


Abstract
The involvement of the canalicular multidrug resistance protein 2 (Mrp2) in the hepatobiliary excretion of acetaminophen (APAP)-glutathione (GSH) conjugate and its derivatives was investigated using transport-deficient (TR- rats. Although no differences in the biliary concentration of APAP itself were detected between normal Wistar and TR- rats, significant differences in the biliary disposition of several conjugated metabolites of APAP were detected. APAP-GSH was virtually absent in bile from TR- rats. Also, biliary concentrations of APAP-mercapturate (NAC; N-acetylated l-cysteine) and APAP-GLU were significantly reduced in TR- rats. No differences in the biliary concentration of APAP-cysteinylglycine/cysteine (CG/CYS) were detected between normal and mutant rats. The cumulative amounts of APAP-CG/CYS and APAP-NAC excreted in urine of mutant rats were decreased, whereas APAP-GLU was markedly increased. Analysis of liver samples revealed that APAP-GSH and APAP-NAC accumulate in mutant rat livers. Our results support the direct involvement of Mrp2 in the hepatobiliary excretion of several conjugated metabolites of APAP, including APAP-GSH and APAP-NAC, and provide relevant information on processes that may be involved with both their hepatic basolateral transport and renal elimination.
Acetaminophen (APAP1) is biotransformed through phase I and phase II reactions into negatively charged, hydrophilic metabolites that are eliminated in urine and bile. Cytochrome P450 converts APAP to the highly reactive electrophile N-acetyl-p-benzoquinoneimine (Dahlin et al., 1984), which produces liver injury unless it is neutralized by conjugation with glutathione (GSH) (Mitchell et al., 1973). The resulting conjugate (APAP-GSH) can be sequentially metabolized to APAP-cysteinylglycine (APAP-CG), -cysteine (APAP-CYS), and -mercapturate (N-acetylated l-cysteine) (APAP-NAC). By contrast, conjugation with glucuronic acid and sulfate produces APAP-glucuronide (APAP-GLU) and APAP-sulfate (APAP-SUL), respectively. In several species, a large fraction of APAP-GSH undergoes biliary excretion in its original form and the rest is excreted in urine and bile as a mixture of thiol-containing derivatives, whereas the majority of APAP-GLU and APAP-SUL is excreted in urine (Gregus et al., 1988).
Measuring the amount of APAP metabolites in urine and/or bile is considered a useful in vivo method for examining the effects of other chemicals on APAP metabolism, because changes in urinary and/or biliary excretion of APAP conjugates correlate well with changes in APAP biotransformation produced by xenobiotics (Jollow et al., 1974; Madhu et al., 1989; Liu et al., 1993). This approach assumes that membrane-associated transport systems for APAP metabolites in hepatocytes are not affected by xenobiotic treatment. With the recent identification and characterization of several apical and basolateral transporters (Muller and Jansen, 1997), it is now clear that the hepatic levels of some transport systems can be modulated by prototypical microsomal inducers such as pregnenolone 16α-carbonitrile (Salphati and Benet, 1998), dexamethasone (Courtois et al., 1999), and phenobarbital (Ogawa et al., 2000). Therefore, these types of APAP disposition studies should be interpreted with respect to both xenobiotic inducibility of hepatic transporters and molecular mechanisms for the hepatic excretion of APAP and its metabolites.
We recently showed that the biliary concentration of APAP-GSH, APAP-NAC, and APAP-GLU, but not that of APAP itself, APAP-SUL or APAP-CG/CYS, was significantly decreased by coadministration of the nonmetabolizable organic anion indocyanine green (ICG) in male CD-1 mice (Chen et al., 2000). These findings suggest that several APAP conjugates share hepatobiliary transport systems with ICG.
