Article Text

Abstract
Despite the success of biological therapies in rheumatoid arthritis (RA), orally active small-molecule drugs are desirable. Signal transduction inhibitors have been the focus of intense efforts, with some recent notable successes and failures. p38α is a signalling molecule that regulates proinflammatory cytokines, which makes it a logical target for RA. Unfortunately, selective p38α inhibitors have limited efficacy. An attempt is made here to put these studies into perspective and offer possible explanations for the failure of p38α blockers. Alternative strategies, such as targeting kinases higher in the signalling cascade or using less selective compounds, might be more successful as suggested by the efficacy seen with Syk and JAK inhibitors.
Statistics from Altmetric.com
Novel biological therapies have revolutionised the treatment of chronic inflammatory diseases such as rheumatoid arthritis (RA), psoriasis and Crohn's disease.1 2 3 In the past decade, the success of tumour necrosis factor (TNF) antagonists combined with advances in the science of signal transduction has underscored the importance of cytokines like TNF, interleukin (IL)1 and IL6 in the pathogenesis of human diseases. The advantages of biological agents are limited, however, owing to cost, long half-lives, the requirement for parenteral administration and the observation that only a fraction of patients have a robust clinical response.4 Treatment of autoimmune diseases remains a significant unmet medical need that could benefit from small-molecule, orally active drugs.
Protein kinases are potential therapeutic targets amenable to this approach. These intracellular enzymes transmit and amplify information by phosphorylating substrates leading to an altered cellular response. A wide range of cellular stresses such as inflammatory cytokines, pathogens, growth factors, ultraviolet irradiation and osmotic stress engage kinases, which, in turn, regulate the expression of key genes.5 The mitogen-activated protein kinases (MAPKs) have attracted considerable attention as potential targets for autoimmunity because they can alter the production of key inflammatory mediators. There are three major families of MAPKs—namely, p38, extracellular-regulated protein kinase (ERK) and c-Jun N-terminal kinase (JNK) (fig 1).6 p38 has four homologous isoforms, α, β, γ and δ. The α and β isoforms are ubiquitous, whereas the γ isoform is found in mainly skeletal muscle and the δ isoform is located in the testes, pancreas and small intestine.7

A simplified mitogen-activated protein kinase (MAPK) pathway: complex parallel and crossover signalling cascades link the three main MAPK families, extracellular-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK) and p38, which are activated by cytokines, stress and growth factors. The top level shows the MAP3Ks, the second tier shows the upstream MAPK kinases (MKKs) and the third tier comprises the MAPKs (ERK, JNK, p38) that regulate various genes through phosphorylation of transcription factors (eg, c-Jun, ATF2) and other kinases (MAPKAPK2/MK2). The primary, but overlapping, responses include cell growth and differentiation (ERK), matrix regulation (JNK) and inflammatory cytokine production (p38). Reprinted by permission from MacMillan Publishers Ltd: Nat Clin Pract Rheumatol 2007;3:651–60, copyright©2007. http://www.nature.com/clinicalpractice/index
MAPK activation is mediated by upstream MAPK kinases (MKKs), which in turn, are activated by MKK kinases (MKKK or MAP3K).6 p38 activation and phosphorylation is regulated by two upstream kinases, MKK3 and MKK6, which are phosphorylated by multiple MAP3Ks.8 The p38-mediated signalling cascade culminates in increased expression of proinflammatory molecules like TNFα, IL6, IL1, cyclo-oxygenase 2 (COX-2) and metalloproteinases (MMPs).9
p38: The holy grail of targets for rheumatoid arthritis?
