


Abstract
Drug-drug interactions involving induction of cytochrome P450 enzymes (P450s) can lead to loss of drug efficacy. Certain drugs, particularly those used to treat mycobacterial and human immunodeficiency virus (HIV) infections, are especially prone to induce P450s. During studies to examine drug-interaction potential of compounds in cultured human hepatocytes, exposure with (S)-1-[(1S,3S,4S)-4-[(S)-2-(3-benzyl-2-oxo-imidazolidin-1-yl)-3,3-dimethyl-butyrylamino]-3-hydroxy-5-phenyl-1-(4-pyridin-2-yl-benzyl)-pentylcarbamoyl]-2,2-dimethyl-propyl-carbamic acid methyl ester (A-792611), a novel HIV protease inhibitor (PI) previously under investigation for the treatment of HIV infection, resulted in significant down-regulation of constitutive CYP3A4 expression. Furthermore, coadministration of A-792611 was found to attenuate CYP3A4 induction mediated by known inducers rifampin and efavirenz. A-792611 also attenuated the rifampin and ritonavir-mediated activation of the human pregnane X receptor (PXR) in luciferase reporter assays. Microarray analysis on cultured human hepatocytes revealed that A-792611 treatment down-regulated the expression of PXR target genes CYP3A4, CYP2B6, CYP2C8, and CYP2C9, whereas there was a lack of inductive effect observed in treated rat hepatocytes. A-792611 did not interact with other ligand-activated nuclear receptors that regulate P450 expression such as constitutive androstane receptor, farnesoid X receptor, vitamin D receptor, and peroxisome proliferator-activated receptor α. These data suggest that A-792611 is a functional and effective human PXR inhibitor. Among the class of HIV-PIs, which are typically PXR activators, A-792611 seems to have a unique property for PXR antagonism and could be a useful tool for studying nuclear receptor pathway regulation.
Hepatic gene induction occurs in response to exposure to various drugs and xenobiotics. It is characterized by an increase in levels of transporters and/or phase I or II metabolizing proteins. The cytochrome P450 (P450) enzymes are phase I enzymes that are importantly involved in the metabolism of a variety of xenobiotics (Handschin and Meyer, 2003). Induction of one or more P450s in response to drug administration is a common cause of drug-drug interactions, alterations in drug response, or adverse drug reactions (Willson and Kliewer, 2002; Plant and Gibson, 2003).
CYP3A4 is an important drug metabolizing enzyme whose hepatic expression is regulated at the transcriptional level by nuclear receptors such as the pregnane X receptor (PXR, also called steroid and xenobiotic receptor) and the constitutive androstane receptor (CAR) (Handschin and Meyer, 2003; Martinez-Jimenez et al., 2007). In its inactive state, PXR is localized in the cytoplasm of cells. Upon compound binding, nuclear translocation and heterodimerization to the retinoid X receptor (RXR) occur (Matic et al., 2007). Likewise, CAR is present in an inactive form in the cytoplasm and, during exposure to an inducing compound, translocates to the nucleus through a phosphorylation-dependent mechanism (Willson and Kliewer, 2002). Upon entry into the nucleus, PXR/RXR and CAR/RXR heterodimers bind to promoter elements of many genes involved in drug metabolism and transport to stimulate gene transcription (Stanley et al., 2006; Urquhart et al., 2007).
Certain drugs used to treat human immunodeficiency virus (HIV) infection are widely known to induce drug-metabolizing enzymes, especially CYP3A4. Nevirapine and efavirenz, two non-nucleoside HIV reverse transcriptase inhibitors, potently up-regulate CYP3A4 (Fichtenbaum and Gerber, 2002; Bergshoeff et al., 2005), producing negative drug-drug interactions with a second drug class, the HIV protease inhibitors (HIV-PIs). The latter are almost exclusively metabolized by CYP3A4, and dose adjustments are often necessary when nevirapine or efavirenz is combined with a protease inhibitor (Bergshoeff et al., 2005).

Structure of A-792611.
The HIV-PI ritonavir is a well known, potent CYP3A4 inhibitor, but this drug also causes up-regulation of CYP3A4 expression by acting as a PXR agonist. However, inhibition of CYP3A4 by ritonavir supersedes induction, resulting in net lowering of CYP3A4 activity (Luo et al., 2002). Because of this, coadministration with ritonavir is a widely used strategy to boost the plasma concentrations of other HIV-PIs, thereby increasing their potency (Zeldin and Petruschke, 2004).