A primary active transporter on the canalicular domain of hepatocytes, known as multidrug resistance protein 2 (Mrp2), mediates the hepatobiliary transport of a wide range of organic anions, including GSH-S-conjugates (e.g., leukotriene C4 and 2,4-dinitrophenyl-S-GSH), oxidized GSH (GSSG), glucuronide conjugates (e.g., glucuronidated bilirubin and bile salts), sulfate conjugates of certain bile acids (e.g., 3α-sulfatolithocholytaurine), and some nonmetabolizable organic anions [e.g., dibromosulfophthalein (DBSP) and pravastatin] (Muller and Jansen, 1997). Transport-deficient (TR-) and Eisai hyperbilirubinemic (EHBR) rats, which are mutant strains derived from Wistar and Sprague-Dawley rats, respectively, lack expression of functional Mrp2 protein (Buchler et al., 1996; Paulusma et al., 1996; Ito et al., 1997) and display a deficiency in the biliary excretion of organic anions. Recently, Brouwer and coworkers (Xiong et al., 2000) reported that the biliary excretion of APAP-GLU and, to a lesser extent, APAP-SUL, was impaired in isolated perfused livers from TR- rats, which indicates the involvement of Mrp2 in the hepatobiliary transport of these two metabolites. Their results are in agreement with our observations that the biliary concentration of APAP-GLU, but not that of APAP-SUL, is significantly decreased by coadministration of the organic anion ICG (Chen et al., 2000). Their studies, however, did not examine the biliary disposition of APAP-GSH and its hydrolytic derivatives, which are reflective of APAP oxidation by cytochrome P450.
APAP-GSH is the major constituent of APAP metabolites in bile of mice and its biliary concentration was decreased the most by ICG treatment (Chen et al., 2000). The main objective of the present study was to further investigate the involvement of Mrp2 in the biliary excretion of APAP-GSH and its thiol-containing derivatives using bile duct-cannulated TR- and normal Wistar rats.
Materials and Methods
Reagents. Authentic standards of APAP metabolites (e.g., APAP-glutathione, -glucuronide, -sulfate, -mercapturate, and -cysteine) were a generous gift from Dr. Sidney Nelson (University of Washington, Seattle, WA). All other chemicals were of reagent grade or higher and obtained from Sigma-Aldrich (St. Louis, MO).
Animals. Male Wistar rats (body weight 395–490 g) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). TR- rats of the same sex (body weight 386–444 g) were obtained from a colony bred in the animal facilities at the University of Connecticut. Breeding pairs were from a colony kept by Dr. Mary Vore at the University of Kentucky, who obtained her breeding pairs from Dr. Ronald Oude Elferink (Academic Medical Center, Amsterdam, the Netherlands). The presence of the spontaneous mutation in the Mrp2 gene in our colony of rats was confirmed using two approaches as previously described by Paulusma et al. (1996): restriction enzyme digestion of an amplified Mrp2 cDNA fragment and Western blotting (data not shown). The mutant rats also had hyperbilirubinemia. Mean plasma conjugated bilirubin levels in TR- rats were 5.4 mg/dl (range, 4.6–6.6), whereas those in the normal Wistar rats were 0.05 mg/dl (range, 0.01–0.07).
Rats were kept individually in polypropylene cages with hard wood shavings and had free access to rodent chow and water. Animal room temperature was maintained at 22–25°C with a 12-h light/dark cycle. The University of Connecticut Institutional Animal Care and Use Committee approved all animal protocols.
APAP Disposition in Bile Duct-Cannulated Normal Wistar and TR- Rats. After overnight fasting, rats were anesthetized (100 mg of ketamine/kg and 10 mg of xylazine/kg i.p.), and the common bile duct was cannulated using 1.2 French polyurethane catheter tubing. The external jugular vein was also catheterized for dosing and blood sampling with 2 French silicone catheter tubing filled with heparinized saline solution (15 U of heparin/ml). Rectal temperature was monitored with a thermistor thermometer unit and maintained between 35 and 37°C with a heat lamp. The urinary bladder was exteriorized through a midline incision of the abdominal wall and carefully pierced with a 30-guage needle attached to a syringe to empty the urine.
Fifteen minutes after completion of these procedures, a nontoxic dose of APAP (1 mmol/kg dissolved with 20% propylene glycol/saline) was administered through the jugular vein catheter. The dead volume in the catheter was displaced with heparinized saline. Bile samples were collected at 20-min intervals for 100 min, whereas blood was withdrawn through the vascular catheter and collected in heparinized tubes every 20 min (total volume of blood samples was less than 1.5 ml). At the end of the experiment, the bladder was punctured again and urine samples were collected. Liver samples were also collected at this time. Blood plasma was separated by centrifugation at 8800g for 5 min using a refrigerated microcentrifuge. All samples were stored at -70°C until assayed.
