


Abstract
Ketoconazole is no longer available for clinical determination of worst-case victim drug-drug interaction (DDI) potential for cytochrome P450 3A (CYP3A)-substrate drugs; clarithromycin and itraconazole are the proposed replacements. Ketoconazole DDIs are described by unbound systemic exposures due to absence of carrier-facilitated hepatic uptake, but this aspect of clarithromycin and itraconazole disposition has not been investigated. At present, transport of clarithromycin, itraconazole, and hydroxyitraconazole by hepatic organic anion transporting polypeptides (OATPs) and organic cation transporter 1 (OCT1) was examined in vitro and in vivo. As for ketoconazole, uptake of clarithromycin, itraconazole, and hydroxyitraconazole into OATP1B1, OATP1B3, OATP2B1, and OCT1 expressing human embryonic kidney 293 (HEK293) cells was not greater than in vector controls. Uptake into these HEK293 cells and human hepatocytes was not impaired by the prototypical OATP, OCT, and sodium/taurocholate cotransporting polypeptide inhibitors bromosulfophthalein, imipramine, and taurocholate, respectively. In contrast, uptake of the positive controls, atorvastatin for OATPs and metformin for OCT1, was significantly enhanced by relevant transporter expression, and uptake into both these HEK293 cells and human hepatocytes was significantly impaired by prototypical inhibitors. In Oatp1a/1b gene cluster knockout mice, which lack the major hepatic Oatps, and in Oct1/2 knockout mice, ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole oral exposure was not increased, and the liver-to-blood partition coefficient (Kp) was not decreased. By contrast relative to wild-type mice, in Oatp1a/1b- and Oct1/2-knockout mice, atorvastatin and metformin oral exposure was significantly increased, and liver Kp was significantly decreased. The present studies provide in vitro and in vivo evidence that, like ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole are not transported into the liver by hepatic uptake transporters, including OATPs and OCT1.
Introduction
High-dose ketoconazole (400 mg by mouth, each day for ≥5 days) has long been the gold-standard cytochrome P450 3A (CYP3A) inhibitor in clinical drug-drug interaction (DDI) studies (Zhao et al., 2009). In 2013, based on emerging clinical safety reports, both the US Food and Drug Administration and European Medicines Agency advised against using ketoconazole in DDI studies (http://www.fda.gov/Drugs/DrugSafety/ucm371017.htm). Withdrawal of oral ketoconazole from the market triggered a comprehensive search for alternatives that could be used for evaluation of victim DDI potential for drugs cleared by CYP3A. Ke et al. (2014) reviewed available CYP3A-inhibitor drugs and proposed clarithromycin and itraconazole as the best clinical alternatives.
Ketoconazole has been favored due to nearly complete CYP3A inhibition in humans at clinically relevant doses, selectivity, and predictability of DDIs based on unbound plasma concentrations (Zhao et al., 2009; Han et al., 2013). The ability to predict DDIs based on unbound circulating exposures is of particular practical importance. Steady-state ketoconazole concentrations available for interaction with hepatic CYP3A enzyme are in equilibrium with plasma unbound concentrations because of ketoconazole’s high passive membrane permeability (Clarysse et al., 2009) and absence of carrier-facilitated hepatic uptake (Zhao et al., 2009). As such, ketoconazole DDIs are accurately predicted by circulating concentrations corrected for plasma protein binding (Smith et al., 2010), which are easily sampled, unlike intracellular unbound liver concentrations which are practically impossible to sample directly in humans.
Although clarithromycin and itraconazole are the best available clinical CYP3A inhibitor alternatives to ketoconazole, both drugs exhibit properties that may be indicative of carrier-facilitated uptake into the liver (Ke et al., 2014). Clarithromycin is a known in vitro and clinical inhibitor of organic anion transporting polypeptide (OATP) hepatic uptake (Jacobson, 2004; Hirano et al., 2006). OATP inhibition can be competitive (Zamek-Gliszczynski et al., 2013), which prompts the question of whether clarithromycin also is an OATP substrate? At a kinetic level, clarithromycin preferentially partitions into suspended rat hepatocytes, with an unbound liver-to-buffer partition coefficient of 6 (Yabe et al., 2011), an observation that could be explained by hepatic uptake (Smith et al., 2010; Kalvass et al., 2013). No mechanistic evidence exists for hepatic uptake of itraconazole or its major metabolite, hydroxyitraconazole, which also is an inhibitor of CYP3A (Templeton et al., 2008). However, physiologically based pharmacokinetic model DDI simulations in which the unbound itraconazole and hydroxyitraconazole hepatic concentrations paralleled unbound plasma concentrations slightly, but consistently, underestimated the clinically observed DDI magnitude, raising the possibility of hepatic uptake of parent and/or metabolite (Ke et al., 2014).