Because of the potential of HIV drugs to cause induction of drug metabolizing enzymes and transporters, screening for induction early in the drug development process has become standard practice. Due to known species differences in the ligand-binding domains of PXR and CAR, induction studies are often performed using functional assays of the human receptors. Moreover, gene expression studies in cultured hepatocytes exposed to drug candidates are often performed to determine the potential for inductive drug interactions.
A-792611 [(S)-1-[(1S,3S,4S)-4-[(S)-2-(3-benzyl-2-oxo-imidazolidin-1-yl)-3,3-dimethyl-butyrylamino]-3-hydroxy-5-phenyl-1-(4-pyridin-2-yl-benzyl)-pentylcarbamoyl]-2,2-dimethyl-propyl-carbamic acid methyl ester] is a novel HIV protease inhibitor (Fig. 1) that was investigated preclinically. Metabolism studies showed that A-792611 is metabolized by and is a functional inhibitor of CYP3A4. In this study, we examined the effect of A-792611 on the regulation of drug metabolizing enzymes and transporters in cultured human and rat hepatocytes by microarray and quantitative gene expression analyses. Furthermore, receptor transactivation assays were performed to examine the interactions of A-792611 with various nuclear receptors. These studies showed that A-792611 caused a decrease in the levels of PXR-regulated genes, including CYP3A4, in human but not rat hepatocytes. Furthermore, the inductive effect of potent activators of PXR could be attenuated by coadministration with A-792611. Although numerous PXR activators have been identified, to date there are few examples of potent inhibitors of human PXR. Here, we establish A-792611 as a novel functional antagonist of human PXR, which can be a useful chemical tool for modulating gene expression in vitro or in vivo.
Materials and Methods
Human Hepatocyte Culture. Plateable cryopreserved human hepatocytes from several donors were purchased from In Vitro Technologies (IVT; Baltimore, MD; lots QKR, 130, NPV, and ZCA, part number F00995-P) and BD Gentest (BD Discovery Labware, Bedford, MA; lot 70, catalog number 454551). Donor demographics are shown in Table 1. Vials of cryopreserved cells were gently shaken in a 37°C water bath until the ice was just melted. The contents of the vials were added to a 37°C hepatocyte thawing medium (IVT; part number Z90019) supplemented with 0.5 g/l d-glucose and 10% fetal bovine serum. Cell suspension was centrifuged at 500 rpm for 5 min to gently pellet the hepatocytes. The supernatant was aspirated, and the cell pellet was resuspended in hepatocyte plating medium (IVT; part number Z90029). Cell count and viability were determined by trypan blue exclusion, and hepatocyte suspensions were diluted to 700,000 viable cells/ml in plating medium. Suspended cells were added to BioCoat (BD Discovery Labware) 12-well plates (1.0 ml/well) and allowed to attach overnight at 37°C in 5% CO2/95% air. After 24 h, plating medium was aspirated, and wells were refilled with 1.0 ml of 37°C hepatocyte incubation medium (IVT; part number Z99009); hepatocytes were then incubated at 37°C in 5% CO2/95% air for an additional 24 h. Cells were then treated for 48 h with fresh dosing solutions added after 24 h.
Demographics of human hepatocyte donors
Cytotoxicity Assessment in Human Hepatocytes. Cytotoxicity for isolated human hepatocytes (donor K.Q.G.) treated with A-792611 by itself or in combination with rifampin or efavirenz was determined using both the lactate dehydrogenase assay (CytoTox 96; Promega, Madison, WI; catalog number 9FB054) and the Cell Titer-Glo cell viability assay (Promega; catalog number G7571) according to the manufacturer's instructions. All compounds were dosed at 10 μM.