HPLC Methods. APAP and its metabolites in bile and urine were analyzed using an HPLC method modified from that of Howie et al. (1977) as previously described (Manautou et al., 1996). Samples were diluted with 2 volumes of ice-cold HPLC-grade methanol and centrifuged at 1200g for 30 min. The resulting supernatant was further diluted with double-deionized water, filtered through a 0.20-μm nylon filter, and analyzed using a Beckman System Gold HPLC system (Beckman Coulter, Inc., Fullerton, CA) equipped with a 128-nm solvent module and a 166-nm detector. Aliquots (20 μl) of the processed samples were injected into a Zorbax SB 5-μm C18 reverse-phase column (4.6 mm × 250 mm). APAP and its metabolites were eluted using a mobile phase composed of 12.5% HPLC-grade methanol, 1% acetic acid, and 86.5% water, run isocratically at a flow rate of 1.2 ml/min. The elution of metabolites was monitored at a wavelength of 254 nm.
Retention times of APAP and its metabolites were determined by comparison with that of authentic standards. Since this HPLC method does not separate the cysteinylglycine and cysteine conjugates of APAP, they were quantitated together as APAP-CG/CYS. Preliminary chromatographic analysis of control bile and urine samples shows no interfering peaks. Quantitation was based on integrated peak areas. The concentration of APAP and its metabolites was calculated using an APAP standard curve since the molar extinction coefficients of APAP and its conjugated metabolites are approximately the same (Howie et al., 1977).
The HPLC method of Moldeus (1978) was followed for determining APAP and its metabolites in plasma and liver samples. Briefly, livers (0.5 g) were homogenized in 9 volumes of ice-cold methanol. Perchloric acid (0.1 ml, 3 N) was added to 2 volumes of plasma samples and liver homogenates to precipitate proteins. After centrifugation, supernatants were filtered through 0.2-μm nylon filter and used for analysis, immediately or after storage at -20°C. A linear gradient was used to separate APAP and its metabolites with a constant flow rate of 1.7 ml/min. Solvent A consisted of 1% aqueous acetic acid; solvent B was composed of 1% aqueous acetic acid/methanol/ethyl acetate (90:15:0.1). The mobile phase was initially kept at 75% A and 25% B for 7 min. This was followed by a 20-min linear gradient that finished at 99% B. The composition of the mobile phase was restored to 75% A and 25% B using an 8-min linear gradient. All other HPLC conditions were the same as those described above for analysis of bile and urine samples.
Statistical Analysis. Results are expressed as means ± S.E.M. of four animals per treatment group. Biliary concentrations of APAP and its metabolites were analyzed using one-way repeated measures analysis of variance followed by the Newman-Keuls test. All other data were analyzed using Student's t test. Differences with p < 0.05 were considered significant.
Results
Biliary Excretion of APAP Metabolites. Biliary concentrations of unchanged APAP in both strains of rats closely matched each other and declined with time exponentially (Fig. 1a). By contrast, striking differences in the biliary concentrations of APAP-GSH and APAP-NAC between mutant and normal rats were observed (Fig. 1, b and c). In normal rats, APAP-GSH biliary concentration was highest 20 min after APAP administration and steadily declined thereafter. However, this conjugate was virtually undetectable in the bile of TR- rats. N-Acetylation of APAP-CYS results in the formation of APAP-NAC. In normal rats, APAP-NAC was undetectable in bile samples collected at 20 min after APAP, with a steady increase in biliary excretion thereafter. In mutant rats, APAP-NAC was not detected until 60 min after APAP. Despite the increase in APAP-NAC biliary excretion at later time points, its biliary concentration in mutant rats was still less than 10% of that detected in normal rats. These findings clearly demonstrate that the biliary excretion of both APAP-GSH and APAP-NAC is dramatically impaired in mutant rats.