The discovery that a p38 inhibitor blocked lipopolysaccharide (LPS)-induced TNFα and IL1β production by monocytes initiated the exploration of p38 as a potential target.10 Investigators in many laboratories provided abundant evidence suggesting that this enzyme has a key role in RA including:
p38α is the key isoform that regulates cytokine expression;
p38 is expressed and activated in the rheumatoid synovium;
inhibition of p38 suppresses numerous cytokines implicated in RA;
p38 blockade decreases fever and cytokine production in a human LPS challenge model;
p38 inhibitors are effective in numerous animal models of arthritis.11 12
Thus, p38, especially p38α, appeared to be a potential “wonder drug” and work began in earnest to synthesise novel inhibitors. These compounds were mainly competitive antagonists that blocked ATP binding to the kinase.13 However, potency, lack of selectivity and toxicity limited their utility.12 These compounds inhibited p38α and β but not the γ or δ isoforms14; at higher concentrations many other kinases were blocked.15 While effective in preclinical models, a variety of toxicity problems, especially affecting the liver, interfered with clinical development.16
Eventually, the chemistry improved and compounds with higher specificity and potency were discovered. Among the first p38 inhibitors to advance to phase IB clinical trials were VX-745 and BIRB 796. VX-745 is more selective for p38α than p38β and is an ATP-competitive antagonist. In a 12-week placebo-controlled trial in RA, a signal for clinical efficacy was seen in the low-dose group.12 Further study of this compound and several others was hampered by hepatotoxicity and preclinical safety studies in dogs, in which a mechanism-based central nervous system (CNS) inflammatory syndrome was observed with chronic dosing. This had a major impact on the design of later compounds to limit CNS penetration.16
BIRB 796 exemplified a new class of allosteric p38 inhibitors.17 Despite this new mechanism, BIRB 796 still inhibited several non-p38 kinases.18 The compound was investigated in healthy humans who were injected with LPS.19 Induction of TNFα, IL6, IL10 and IL1 receptor antagonist (IL1Ra) was significantly attenuated in the BIRB 796-treated group compared with placebo. A randomised placebo-control trial was conducted to investigate the efficacy of BIRB 796 in Crohn disease.20 No efficacy was seen and the liver toxicity prevented sustained exposure. One curious observation was that the initial rapid decreases in acute phase reactants like C-reactive protein (CRP) was transient, with a return to baseline by 8 weeks. A third compound, SCIO-469, had a very similar profile in an RA study—that is, limited or no efficacy, liver enzyme abnormalities and transient decrease in acute phase reactants.21 Although SCIO-469 efficacy in RA was disappointing, the compound was effective in a dental pain model, suggesting that p38 is a reasonable target for pain.22
More clinical development and more frustration
At least 22 different p38 inhibitors have been investigated in phase I/II clinical trials for a variety of clinical indications and none have progressed to phase III. The newest generation of compounds has greater selectivity, less CNS penetration and less toxicity (liver and other organ systems). Some of these have been thoroughly tested in RA. For instance, two phase II clinical trials evaluated the safety and efficacy of VX-702 in RA.23 24 In the VeRA trial, VX-702 was administered daily to patients as monotherapy and compared with methotrexate (MTX). At week 12, a modest response (44%) to VX-702 was seen in the 10 mg group compared with placebo (32%).
In study 304, VX-702 was administered to MTX partial or non-responders to determine potential synergy.23 Patients received either daily VX-702, the drug twice a week, or placebo in addition to continuing MTX, with the twice a week group providing an opportunity to determine if this dosing regimen prevented the acute phase reactant “escape”. As in the VeRA trial, modest response was noted with the group treated daily. Forty-four per cent of the intermittent-treatment group achieved an ACR20 response at week 12, which was significantly greater than the 22% in the placebo control. Overall, the compound was well tolerated. In both studies, a transient decrease in CRP and serum amyloid A levels was seen, with a return to baseline levels by week 12. Surprisingly, the escape phenomenon was also seen in the intermittent-treatment group. The CRP phenomenon was not due to altered drug metabolism because VX-702 plasma levels at steady state (week 4) were deemed sufficient.
Another (perhaps final) nail in the p38 coffin for RA involved pamapimod (RO4402257), a highly selective p38α inhibitor that demonstrated efficacy in multiple animal models of inflammatory arthritis.25 It was evaluated in two trials—as monotherapy and as an add-on to patients with partial responses to MTX. After 12 weeks of monotherapy, approximately 23–31% of patients fulfilled ACR20 criteria compared with 45% of those receiving MTX.26 In the combination therapy study, there was a modest trend towards improvement in the pamapimod group that did not reach statistical significance, despite a transient decrease in CRP. Adverse events included elevated liver enzymes, skin rash and dizziness.
Additional p38 inhibitors are being evaluated for RA as well as other conditions. ARRY-797 significantly decreased post-surgical pain in a dental extraction model compared with placebo.27 The inhibitor also reduced serum CRP peri- and postoperatively at 24 h. These data confirm a role of p38 in pain processing and might be an alternative development pathway. Clinical trials to determine the efficacy of ARRY-797 in RA are in progress.28
What has gone wrong?
While many p38α inhibitors have been evaluated in phase I/II clinical trials, the relative lack of therapeutic efficacy in RA was a major surprise and unexpected. It is not clear why they have failed, but some potential explanations are offered below.
Dosing
One obvious explanation is inadequate exposure due to dose limitations imposed by toxicity. This seems less likely for the latest generation of p38α inhibitors. Compounds like VX-702 and pamapimod are reasonably well tolerated and the half maximal inhibitory concentrations can be readily achieved in patients for a prolonged period of time. The effect of the compounds on acute phase reactants (at least initially) is reasonable evidence that the therapeutic concentration was reached.