Rat Hepatocyte Culture. Isolated rat hepatocytes for the microarray experiment were prepared as follows. Whole liver was removed from a naive Sprague-Dawley rat, and the periportal vein was cannulated with an 18-guage catheter. The liver was perfused for 4 to 5 min with perfusion buffer at pH 7.4 containing 148 mM NaCl, 9.2 mM Na-HEPES, 16.7 mM d-fructose, 0.5 mM EGTA, and 1000 units/l heparin (Abbott Laboratories, Abbott Park, IL) at a flow rate of 25 ml/min with the Masterflex L/S (Cole-Parmer, Vernon Hills, IL) peristaltic pump then perfused for 6 to 10 min with collagenase buffer at pH 7.4 containing 6.7 mM KCl, 142 mM NaCl, 9.2 mM Na-HEPES, 5.8 mM CaCl2-H20, 16.7 mM d-fructose, 30 μM phenol red, 0.57% of the fraction V bovine serum albumin, 170 U/ml trypsin inhibitor (Sigma-Aldrich, St. Louis, MO), 100 U/ml collagenase (Sigma-Aldrich), and 93 U/ml hyaluronidase (Sigma-Aldrich). The liver was excised and transferred to a Petri dish containing collagenase buffer. The capsule was mechanically broken up with a comb, and hepatocytes were collected in a 50-ml conical tube and placed on ice. Cell suspension was drawn through a wide-bore pipette to break up clumps then passed through a 100-μm nylon mesh sleeve and collected in a 250-ml sterile centrifuge tube. IVT hepatocyte incubation medium was added to a total volume of 125 ml, and the suspension was centrifuged for 3 min at 50g. Supernatant was aspirated, and rinse was repeated with hepatocyte incubation medium. Cell pellet was resuspended in 40 ml of hepatocyte incubation medium and passed through a 106-μm sleeve. Cell count and viability were determined by trypan blue exclusion, and cells were plated onto a six-well plate at 1 million cells/well. Plated cells were incubated overnight in a 37°C, 5% CO2 incubator. Rat hepatocyte dosing solutions were prepared so that they contained the desired concentration of compound and 0.1% DMSO. One well was treated for each treatment group. Cells were treated for 48 h, and fresh dosing solutions were added after 24 h.
RNA Isolation. After 48 h of treatment, dosing solutions were aspirated off of the cells, and cells were washed with phosphate-buffered saline. TRIzol (800 μl) (Invitrogen, Carlsbad, CA) was added to each well, and cell suspensions were aliquoted into 1.5-ml microfuge tubes. Chloroform (200 μl) was added to each tube, and tubes were shaken by hand for 15 s. Samples were centrifuged at 12,200g at 4°C for 15 min. The upper aqueous phase of each sample was pipetted into a clean microfuge tube, and the lower organic phase was discarded. Glycogen (2 μl) and isopropyl alcohol (500 μl) were added to each sample, and samples were allowed to incubate at room temperature for 10 min. Samples were centrifuged at 4°C for 10 min, and supernatant was discarded. The RNA pellet was washed with 1 ml of 75% ethanol and then resuspended in a small volume of RNase-free H2O. Total RNA concentration was assessed by spectrophotometry, and RNA quality was assessed on the 2100 Bioanalyzer (Agilent, Santa Clara, CA).
Quantitative Real-Time PCR Analysis. Samples were diluted with RNase-free H2O so that they contained 20 ng/μl total RNA. Five microliters of each sample was pipetted in duplicate into a 96-well optical reaction plate (Applied Biosystems, Foster City, CA). Reagent mix was made using the TaqMan EZ RT-PCR kit (Applied Biosystems). The following probe sets were used:
-
Human CYP3A4: forward, 5′-CTT CAT CCA ATG GAC TGC ATA AAT-3′; reverse, 5′-TCC CAA GTA TAA CAC TCT ACA CAG ACA A-3′; TaqMan, 5′-/5-FAM/CCG GGG ATT CTG TAC ATG CAT TG/3-BHQ-1/-3′;
-
Human CYP2B6: forward, 5′-AAG CGG ATT TGT CTT GGT GAA-3′; reverse, 5′-TGG AGG ATG GTG GTG AAG AAG-3′; TaqMan, 5′-/5-FAM/CGC CCG TGC CGA ATT GTT CC3-BHQ-1/-3′;
-
Human 28S (control to account for any variability in RNA levels): forward, 5′-TTC ACC AAG CGT TGG ATT GTT-3′; reverse, 5′-TGT CTG AAC CTG CGG TTC CT-3′; TaqMan, 5′-/5-HEX/TCA CGA CGG TCT AAA CCC AGC TCA CG/3-BHQ-1/-3′.
RT-PCR was performed on the ABI Prism 7700. Results were analyzed by calculating the fold change of P450 induction caused by each compound compared with vehicle levels.