Biliary concentration of APAP and its metabolites in Wistar and TR-rats.
a, APAP; b, APAP-GSH; c, APAP-NAC; d, APAP-CG/CYS; e, APAP-GLU; and f, APAP-SUL. After overnight fasting, rats were anesthetized and the common bile duct was cannulated. Fifteen minutes later, APAP (1 mmol/kg) was given i.v. Bile samples were then collected and analyzed as described under Materials and Methods. Results are expressed as means ± S.E.M. (n = 4). *, significantly different (p < 0.05) from Wistar rats.
Sequential hydrolysis of APAP-GSH gives rise to APAP-CG/CYS. The biliary concentration of APAP-CG/CYS in mutant rats was equal to that in normal rats over the course of the experiment (Fig. 1d). These results indicate that the biliary excretion of APAP-CG/CYS remains normal in Mrp2-deficient TR- rats.
APAP-GLU was the most abundant metabolite detected in the bile of normal rats. In mutant rats, only trace amounts of APAP-GLU were detected in bile (Fig. 1e). Additionally, APAP-SUL biliary concentration in TR- rats was lower than in normal rats, but significantly different only at 60 min after APAP challenge (Fig. 1f). These results are consistent with impaired biliary excretion of these two metabolites in isolated perfused livers from mutant rats (Xiong et al., 2000)
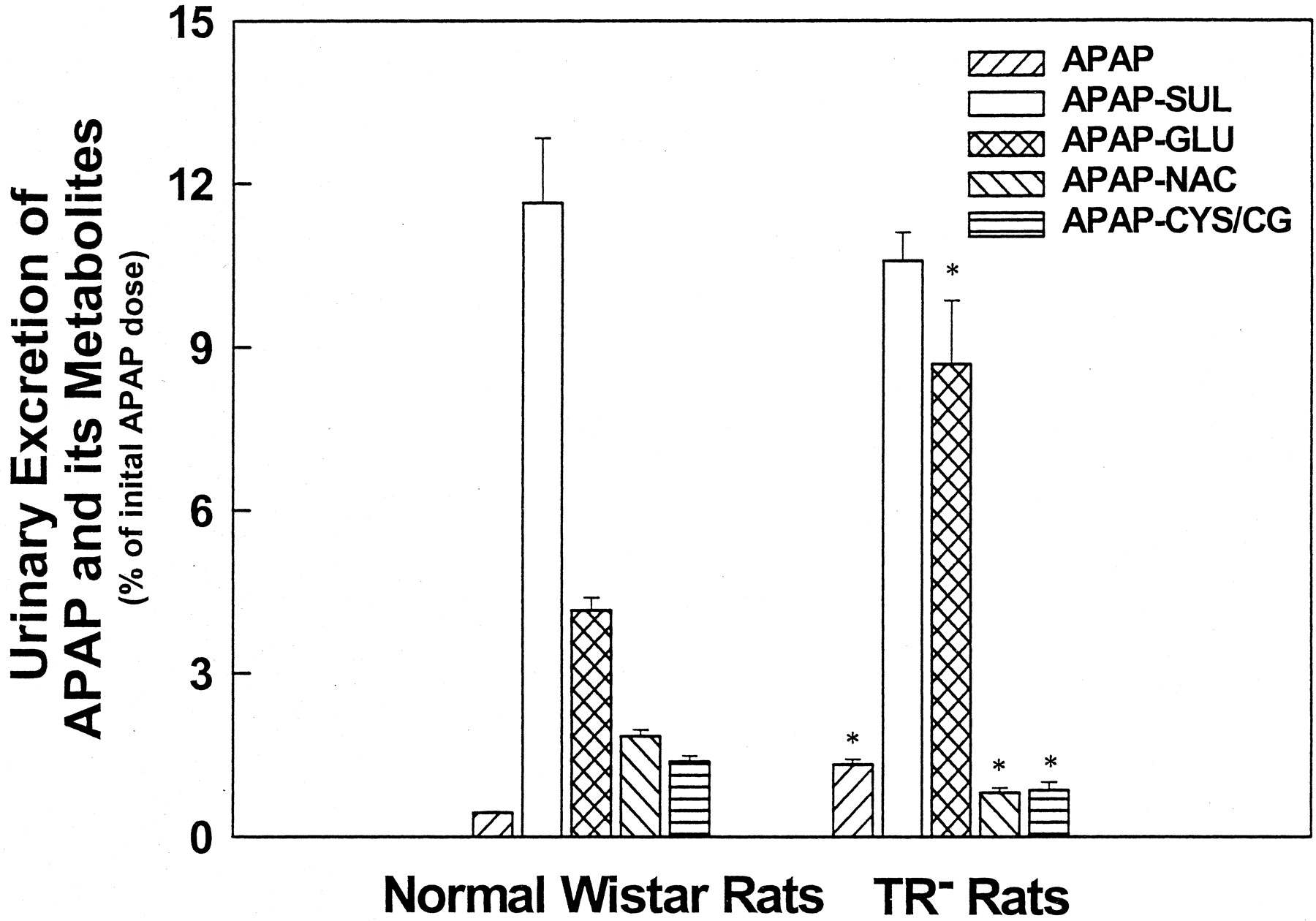
Cumulative Urinary Excretion of APAP Metabolites. In normal rats, 11.64% and 4.15% of the initial APAP dose were excreted in urine as APAP-SUL and APAP-GLU, respectively (Fig. 2). In mutant rats, the amount of APAP-SUL excreted in urine (10.57%) was similar to that in normal rats, whereas APAP-GLU urinary excretion (8.68%) was twice that of normal rats. This increase in urinary excretion of APAP-GLU in TR- rats is confirmatory of the elevated basolateral egress for this conjugate reported in the isolated perfused liver (Xiong et al., 2000). APAP-GSH is normally undetectable in urine samples of rats, but its derivatives can be readily detected (Siegers et al., 1983). We observed that the cumulative urinary excretions of APAP-CG/CYS and APAP-NAC were significantly reduced in mutant rats, despite the impaired biliary excretion of APAP-GSH and APAP-NAC. Urinary excretion of APAP itself was significantly increased in mutant rats (1.33 ± 0.1% versus 0.44 ± 0.02%; p < 0.05). These data indicate that urinary disposition of APAP, APAP-GLU, APAP-CG/CYS, and APAP-NAC is altered in TR- rats.