CYP3A inhibitors can be taken up into the liver by OATPs (Liu and Unadkat, 2013), in which case hepatic unbound inhibitor concentrations are higher than plasma unbound concentrations (Smith et al., 2010), and the DDI based on systemic inhibitor exposure is underpredicted without accounting for hepatic uptake (Maeda et al., 2011). In addition to OATPs, organic cation transporter 1 (OCT1) is a hepatic uptake mechanism for small type I organic cations such as metformin; however, it is unlikely to transport drugs with physicochemical properties such as these CYP3A inhibitors (Giacomini et al., 2010). Nonetheless, investigation of OCT1 was included in the present study for the sake of completeness. Likewise, potential uptake by the sodium/taurocholate cotransporting polypeptide (NTCP) was investigated in hepatocytes.
To enable quantitative predictions of the DDI magnitude with proposed clinical CYP3A inhibitor replacements for ketoconazole (Ke et al., 2014), the possibility of clarithromycin, itraconazole, and hydroxyitraconazole uptake into the liver is an important issue to investigate and document in the literature. The present studies provide convincing in vitro and in vivo evidence that, like ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole are not transported into the liver by hepatic OATPs or OCT1.
Materials and Methods
Ketoconazole, clarithromycin, itraconazole, hydroxyitraconazole, and atorvastatin as well as their deuterated internal standards bromosulfophthalein and imipramine were purchased from Sigma-Aldrich (St. Louis, MO) and Toronto Research Chemicals (North York, ON, Canada). [14C]metformin and [14C]tetraethylammonium were purchased from American Radiolabeled Chemicals (St. Louis, MO); [3H]estrone-3-sulfate, [3H]cholecystokinin octapeptide, and [3H]taurocholate were obtained from PerkinElmer (Waltham, MA). Cryopreserved human hepatocytes [lot NRJ (female), lot KQN (female), and lot YUA (male)] and all hepatocyte thawing and plating media were procured through Celsis IVT (Baltimore, MD).
Expressed Transporter Studies.
SLCO (OATP) 1B1, 1B3, 2B1, SLC22A1 (OCT1) cDNA (Thermo, Waltham, MA) were individually inserted into EW1969 plasmid vectors. HEK293 cells stably expressing the EBNA1-gene (i.e., PEAKSTABLE cells; Edge Biosystems, Gaitherburg, MD) (Godinot et al., 2003) were transfected with DNA vector (1 µg DNA/5 × 106 cells) following the standard Effectene protocol (Qiagen, Venlo, Netherlands). The following day, cells were lifted with trypsin and moved to a flask for selection in complete medium: 10% fetal bovine serum Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT) with 50 µg/ml gentamicin and 0.5 µg/ml puromycin. After selection, vector control, OATP2B1, and OCT1 cells were used as pooled stable transfections, while OATP1B1 and OATP1B3 cells were dilution cloned and selected for optimal activity. All cell types were plated at 75,000 cells/cm2 in 12-well BioCoat poly-d-lysine plates (Corning, Tewksbury, MA) and cultured for 3 days in complete media with the addition of 5 mM sodium butyrate in medium on the final day of culture.
Cells were incubated with 0.5 µM test article in the absence or presence of prototypic inhibitor (25 µM bromosulfophthalein for OATPs; 100 µM imipramine for OCT1) in uptake buffer (HBSS supplemented with 10 mM HEPES, pH 7.4, 37°C) for 2.5 minutes. Uptake reactions were stopped by addition of ice-cold phosphate-buffered saline, and cells were washed 3 times before lysing with a 50:50 methanol:water (v/v) solution containing internal standard for liquid chromatography with tandem mass spectrometry analysis. Cells incubated with [14C]metformin were lysed in 1% Triton X (Sigma-Aldrich, St. Louis, MO) and mixed with scintillation fluid (ScintSafe 30%; Fisher Scientific, Waltham, MA) for scintillation counting. Protein concentrations were determined using standard BCA assay methodologies (Sigma-Aldrich). Uptake velocities were calculated as the accumulation of test article per well normalized to total protein and incubation time.
Hepatocyte Uptake Studies.