Microarray Analysis. Microarray analysis was performed using the standard protocol provided by Affymetrix (Santa Clara, CA). Briefly, approximately 5 μg of total RNA was reverse transcribed into cDNA using a Superscript II Double-Strand cDNA synthesis kit (Invitrogen), according to the manufacturer's instructions, with the exception that the primer used for the reverse transcription reaction was a modified T7 primer with 24 thymidines at the 5′ end (Affymetrix). The sequence was 5′GGCCAGTGAATTGTAATACGAC-TCACTATAGGGAGGCGG-(dT)24–3′. cDNA was purified via phenol/chloroform/isoamylalcohol (Invitrogen) extraction and ethanol precipitation. The purified cDNA was resuspended in molecular biology-grade water. After this procedure, biotin-labeled cRNA was synthesized according to the manufacturer's instructions from the cDNA using the Enzo RNA Transcript Labeling Kit (Enzo Life Sciences, Farmingdale, NY). The labeled cRNA was then purified using RNeasy kits (QIAGEN, Valencia, CA). Subsequently, cRNA concentration and integrity were evaluated. Approximately 20 μgof cRNA was then fragmented in a solution of 40 mM Tris-acetate, pH 8.1, 100 mM potassium acetate, and 30 mM magnesium acetate at 94°C for 35 min. Fragmented, labeled cRNA was hybridized to an Affymetrix rat genome RAE230A or human genome U133A array at 45°C overnight using an Affymetrix Hybridization Oven 640. The array was subsequently washed and stained twice with streptavidin-phycoerythrin (Invitrogen) using the GeneChip Fluidics Workstation 400 (Affymetrix). The array was then scanned using the Affymetrix GeneChip scanner (part 900154). The microarray scanned image and intensity files (.cel files) were imported into Rosetta Resolver gene expression analysis software version 3.2 (Rosetta Inpharmatics, Seattle, WA), and ratios were built between the protease inhibitor-treated and the vehicle-treated controls. The p values for each gene expression change were calculated according to the Rosetta Resolver analysis software.
Reporter Constructs and Cell-Based Transactivation Assays. All assays involving reporter constructs were performed at Vanderbilt University in the laboratory of Dr. Richard Kim. A CYP3A4-XREM-Luc reporter plasmid driven by CYP3A4 regulatory elements (-362/+53 and -7836/-7208) in pGL3 Basic Vector (Promega) was prepared as described previously (Zhang et al., 2001) based on the report of Goodwin and colleagues (Goodwin et al., 1999). The BSEP-luc reporter plasmid was prepared by cloning the -1.6-kilobase promoter of the BSEPgene (-1540/+75) into pGL3 Basic (Marzolini et al., 2007). A PPAR-responsive luciferase reporter plasmid and a PPARα expression plasmid were kindly provided from Dr. Hong-Guang Xie (Vanderbilt University). PXR, CAR, VDR, and FXR cDNAs cloned in pEF6/V5-His expression vector (Invitrogen) were assembled as described previously (Tirona et al., 2003). Human hepatocarcinoma cells, HepG2, were seeded at 0.6 × 106 cells/well into 12-well plates. After 24 h, cells were transfected with 250 ng/well of luciferase reporter plasmid, 250 ng/well of the nuclear receptor expression plasmid, and 25 ng/well pCMV-TK (Promega) to control for transfection efficiency using Lipofectin (Invitrogen). Sixteen hours later, cells were treated with compounds or vehicle (DMSO, 0.1%) for 24 h. Luciferase activities were determined with the Dual Luciferase Assay Kit (Promega).
Results
Regulation of Hepatocyte CYP3A4 and CYP2B6 Expression by A-792611. Rifampin (20 μM) caused a marked increase in CYP3A4 mRNA levels in hepatocytes from donors 130 and Q.K.R. and a moderate increase in CYP2B6 mRNA levels (Table 2). These observations are consistent with literature reports of rifampin as an inducer of CYP3A, and to a lesser extent CYP2B, in humans. Ritonavir (10 μM) also induced CYP3A4 and CYP2B6 mRNA in both donors, consistent with literature reports of ritonavir as a transcriptional inducer (Table 2). Experimental compound A-792611 caused a decrease in CYP3A4 mRNA levels in both donors. At a concentration of 10 μM, A-792611 caused CYP3A4 levels to drop to 6% of vehicle in donor 130 and 3% of vehicle in donor Q.K.R. A-792611 also caused a slight decrease in CYP2B6 levels in both donors (Table 2).
CYP3A4 and CYP2B6 expression in cultured human hepatocytes treated with rifampin, ritonavir, and A-792611 Results are expressed as fold induction relative to vehicle-treated control for human donors 130 and Q.K.R.
The unusual down-regulation of CYP3A4 and CYP2B6 mRNA by A-792611 caused us to investigate whether A-792611 could attenuate mRNA induction caused by known inducers. In donor Q.K.R., rifampin and efavirenz caused dose-dependent up-regulation of CYP3A4 mRNA levels. Codosing A-792611 with rifampin and efavirenz attenuated the CYP3A4 mRNA induction caused by the positive controls (Fig. 2, A and B; Table 2). Cytotoxicity assays performed on isolated hepatocytes treated with A-792611 alone or in combination with rifampin or efavirenz did not induce any detectable cell death (data not shown). Rifampin and efavirenz also caused dose-dependent up-regulation of CYP2B6 mRNA in hepatocytes from donor Q.K.R. As was the case with CYP3A4, codosing with A-792611 attenuated this induction. A-792611 (10 μM) alone caused levels of CYP2B6 mRNA that were 35% of vehicle level (data not shown).