Cumulative urinary excretion of APAP and its metabolites in Wistar and TR-rats.
After overnight fasting, rats were anesthetized and the common bile duct was cannulated. Fifteen minutes later, APAP (1 mmol/kg) was given i.v. Urine samples were collected and analyzed as described under Materials and Methods. Results are expressed as means ± S.E.M. (n = 4). *, significantly different (p < 0.05) from Wistar rats.
Although the total (biliary plus urinary) excretion of APAP-GSH and its derivatives was significantly lower in mutants, compared to normal rats (1.72 ± 0.15% versus 3.61 ± 0.14%; p < 0.05), no difference in the percentage of the initial APAP dose that was recovered in bile and urine during the experiment was observed between these two groups of rats (mutant rats, 22.16 ± 1.60%; normal rats, 22.42 ± 1.50%).
Hepatic Concentration of APAP Metabolites in Normal andWistar TR- Rats.Table 1 shows the hepatic concentration of APAP and its metabolites in normal and mutant rats at the completion of the study. APAP-SUL and APAP-CG/CYS were undetectable in livers from both strains of rats. Notably, there was an accumulation of APAP-GSH and APAP-NAC in mutant rat livers (460 ± 205 and 404 ± 274 nmol/g liver, respectively), whereas these two APAP metabolites were undetectable in normal rats. This is consistent with a greatly impaired biliary excretion of these conjugates in mutant rats. The hepatic concentration of APAP-GLU in mutant rats was lower than that in normal rats (512 ± 209 versus 426 ± 28 nmol/g liver), but the difference was not significant.
Hepatic concentration of APAP and its metabolites in the bile duct-cannulated Wistar and TR- rats at the end of the experiment
Liver samples were collected at the end of the experiment and stored at -70°C before analysis by HPLC as described under Materials and Methods. The results are expressed as means ± S.E.M. for four animals per group.