Cryopreserved human hepatocytes were thawed per vendor protocol and plated at 350,000 cells/1.9 cm2 in 24-well collagen-coated plates. Cells were incubated for 2 hours at 37°C with 5% CO2 and 95% relative humidity in plating medium to allow cells to attach. Before uptake study initiation, cells were rinsed twice with prewarmed 37°C uptake buffer (Hanks’ buffered saline solution supplemented with 10 mM HEPES, pH 7.4). Uptake reactions were initiated by the addition of uptake buffer containing 0.5 µM test article in the absence or presence of prototypic inhibitors (5 µM bromosulfophthalein for OATPs, 100 µM imipramine for OCTs, 25 µM taurocholate for NTCP). Bromosulfophthalein at 5 µM concentration was used for pan OATP inhibition (Sai et al., 2006; Izumi et al., 2013), while minimizing the inhibition of other transporters (e.g., NTCP) in human hepatocytes (Kim et al., 1999). Uptake studies in hepatocytes from each donor were performed in triplicates at the 1.5-minute time point. Uptake reactions were stopped with the addition of ice-cold phosphate-buffered saline (PBS) and washed 2 times before quench with a 50:50 methanol:water (v/v) solution containing internal standard. Uptake of radiolabeled positive control substrates (4.4 nM [3H]estrone-3-sulfate for OATP1B1, 2.5 nM [3H]cholecystokinin octapeptide for OATP1B3, 3.6 µM [14C]tetraethylammonium for OCT1, and 13.0 nM [3H]taurocholate for NTCP) were parallel tested in each lot of hepatocytes in the absence or presence of inhibitors (5 µM bromosulfophthalein for OATPs; 100 µM imipramine for OCTs; 25 µM bromosulfophthalein for both NTCP and OATPs). Uptake reactions were stopped with the addition of ice-cold PBS, and cells were washed twice before cell lysis with 1% Triton-X in PBS. Uptake velocities were determined as the total accumulation of test article per well normalized to average total protein and reaction duration.
In Vivo Transport Studies.
Age-matched Oatp1a/1b cluster-knockout, Oct1/Oct2 double-knockout, and wild-type FVB male mice were purchased from Taconic Farms (Germantown, NY). Mouse 100 mg/kg oral doses of ketoconazole, itraconazole, clarithromycin, atorvastatin, and metformin were selected to fall within the human dose range based on body surface area scaling. Drugs were administered by oral gavage as suspensions (10 ml/kg of 1% hydroxyethyl cellulose, 0.25%, polysorbate-80, 0.05% antifoam in water). Blood spots were collected via tail bleeds at 0.08, 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, and 6 hours after dosing (blood was sampled up to 4 hours for ketoconazole and clarithromycin); livers were collected at the final 4- or 6-hour time point. After metformin administration, plasma samples were collected at the following time points: 5, 10, 20, 30, 45, 60, 90, 120, and 150 minutes; liver-to-plasma concentration ratios were determined 1.5 hours after oral metformin administration.
Bioanalysis.
Ketoconazole, clarithromycin, itraconazole, hydroxy-itraconazole, metformin, and atorvastatin in relevant matrices [blood spots (3-mm punch), plasma, liver homogenates, cell buffers and lysates] were quantified by liquid chromatography with tandem mass spectrometry. All samples were mixed with an organic internal standard solution to precipitate protein, centrifuged, and the resulting supernatants were directly analyzed. Analytes and their deuterated internal standards were separated using reverse-phase chromatography with gradient elution and detected using selected reaction monitoring [Sciex API 4000 triple quadrupole mass spectrometer equipped with a TurboIonSpray interface (Applied Biosystems/MDS, Foster City, CA)]: ketoconazole, [M+H]+ m/z 531.1 → 489.2; clairthromycin, [M+H]+ m/z 748.3 → 158.3; itraconazole, [M+H]+ m/z 705.3 → 392.3; hydroxyitraconazole, [M+H]+ m/z 721.2 → 408.2; atorvastatin, [M+H]+ m/z 560.1 → 440.1; metformin metformin [M-H]− m/z 130.1 3 → 71.1. The dynamic range of the assays was 1–5,000 ng/ml for in vitro samples and plasma, 1–10,000 ng/ml in blood spot samples, and 1–50,000 ng/ml in liver homogenate samples.
Data Analysis.
