp, Bcrp, and P-gp/Bcrp knockout mouse. Its brain concentrations in P-gp and P-gp/Bcrp mouse brains were approximately 3-fold higher than that in Bcrp mouse brains, indicating that Ko143 is a P-gp and Bcrp dual substrate and P-gp may be more important than Bcrp in limiting its brain penetration.
In summary, the present study demonstrates that inhibition of P-gp or Bcrp at the BBB is unlikely in cassette dosing for the compounds dosed at 1 to 3 mg/kg. Furthermore, the Kp can be estimated from the mass spectrometer response without using standard curves. Finally, use of the cassette dosing approach can correctly identify the impairing effects of P-gp or Bcrp or both P-gp and Bcrp on brain penetration. The results from the present study support the use of the cassette dosing approach to screen brain penetration in drug discovery. It may also be used to study drug transport at the BBB when the number of available animals such as transgenic mice is limited.
Authorship Contributions
Participated in research design: Liu, Ding, Deshmukh, Liederer, and Hop.
Conducted experiments: Ding and Deshmukh.
Contributed new reagents or analytic tools: Ding.
Performed data analysis: Liu and Deshmukh.
Wrote or contributed to the writing of the manuscript: Liu, Ding, Deshmukh, Liederer, and Hop.
Acknowledgments
We thank Kirsten Messick, Linda Bao, Po-Chang Chiang, Jason Boggs, and Brian Dean for their contributions to this work.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- BBB
- blood-brain barrier
- P-gp
- P-glycoprotein
- Bcrp
- breast cancer resistance protein
- Kp
- brain-to-plasma concentration ratio
- Kp,uu
- unbound brain-to-plasma concentration ratio
- AUC
- area under the curve
- Ko143
- (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester
- LLOQ
- low limit of quantitation
- AUCp,R
- area under the curve values calculated by using the mass spectrometer response for plasma
- AUCb,R
- area under the curve values calculated by using the mass spectrometer response for brain
- KO/WT ratio
- Kp in knockout mice versus the Kp from wild-type mice
- PF-407288
- 2-(4-(2-(2-(4-methoxyphenyl)-5-methyloxazol-4-yl)ethoxy)benzyl)-tetrahydrofuran-2-carboxylic acid.
- Received December 23, 2011.
- Accepted February 10, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}