Plasma Concentrations of APAP Metabolites in Normal Wistar and TR- Rats. As shown in Fig. 3a, no significant differences in the plasma concentration of unchanged APAP were observed between normal Wistar and TR- rats throughout the experiment, indicating that these two strains of rats have similar plasma disappearance rates for APAP itself. In contrast, plasma levels of APAP-GSH, APAP-NAC, and APAP-CG/CYS in mutant rats were lower than those in normal rats at most of the time points examined (Fig. 3, b–d). Plasma levels of APAP-GLU and APAP-SUL were much higher than those of thiol-containing metabolites in both strains of rats. Plasma concentration of APAP-GLU in TR- rats at 20 min after APAP administration was 1.5-fold higher than in normal rats and remained higher for the next 60 min (Fig. 3e), whereas APAP-SUL plasma concentration in mutant rats was approximately 40 to 60% of normal rats (Fig. 3f).

Plasma concentrations of APAP and its metabolites in Wistar and TR-rats.
After overnight fasting, rats were anesthetized and the common bile duct was cannulated. Fifteen minutes later, APAP (1 mmol/kg) was given i.v. Blood samples were collected and analyzed as described under Materials and Methods. Results are expressed as means ± S.E.M. (n = 4). *, significantly different (p < 0.05) from Wistar rats.
Discussion
APAP biotransformation occurs mainly in the liver. APAP-GLU, APAP-SUL, and APAP-GSH are the major metabolites identified in cultured hepatocytes and the isolated perfused liver (Moldeus, 1978; Grafstrom et al., 1979). In the latter model, these three APAP metabolites are secreted into the perfusate and bile across basolateral and apical membranes of hepatocytes, respectively (Grafstrom et al., 1979). Furthermore, APAP-GLU, APAP-SUL, and APAP-GSH added to perfusate are barely taken up by the liver, suggesting the existence of a diffusion barrier on the basolateral membrane for these hydrophilic metabolites (Grafstrom et al., 1979). In contrast, the biliary and urinary disposition of APAP metabolites in the whole animal is more complex. APAP-GSH can be rapidly degraded into APAP-CG, APAP-CYS, and APAP-NAC by extrahepatic organs such as the kidneys and intestine (Moldeus et al., 1978; Moldeus, 1978). Thus, APAP-GSH is usually undetectable in urine. Instead, its hydrolytic products, APAP-CG, APAP-CYS and APAP-NAC are excreted in urine (Gregus et al., 1988).
A reduction in the biliary concentration of APAP-GLU, APAP-GSH, and APAP-NAC produced by coadministration of ICG (Chen et al., 2000) suggests that these APAP metabolites and the model organic anion may be excreted in bile through common transport systems. Since the biliary excretion of ICG is partially impaired in Mrp2-deficient EHBR and TR- rats (Jansen et al., 1993; Johnson and Klaassen, 2002), it is possible that Mrp2 mediates, at least in part, the biliary excretion of conjugated metabolites of APAP. The involvement of this transporter in the biliary excretion of APAP-GLU and APAP-SUL was recently investigated in TR- rats (Xiong et al., 2000). In that study, isolated livers from normal and mutant rats were perfused with the individual conjugates and their transport was examined. The results showed that the biliary excretion of APAP-GLU was reduced to negligible levels in mutant rat livers, whereas that of APAP-SUL was partially preserved.
The present studies expand the observations from the isolated perfused organ system and give us important insights on the fate of these metabolites once they exit the liver. Furthermore, APAP administration to normal and mutant rats allows for the simultaneous generation of multiple metabolites that subsequently will undergo hepatic disposition. The importance of our studies is evident when considering that multiple APAP metabolites are presented simultaneously at sites of transport (canalicular and/or basolateral membranes of hepatocytes) and that the overall disposition of each metabolite will be affected by the presence of other metabolites with similar requirements for transport.
In the current study, similar alterations in the biliary excretion of APAP-GLU and APAP-SUL were observed in intact mutant rats to those reported previously in isolated perfused livers. Collectively, data obtained from in vitro and in vivo studies using TR- rats indicate that APAP-GLU is a substrate for Mrp2, and that more than one transporter, one being Mrp2, contributes to the biliary excretion of APAP-SUL.