Statistical significance was determined by the Student’s t test, corrected for unequal variance, where applicable. In all cases, P < 0.05 was considered statistically significant. Data are reported as mean ± S.E.M., with the associated n reported in all cases, unless otherwise indicated.
Results/Discussion
Uptake of ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole was examined in HEK293 cells expressing OATP1B1, OATP1B3, OATP2B1, or OCT1 (Fig. 1, A–D). OATP uptake of the positive control, atorvastatin, was 4.5- to 7.7-fold enhanced in OATP-transfected cells relative to vector controls and was significantly 55–60% inhibited by bromosulfophthalein. Likewise, uptake of the OCT1 positive control metformin was 6.9 ± 0.2-fold enhanced in OCT1 cells and was significantly 82% impaired by imipramine. In contrast, uptake properties of CYP3A inhibitors were generally consistent with compounds not transported by hepatic OATPs and OCT1: 1) uptake activity in transporter-expressing cells was not enhanced relative to vector controls, and 2) prototypical OATP and OCT inhibitors did not impair uptake. Raw uptake velocity values are summarized in Supplemental Table 1.
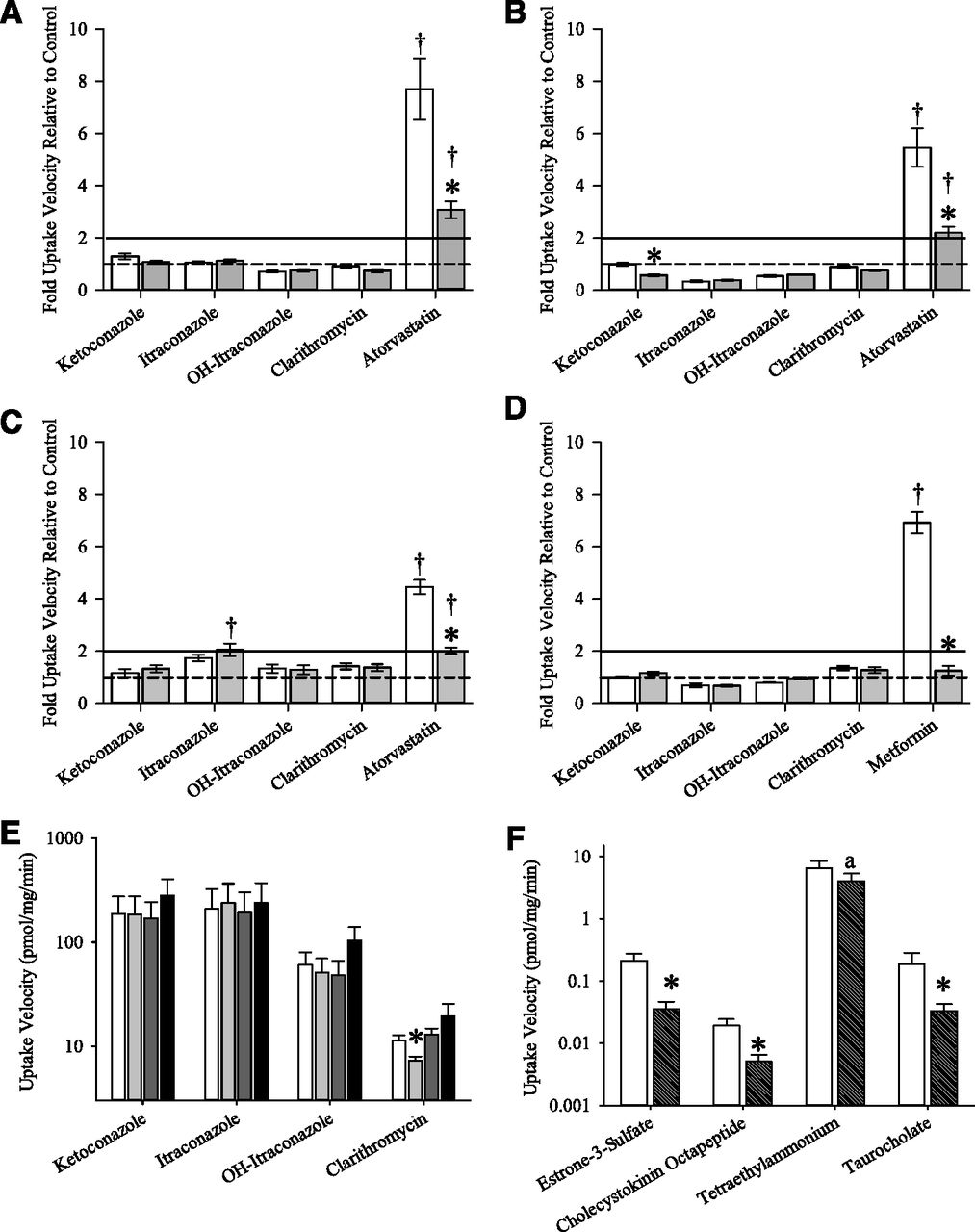
In vitro uptake of CYP3A inhibitors by OATP1B1 (A), OATP1B3 (B), OATP2B1 (C), OCT1 (D), and human hepatocytes (E and F). In panels A–D, transport activity is presented as the transporter-to-vector control transfected cell ratio of uptake velocity and is shown in the absence (open bars) or presence of uptake inhibitors (gray bars): the OATP inhibitor bromosulfophthalein (A–C; 25 μM), or the OCT inhibitor imipramine (D, 100 μM); corresponding raw uptake velocity values are reported in Supplemental Table 1. The dashed line of unity denotes the same uptake velocity in transporter and vector control transfected cells, and the solid line at uptake ratio of 2 is the commonly-accepted transport activity exceeded by substrate drugs. Mean ± S.E.M., n = 3, †P < 0.05: enhanced uptake in transporter-expressing cells relative to vector controls when