Interactions of A-792611 with Human PXR. To determine whether the down-regulation of CYP3A4 and CYP2B6 was PXR-mediated, activation of PXR in transfected HepG2 cells was measured by luciferase reporter assay. Treatment of transfected HepG2 cells with rifampin resulted in dose-dependent activation of PXR (Fig. 3A). When rifampin was codosed with A-792611, the activation of PXR decreased, falling almost to vehicle levels, as the dose of A-792611 increased (Fig. 3A). The IC50 for A-792611 inhibition of rifampin-mediated PXR activation was approximately 2 μM. Treatment of transfected HepG2 cells with ritonavir also resulted in dose-dependent activation of PXR. When ritonavir was codosed with A-792611, the activation of PXR again decreased as the dose of A-792611 increased (Fig. 3B). These results suggested that PXR antagonism plays a role in the regulation of P450 enzymes by A-792611.

CYP3A4 expression in cultured human hepatocytes treated with drugs for 48 h. Real-time PCR analysis of CYP3A4 mRNA in cultured human hepatocytes (donor Q.K.R.) treated with the PXR agonists rifampin (A) or efavirenz (B) in combination with A-792611 (n = 2). The error bars display standard deviation.
Genomic Analysis of A-792611-Treated Hepatocytes. To further investigate the possibility that inhibition of PXR was causing the down-regulation of CYP3A4 and CYP2B6, microarray analysis was performed on human hepatocytes from donor 70. Microarray analysis confirmed that A-792611 caused the down-regulation of CYP3A4, CYP2B6, CYP2C8, CYP2C9, and the liver transporters MDR1 and Na+/taurocholate cotransporting polypeptide (Fig. 4A). Hepatocytes codosed with A-792611 and ritonavir also showed down-regulation of P450 enzymes and transporters. By contrast, ritonavir, atazanavir, and amprenavir caused an induction of P450 enzymes and did not affect the expression of liver transporters, with the exception of multidrug resistance protein 2, which was up-regulated (Fig. 4A). A-792611 did not affect the expression of the nuclear receptors PXR, RXR, CAR, aryl hydrocarbon receptor (AhR), PPARα, FXR, VDR, glucocorticoid receptor, and estrogen receptor (data not shown). A lack of PPARα agonist effects by A-792611 is also evidenced by unchanged CYP4A mRNA levels in human hepatocytes after treatment (data not shown). It is also unlikely that A-792611 is an AhR antagonist, as no decreases in expression levels were observed for CYP1A1 (Fig. 4A). However, antagonism of the AhR cannot be entirely ruled out, because down-regulation of the CYP1A2 gene was observed (Fig. 4A).
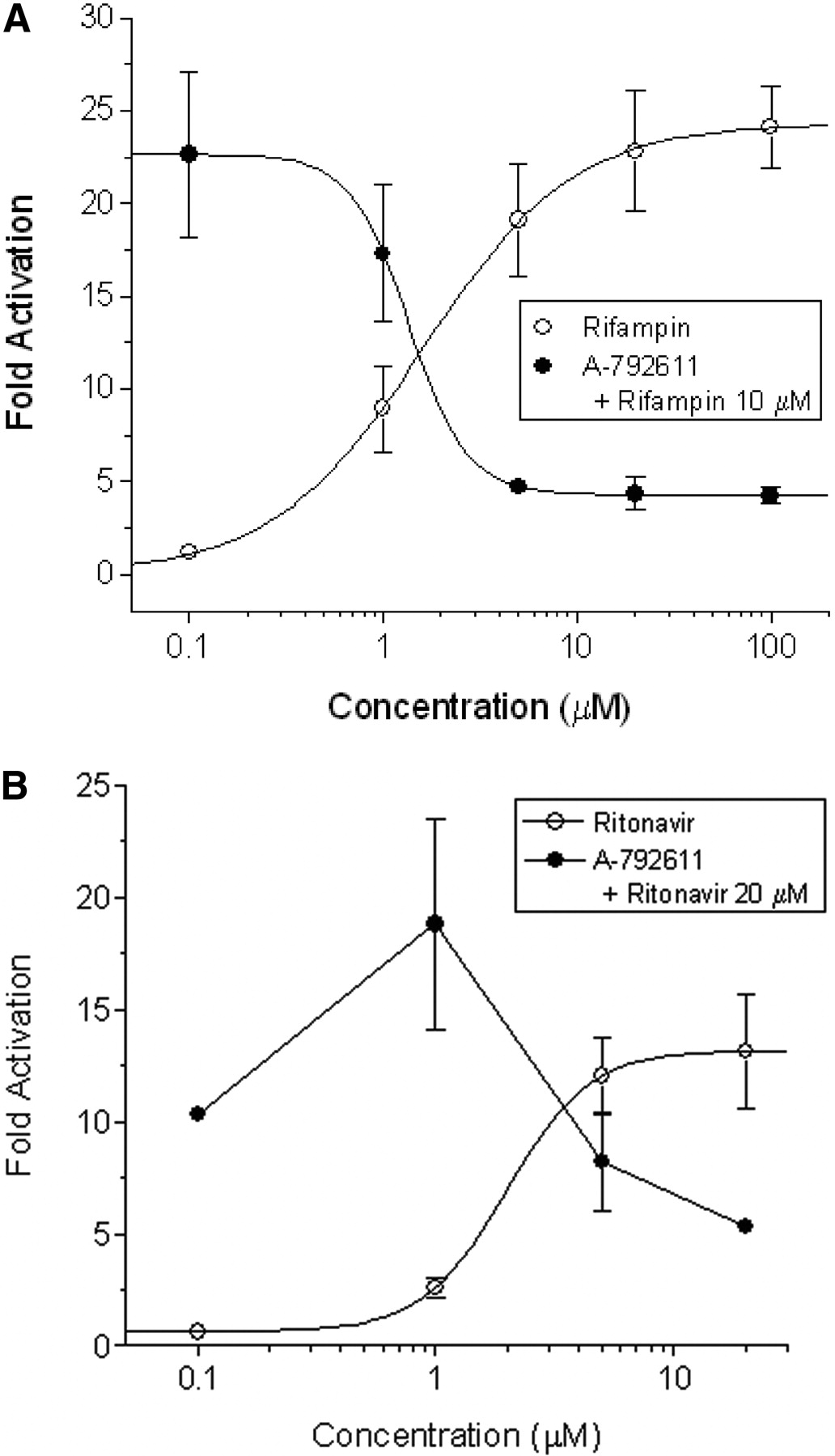
Transactivation assays in HepG2 cells transiently transfected with CYP3A4-XREM-Luc reporter and PXR expression plasmids. After 24 h of drug treatment, luciferase activities were determined. A, concentration-dependent activation of human PXR by rifampin and attenuation of rifampin-mediated (10 μM) PXR activation by escalating levels of A-792611 (n = 4). B, A-792611 attenuates PXR activation mediated by ritonavir treatment (n = 3). Data are shown for a single experiment.