Biodistribution
Engineering of compound design to lower lipophilicity and prevent CNS penetration might potentially limit efficacy. Recent studies show that selective p38 blockade in the spinal cord reduces inflammation in a rat model of arthritis.29 Direct intrathecal administration of a p38 inhibitor not only decreased synovial inflammation but also suppressed articular cytokine and protease expression as well as joint destruction. The mechanism appears to be related to enhanced vagal outflow,30 which can activate α7 nicotinic receptors in the periphery.31 In addition, p38 plays an important part in pain perception and behaviour.32 Blockade of spinal p38 has potent anti-nociceptive effects and p38 inhibitors block acute pain in humans.33 Decreasing CNS penetration to avoid side effects, such as dizziness in humans or the unexpected CNS inflammatory effects in dogs (not seen in any other species), might have an impact on clinical end points relevant to RA.
Targeting the wrong isoforms
Although p38α is the best characterised isoform because it is a key regulator of the immune and inflammatory response, perhaps other isoforms are important. Each of these isoforms is expressed and activated in human synoviocytes and in RA synovium.9 34 Although p38β-deficient mice are not resistant to arthritis,35 recent studies suggest that p38β might have a proinflammatory role by regulating endothelial derived chemokine production.36 Furthermore, the β isoform has been implicated in spinal-mediated pain responses. By “dialling out” CNS penetration and p38β activity, drug development programmes might be limiting potential efficacy. Activated p38γ inhibits c-Jun phosphorylation thus antagonising p38α, which stimulates c-Jun activation.37 In some cells, p38γ also inhibits transcription mediated by activated protein-1, a key regulator of cytokine production, suggesting a potential anti-inflammatory function.38
Anti-inflammatory effects of p38α
Recent data suggest that p38α regulates anti-inflammatory cytokines in addition to the well-known effects on proinflammatory factors.39 For instance, p38α is required for IL10 production by macrophages. Genetic deletion of the p38α gene in macrophages increases skin oedema after exposure to ultraviolet light.40 Thus, it is possible that the beneficial effects of p38 blockade are counterbalanced. p38α also participates in negative feedback loops that inhibit the activities of upstream MAP3Ks.41 By blocking this effect, p38α inhibitors can potentially divert the signalling flux to other MAPKs such as JNK and ERK.
Reliance on animal models
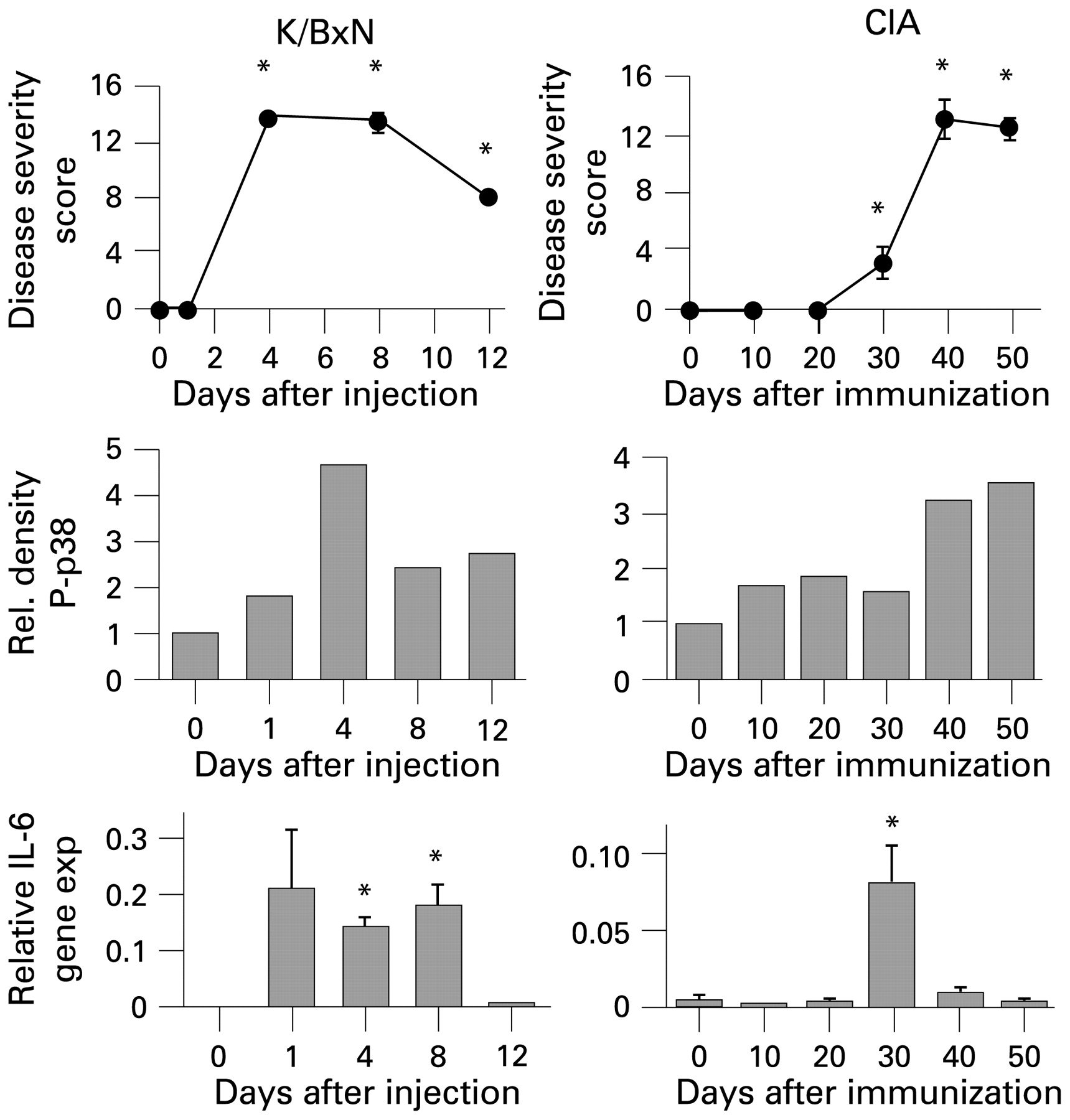
Animal models are vital when evaluating potential therapeutic compounds. However, they do not replicate the complexities of human disease.42 Careful selection of animal models and interpretation of preclinical data must be performed in the context of the way in which the models relate to RA.43 Recent studies suggest that the kinetics of MAPK activation in mice does not always correlate with RA..44 In the collagen-induced arthritis model, p38 activation has an early minor peak followed later by a peak during the plateau and regression phases of disease. Selection of the appropriate model and the timing of drug administration can have a major impact on the outcomes and relevance to human disease (fig 2).44