the uptake ratio is >2; *P < 0.05: inhibition of uptake by the relevant transport inhibitor. Panels E and F summarize uptake of CYP3A inhibitors (E) and positive control substrates (F) in cryopreserved primary human hepatocytes (n = 3 donors, triplicate measurements/donor). Uptake velocity of CYP3A inhibitors in the absence of uptake inhibitors (open bars), or in the presence of 5 μM bromosulfophthalein (light gray bars), 100 μM imipramine (dark gray bars), or 25 μM taurocholate (black bars) (E). Uptake of positive control substrates in the absence (open bar) or presence (dashed bar) of prototypic inhibitors: OATP1B1 substrate, 4.4 nM estrone-3-sulfate ± 5 μM bromosulfophthalein; OATP1B3 substrate, 2.5 nM cholecystokinin octapeptide ± 5 μM bromosulfophthalein; OCT1 substrate, 3.6 μM tetraethylammonium ± 100 μM imipramine; NTCP substrate, 13 nM taurocholate ± 25 μM bromosulfophthalein (F). Mean ± S.E.M., n = 3 donors, *P < 0.05: inhibition of uptake by the relevant transport inhibitor. aP < 0.05: inhibition of tetraethylammonium uptake by imipramine in hepatocyte preparations from 2/3 donors.
Hepatic uptake of ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole was subsequently examined in cryopreserved human hepatocytes from three donors (Fig. 1, E and F). Uptake of the positive controls, estrone-3-sulfate for OATP1B1, cholecystokinin octapeptide for OATP1B3, tetraethylammonium for OCT1, and taurocholate for NTCP, was significantly inhibited to 83 ± 7%, 71 ± 12%, 32 ± 23%, 74 ± 15% of control values by OATP1B1 and 1B3 inhibitor, bromosulfophthalein (5 µM), the OCT1 inhibitor, imipramine (100 µM), and NTCP inhibitor bromosulfophthalein (25 µM), respectively. In contrast, the uptake activity of CYP3A inhibitors ketoconazole, itraconazole, and hydroxyitraconazole was not significantly impaired by prototypical OATP, OCT, or NCTP inhibitors, with the exception of clarithromycin, whose uptake activity was, on average, 37 ± 10% decreased by 5 µM bromosulfophthalein (statistically significant in hepatocyte preparations from 2/3 donors).