Microarray analysis of cultured human hepatocyte gene expression after 48 h of treatment with drugs. Drug treatment included ritonivir (RTV), atazanavir (ATZ), amprenavir (AMP), and A-792611 (611) alone or in combination. A, heat map showing P450 and transporter gene changes in human hepatocytes from donor 70. B, heat map showing P450 and transporter gene changes in isolated rat hepatocytes. Red, green, and black represent genes that are up-regulated, down-regulated, and unchanged, respectively, compared with the vehicle control. Genes were considered to be significantly regulated if the calculated p value was less than or equal to 0.05. All compounds were dosed at 10 μM.
To determine whether the P450 down-regulation effect caused by A-792611 was species-specific, microarray analysis was also performed on primary rat hepatocytes treated with A-792611 and ritonavir. A-792611 and ritonavir both caused down-regulation of Cyp2b3, Cyp2c6, and Ntcp and up-regulation of Cyp3a3 and Mdr1 (Fig. 4B). The expression of other P450s and transporters was largely unchanged by treatment with these two compounds. The gene expression changes caused by dosing A-792611 together with ritonavir were very similar to those caused by each compound alone (Fig. 3B). This is in sharp contrast to the primary human hepatocytes, where a distinct P450 and transporter gene expression pattern was observed in cells dosed with A-792611.
Lack of Interaction Between A-792611 and FXR, CAR, VDR, and PPARα. Results from human hepatocytes and PXR activation assays supported the notion that A-792611 causes down-regulation of P450 enzymes through functional inhibition of human PXR. Other nuclear receptors including FXR, CAR, VDR, and PPARα are known to regulate P450 expression (Tirona and Kim, 2005). Therefore, we investigated whether the effects of A-792611 on P450 expression were the result of functional interaction with nuclear receptors other than PXR. In cell-based nuclear receptor transactivation assays, A-792611 did not activate human FXR, and a 20-μM dose only slightly attenuated the FXR activation caused by positive control chenodeoxycholate (CDCA) (Fig. 5A). In a CAR reporter assay, CITCO (10 μM), a CAR-specific agonist, caused a 2.5-fold activation of human CAR. A-792611 did not cause significant CAR activation at a concentration of 20 μM, nor did it antagonize the effect of CITCO (Fig. 5B). Experiments in the reporter assays further showed that A-792611 was not an activator of VDR, nor did it antagonize the inductive effects of 1α,25-dihydroxyvitamin D3 (Fig. 5C). Lastly, we examined the interaction of A-792611 with PPARα and observed that it was not an agonist or antagonist of this receptor (Fig. 5D).