The disposition of APAP-GSH and its derivatives in the isolated perfused liver from mutant rats was not investigated in the study by Brouwer and coworkers (Xiong et al., 2000). Since our previous studies in mice using ICG suggested that the biliary excretion of APAP-GSH is dependent on canalicular transport process(es) for organic anions, the main objective of the present study was to determine the involvement of Mrp2 in the biliary excretion of APAP-GSH and its derivatives. APAP-GSH was virtually undetectable in the bile of TR- rats. Furthermore, biliary excretion of APAP-NAC was dramatically impaired also. By contrast, APAP-CYS/CG biliary concentration was normal in mutant rats, which is consistent with the observations that a bolus dose of ICG has little effect on its biliary concentration (Chen et al., 2000). These results suggest that the biliary excretion of APAP-GSH and APAP-NAC involves the action of Mrp2. The apparently normal APAP-CG/CYS biliary excretion in mutant rats was unexpected since other compounds containing cysteinylglycine and cysteinyl moieties, such as leukotriene D4 and leukotriene E4, are high-affinity substrates for Mrp2 (Huber et al., 1987; Ishikawa et al., 1990). It remains possible that when Mpr2 is not present in the liver, other canalicular transporters may be transporting this GSH-derived conjugate into the bile and not the other conjugates whose biliary excretion was reduced significantly in TR- rats. Further studies are necessary to elucidate the role, if any, of Mrp2 in the biliary excretion of APAP-CYS and APAP-CG. For example, in vitro studies examining the uptake of APAP-CG/CYS by canalicular membrane vesicles should help clarify this.
Besides the well known deficiency in Mrp2 in EHBR and TR- rats, a marked increase in the expression of a basolateral MRP homolog, Mrp3, was observed in liver of these mutant rats (Ogawa et al., 2000; Xiong et al., 2002). Increased Mrp3 expression was shown to be associated with an enhanced basolateral egress of APAP-GLU in isolated perfused livers from TR- rats (Xiong et al., 2000). In vitro transport studies using membrane vesicles prepared from Mrp3-expressing Sf9 insect cells further indicate that APAP-GLU is a lowaffinity substrate of Mrp3 (Xiong et al., 2002). In the present study, we observed a marked increase in urinary excretion of APAP-GLU along with an early increase in plasma levels of this conjugate in TR- rats. This confirms that the higher basolateral egress of APAP-GLU reported by Brouwer and associates (Xiong et al., 2000) translates into a higher urinary output for this metabolite.
Interestingly, the urinary excretion of APAP-CG/CYS and APAP-NAC was significantly lowered in mutant rats, which correlated with lower plasma concentrations for these thiol-containing APAP metabolites and APAP-GSH. In bile duct-cannulated rats, the enterohepatic circulation of APAP-GSH is interrupted. Consequently, amino acid-containing APAP metabolites detected in the urine (i.e., APAP-CG/CYS and APAP-NAC) are mostly derived from APAP-GSH that is secreted from hepatocytes into sinusoidal blood and then hydrolyzed by extrahepatic organs (Moldeus 1978; Moldeus et al., 1978; Fischer et al., 1985). Thus, a lower urinary output of APAP-GSH derivatives suggests that the basolateral secretion of this conjugate is decreased in TR- rats. If the transport machinery for conjugates of xenobiotics on the basolateral domain is unchanged or perhaps increased, as suggested by Mrp3 up-regulation in mutant rats, then the basolateral egress of APAP-GSH should remain similar or be elevated in TR- rats.
Oude Elferink et al. (1989) reported that GSH and GSSG concentrations in the livers of TR- rats are significantly higher than those in normal rats. We also observed an accumulation of APAP-GSH in livers from mutant rats. APAP-GSH may be competing with the high levels of GSH, GSSG, and other retained Mrp2 substrates for common basolateral transport in mutant rats, which may result in a reduction in the urinary excretion of APAP-CG/CYS and APAP-NAC. This, combined with a significantly impaired biliary secretion, could explain the pronounced retention of APAP-GSH seen in livers of TR- rats. The inducible basolateral organic anion transporter Mrp3 is probably not involved in the basolateral egress of APAP-GSH or GSH and GSSG, due to the apparent dissociation between Mrp3 up-regulation in TR- rats and the decrease in urinary excretion of the hydrolytic derivatives of APAP-GSH in the present study. In support of this conclusion, functional characterization of Mrp3 revealed that Mrp3 accepts several kinds of organic anions, but GSH conjugates are poor substrates for this protein (Hirohashi et al., 1999). Another basolateral transporter for organic anions, Mrp1, is expressed in low levels in liver, but its substrate specificity is similar to that of Mrp2 (Keppler et al., 1998). Since the present study indicates that Mrp2 is involved in the biliary excretion of APAP-GSH, it is conceivable that Mrp1 may be involved in basolateral egress of this conjugate.