To confirm the in vivo relevance of these negative in vitro transport findings, oral pharmacokinetics and hepatic distribution of the CYP3A inhibitors were studied in Oatp1a/1b gene cluster knockout mice, which lack the three major hepatic Oatps (Higgins et al., 2014), and in Oct1/2 knockout mice, which are deficient in both hepatic and renal Oct function (Higgins et al., 2012). Atorvastatin and metformin oral exposure was significantly increased (2.4- to 2.9-fold), and liver Kp was significantly decreased (84–99%), in Oatp1a/1b and Oct1/2 knockout mice relative to wild-type controls, respectively (Fig. 2). In contrast, ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole oral exposure (AUC0–last and Cmax) was not increased and the liver-to-blood partition coefficient (Kp) was not decreased in either Oatp1a/1b or Oct1/2 knockout mice (Fig. 2; Supplemental Figs. 1–3)

Oral exposure (A, C, E) and liver-to-blood partition coefficient (liver Kp; B, D, E) of CYP3A inhibitors in Oatp1a/1b gene cluster knockout mice lacking the major hepatic Oatps (red bars), Oct1/2 knockout (KO) mice (green bars), and wild-type (WT) male FVB control mice (open bars). The positive-control substrates atorvastatin and metformin exhibited significantly increased oral exposure (2.4- to 2.9-fold) and significantly decreased liver Kp (84–99%) in Oatp1a/1b- and Oct1/2-knockout mice, respectively. In contrast, CYP3A inhibitor oral exposure was not increased and liver Kp was not decreased in either Oatp1a/1b- or Oct1/2-knockout mice. Mean ± S.E.M., n = 4-6, *P < 0.05: oral exposure increase or liver Kp decrease in knockout versus wild-type mice.
For the first time, the present studies provided direct in vitro and in vivo evidence that, like ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole are not transported into the liver via OATPs or OCT1. These data are of fundamental importance to quantitative DDI predictions as clarithromycin and itraconazole replace ketoconazole as the default clinical CYP3A inhibitors, particularly in light of reports that have suggested the possibility of carrier-facilitated hepatic uptake for these replacement inhibitors (Ke et al., 2014). Specifically, clarithromycin is a known hepatic OATP inhibitor (Hirano et al., 2006; Markert et al., 2014), and it exhibits preferential distribution into suspended rat hepatocytes, with an unbound liver-to-buffer partition coefficient of 6 (Yabe et al., 2011). The current dataset directly demonstrated that clarithromycin is neither transported by hepatic OATPs or OCT1 in vitro, nor taken up into the liver by hepatic Oatps or Oct1 in vivo.
Analysis by Ke et al. (2014) demonstrated that all simulated clinical trials involving multiple dosing of itraconazole slightly (<2-fold), but consistently, underpredicted the victim DDI magnitude. Carrier-mediated hepatic uptake of itraconazole and/or hydroxyitraconazole was one potential explanation for this underprediction. However, hepatic uptake of parent and/or metabolite would have also resulted in underprediction of acute DDIs, which was not observed (Ke et al., 2014). These gross pharmacokinetic findings, combined with the present data rule out hepatic uptake of itraconazole and hydroxyitraconazole as the reason for the steady-state DDI underprediction. Instead, DDI underprediction more likely reflects that Ke et al. (2014) did not account for metabolites like N-desalkylitraconazole, which has a longer half-life and accumulates upon multiple dosing and contributes up to 20% of steady-state itraconazole CYP3A inhibition (Templeton et al., 2010).
In summary, the present studies provide in vitro and in vivo evidence that, like ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole are not transported into the liver by hepatic OATPs or OCT1. Clarithromycin is an inhibitor of hepatic OATP uptake (Hirano et al., 2006; Markert et al., 2014), but it is not taken up into the liver by hepatic OATPs. Steady-state itraconazole DDI underprediction is not caused by hepatic OATP or OCT uptake of parent or hydroxyl metabolite, and is instead more likely due to the accumulation of other inhibitory itraconazole metabolites (Templeton et al., 2010; Ke et al., 2014). In conclusion, like ketoconazole, clarithromycin, itraconazole, and hydroxyitraconazole are not transported into the liver by hepatic OATPs or OCT1.
Acknowledgments
The authors thank Jing Q. Bao for his assistance with the animal studies.
Authorship Contributions
Participated in research design: Higgins, Ke, Zamek-Gliszczynski.
Conducted experiments: Higgins.
Performed data analysis: Higgins, Ke, Zamek-Gliszczynski.
Wrote or contributed to the writing of the manuscript: Higgins, Ke, Zamek-Gliszczynski.
Footnotes
- Received April 26, 2014.
- Accepted August 8, 2014.
↵1 Current affiliation: SimCYP, Certara, Cary, North Carolina.
↵2 Current affiliation: Drug Metabolism and Pharmacokinetics, GlaxoSmithKline, Research Triangle Park, North Carolina.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CYP
- cytochrome P450
- DDI
- drug-drug interaction
- HEK293
- human embryonic kidney 293
- NTCP
- sodium/taurocholate cotransporting polypeptide
- OATP
- organic anion transporting polypeptide
- OCT
- organic cation transporter
- PBS
- phosphate-buffered saline
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}