Discussion
Many peptidomimetic HIV-PIs including amprenavir, atazanavir, lopinavir, nelfinavir, and ritonavir are known for their capacity to induce drug metabolizing enzymes via a PXR activation mechanism. In this report, we demonstrated that A-792611, an HIV-PI that was previously examined preclinically, down-regulates constitutive expression of PXR-regulated genes CYP3A4, CYP2B6, CYP2C8, CYP2C9, and MDR1 in cultured human hepatocytes. These results are in contrast to the PXR agonists ritonavir, atazanavir, and amprenavir, which caused a general up-regulation of P450 mRNA. These results suggest that constitutive CYP3A4 expression in hepatocytes may be controlled by endogenously synthesized PXR agonists. Moreover, the inductive responses caused by treatment of the xenobiotic PXR agonists rifampin and ritonavir could be attenuated by cotreatment of human hepatocytes with A-792611. Cell-based transactivation experiments confirmed that A-792611 inhibits the activity of human PXR but not other nuclear receptors that regulate CYP3A4. The effects of A-792611 seem to be largely species-specific, as we did not observe gene expression changes consistent with PXR antagonism in rat hepatocytes. Although a luciferase reporter assay did show weak antagonism of pregnenolone-16α-carbonitrile-induced rat PXR activation by A-792611, this response was not as robust as that observed in the human PXR assay (data not shown).
Because A-792611 is metabolized in hepatocytes, down-regulation of CYP3A4 by parent compound versus a metabolite has not been definitively established. However, the half-life of A-792611 in human hepatocytes is relatively long at 6 h. Moreover, inhibition of PXR activation was demonstrated in HepG2 cells, which have been shown to have little to no metabolic capability. Thus, it is likely that most, if not all, of the activity of A-792611 observed in this study reflects the action of the parent compound.

Lack of interaction between A-792611 and various nuclear receptors. HepG2 cells were transfected with luciferase reporter constructs as well as expression plasmids for nuclear receptors and treated with A-792611, known inducers, or vehicle (0.1% DMSO). Luciferase activities were determined after 24 h of treatment. A, FXR reporter assay using BSEP-Luc reporter and the bile acid CDCA as positive agonist control (n = 6–12). B, CAR assay using CYP3A4-XREM-Luc reporter and CITCO as agonist (n = 6). C, VDR luciferase reporter assay with CYP3A4-XREM-Luc reporter and 1α,25-dihydroxvitamin D3 as positive agonist (n = 6). D, PPARα assay using PPRE-Luc reporter and Wy-14643 as positive control (n = 6–9). Data are shown for experiments performed on 2 to 3 occasions. *, significantly different from cells expressing FXR and treated with CDCA (Student's t test).
Results from cell-based nuclear receptor transactivation assays indicate that A-792611 selectively antagonizes PXR and does not interact with other nuclear receptors tested, such as FXR, CAR, VDR, and PPARα. Interestingly, microarray analysis in isolated human hepatocytes showed that treatment with A-792611 did cause a down-regulation of CYP2B6, which is largely regulated by CAR (Faucette et al., 2006). However, extensive cross talk exists between the nuclear receptors, and studies have shown that CYP2B6 is also regulated by PXR (Goodwin et al., 2001). Thus, quite possibly, the down-regulation of CYP2B6 mRNA by A-792611 may be due to antagonism of PXR.
An interesting finding that emerged from the microarray analysis of human hepatocytes is that treatment with A-792611 resulted in an up-regulation of CYP1A1 and a down-regulation of CYP1A2. This result is somewhat unusual, because studies have shown that both of these genes can be regulated by the AhR (Ueda et al., 2006). It should be noted that the up-regulation of CYP1A1 by A-792611 is relatively small (∼4-fold). Treatment with classic activators of the AhR usually results in significantly higher levels of induction [see, for instance, Zhang et al. (2006)]. Nonetheless, both the induction of CYP1A1 and repression of CYP1A2 were considered significant based on the Rosetta Resolver error model. Previous studies have shown that some differential response occurs between CYP1A1 and -1A2 and that CYP1A2 can be regulated by mechanisms independent of the AhR (Zaher et al., 1998). Thus, it may be that the response of CYP1A1 and -1A2 to A-792611 is due to AhR-independent mechanisms.