In principle, alterations in hepatobiliary and urinary excretion of APAP metabolites, as determined in in vivo animal studies, are indicative of changes in either APAP biotransformation or transport processes for these metabolites. If the drastic reduction in biliary excretion of APAP-GSH and APAP-GLU seen in TR- rats was the result of decreased APAP metabolism, then negligible levels of thiolcontaining APAP metabolites and APAP-GLU would also be expected to appear in urine. However, the presence of considerable amounts of these APAP metabolites in the urine of both groups of rats indicates that the impaired biliary excretion of APAP-GSH and APAP-GLU is primarily due to the absence of functional transporter on the apical membrane of hepatocytes for these metabolites.
We also observed significant differences in the urinary excretion of thiol-containing metabolites and APAP-GLU between TR- and normal Wistar rats. To investigate the involvement of possible changes in APAP metabolism in these differences, we measured hepatic mRNA levels of phase I and phase II enzymes that are known to catalyze the biotransformation of APAP in rats (i.e., CYP1A2, CYP2E1, and CYP3A1/23, as well as UGT1A6 and UGT1A7) in male TR- and normal Wistar rats using the QuantiGene bDNA Signal Amplification Assay (Hartley and Klaassen, 2000). The steady-state levels of CYP3A1/23 and UGT1A6 mRNA in TR- rats were approximately 300% and 200% of those in normal Wistar rats, respectively, whereas no significant differences in the mRNA levels of CYP1A2, CYP2E1, and UGT1A7 were detected. These results suggest that both APAP bioactivation and glucuronidation pathways are induced in mutant rats in comparison to normal rats.
Higher APAP bioactivation in livers of mutant rats should have resulted in more N-acetyl-p-benzoquinoneimine generation. This, combined with the higher hepatic GSH content in these rats, should lead to more APAP-GSH formation and excretion into the bile. However, our data show that this is not the case. Therefore, it can be concluded that the decrease in biliary and urinary output of thiolcontaining APAP metabolites in TR- rats is primarily due to changes in transport rather than altered APAP bioactivation. In contrast, it is possible that increased APAP glucuronidation in mutant rat liver may contribute to the 2-fold increase in the urinary excretion of APAP-GLU.
In conclusion, the present data clearly demonstrate that APAP-GSH and APAP-NAC are substrates for Mrp2. We provide additional evidence for the involvement of this transport protein in the biliary excretion of APAP-GLU and APAP-SUL. Furthermore, alterations in urinary excretion of APAP-GLU and the derivatives of APAP-GSH in TR- rats present some insightful clues about basolateral transport systems for these APAP metabolites.
Acknowledgments
We thank Dr. Curtis D. Klaassen at the University of Kansas Medical Center for allowing us to perform the QuantiGene bDNA Signal Amplification Assay in his laboratory and Dr. Paul A. Kramer (University of Connecitcut) for reviewing the manuscript.
Footnotes
-
↵ 1 Abbreviations used are: APAP, acetaminophen; GSH, glutathione; APAP-GSH, acetaminophen-glutathione; APAP-CG/CYS, acetaminophen-cysteinylglycine/cysteine; APAP-NAC, acetaminophen-mercapturate (N-acetylated l-cysteine); APAP-GLU, acetaminophen-glucuronide; APAP-SUL, acetaminophen-sulfate; ICG, indocyanine green; Mrp1, 2, and 3, multidrug resistance protein 1, 2, and 3; GSSG, glutathione disulfide; TR-rat, transport-deficient rat; EHBR, Eisai hyperbilirubinemic rat; HPLC, high-performance liquid chromatography.
-
This work was supported in part by National Institutes of Health Grant ES10093 and a Boehringer Ingelheim Pharmaceutical, Inc. Predoctoral Fellowship in Toxicology to C.C. This work was presented at the 40th Annual Meeting of the Society of Toxicology, 2001 Mar 16–29; San Francisco, CA.
- Received June 21, 2002.
- Accepted March 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}