Studies have shown that the antineoplastic agent ET-743 (Synold et al., 2001), the antifungal agent ketoconazole (Takeshita et al., 2002), and the dietary isothiocyanate sulforaphane (Zhou et al., 2007) are also functional antagonists of PXR. Although ET-743 is a potent inhibitor of PXR activation by SR12813 (IC50 = 2nM) (Synold et al., 2001), its utility as an in vitro or in vivo tool may be hampered by its innate toxic actions as a DNA minor groove binder. Sulforaphane antagonizes rifampin-mediated PXR activation with an IC50 of 12 μM and probably interacts with the receptor through covalent interactions from the chemically reactive isothiocyanate group (Zhou et al., 2007). Aside from PXR antagonist properties, sulforaphane is an inducer of several drug metabolizing and antioxidant enzymes acting via the Nrf2/Keap1 pathway (Morimitsu et al., 2002). Although ketoconazole inhibits PXR with an IC50 of 20 μM (data not shown), it is also a well known antagonist of the glucocorticoid (Duret et al., 2006) and androgen receptors (Eil, 1992). Here, we describe a novel, effective PXR antagonist among the HIV protease inhibitor class of drugs that are largely known for their agonist effects toward this receptor. Aside from the anticancer compound ET-743, A-792611 is the most potent of the described PXR antagonists, with an IC50 of 2 μM toward rifampin-mediated receptor activation.
Recently, molecular approaches that consider the three-dimensional structures of PXR have been used to understand the molecular determinants of receptor-ligand interactions with the intent of modulating agonist effects or creating antagonists (Gao et al., 2007; Xue et al., 2007). This strategy has been somewhat ineffective in producing antagonists owing to the fact that the receptor seems to have conformational flexibility (Xue et al., 2007). Our results indicate that such antagonists can be identified. However, additional investigations will be required to understand the structure-activity relationships that determine the PXR agonist/antagonist behavior of HIV-PIs. Such information may have utility in the development of new HIV-PIs with minimal propensity for causing induction-type drug interactions, some of which have been recently described (Hogeland et al., 2007).
Aside from the important role in xenobiotic defense, PXR has been implicated in a number of physiological and pathophysiological processes. For example, PXR signaling contributes to bone (Tabb et al., 2003; Zhou et al., 2006a), cholesterol (Sporstol et al., 2005), lipid (Zhou et al., 2006c), mineralocorticoid (Zhai et al., 2007), glucocorticoid (Zhai et al., 2007), and thyroid hormone homeostasis (Wong et al., 2005), inflammatory responses (Gu et al., 2006; Zhou et al., 2006b), and uterine contractility (Mitchell et al., 2005). Hence, PXR antagonists such as A-792611 may have utility as chemical modulators to interrogate the physiological roles of PXR.
In conclusion, we report the identification of a novel, selective, and functional inhibitor of human PXR. A-792611 should prove to be an important tool for understanding pharmacology and physiology of PXR-regulated signaling pathways.
Footnotes
-
This work was supported by U.S. Public Health Service Grant GM31304 (to R.B.K.).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019547.
-
ABBREVIATIONS: A-792611, (S)-1-[(1S,3S,4S)-4-[(S)-2-(3-benzyl-2-oxo-imidazolidin-1-yl)-3,3-dimethyl-butyrylamino]-3-hydroxy-5-phenyl-1-(4-pyridin-2-yl-benzyl)-pentylcarbamoyl]-2,2-dimethyl-propyl-carbamic acid methyl ester; PXR, pregnane X receptor; RXR, retinoid X receptor; CAR, constitutive androstane receptor; HIV, human immunodeficiency virus; PI, protease inhibitor; P450, cytochrome P450; DMSO, dimethyl sulfoxide; BSEP, bile salt export pump; FXR, farnesoid X receptor; VDR, vitamin D receptor; PPARα, peroxisome proliferator-activated receptor α; MDR, multidrug resistant; AhR, aryl hydrocarbon receptor; PCR, polymerase chain reaction; CDCA, chenodeoxycholate; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl)oxime; Wy-14643, [4-chloro-6-(2,3-xylidino)-2-pyrimidylthio]acetic acid; ET-743, ecteinascidin 743; SR12813, 3,5-di-tert-butyl-4-hydroxystyrene-β,β-diphosphate acid tetraethyl ester.
- Received October 31, 2007.
- Accepted December 19, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}