{kind=link}
{kind=link}
Kinetic analysis of synovial signalling and gene expression in animal models of arthritis. (Top) Disease severity scores in K/BxN and collagen-induced arthritis (CIA) models. Passive K/BxN arthritis was induced by injecting mice with arthritogenic serum from K/BxN mice (n = 4/time point) on day 0. For CIA, mice were immunised on day 0 and day 20 (n = 6/time point). Arthritis was quantified using a clinical scoring system with maximum score of 16. (Middle) Kinetics of p38 activation in arthritic mice; (Bottom) interleukin 6 (IL6) gene expression in arthritic synovium. *p<0.05. Joints from mice were collected at the indicated time points and assayed for phospho-p38 (P-p38) by western blot analysis and for IL6 by quantitative PCR. Note that the time course for IL6 expression, p38 activation and clinical evidence of arthritis do not always correlate. The interpretation of animal model data requires careful consideration of the differences with rheumatoid arthritis. Data modified from Fukushima et al.44
Redundant signalling networks
Like cytokine networks, signalling cascades are highly redundant and complex. It is naïve to think that blocking one kinase, especially downstream in the pathway, would not lead to compensatory effects in other kinases that can regulate the same genes. For example, the kinases upstream from p38, such as MKK3, MKK6 and TAK1 can regulate NF-κB and redirect the signalling flow.45 46
Physiological “escape” from p38 regulation
The surprising observation that CRP decreases are transient despite adequate drug levels suggests that humans have a physiological escape mechanism from p38 inhibition. CRP production in the liver is regulated by p38-dependent cytokines like IL1 and IL6.47 One possible explanation is that CRP production is not impaired in tissues with minimal or reduced exposure to p38α inhibitors such as the CNS where glial cells can synthesise CRP.48 However, it is unclear if CRP production in the brain contributes significantly to the levels in plasma.
Another interesting possibility is that CRP is produced independently of p38α. For instance, Toll-like receptor 4-induced IL6 production in macrophages is independent of p38 or NF-κB activity.49 IL6 is a potent activator of acute phase protein production by hepatocytes. Another potential inducer of CRP and serum amyloid A is endoplasmic reticulum (ER) stress. This stress response can be induced by low cellular calcium or ATP as well as exposure to LPS.50 These factors interfere with ER function leading to an accumulation of aggregated or unfolded proteins. The ER then initiates an acute phase response that requires the liver-specific transcription factor CREBH.51 Prolonged ER stress has been associated with systemic inflammation in Crohn disease, heart disease, diabetes and hepatitis.52 p38-Independent pathways can, therefore, lead to higher CRP levels, especially when hepatocytes are stressed. Since many p38 inhibitors are hepatotoxic, ER stress is a potential escape mechanism that does not require MAPKs. The observation that clinical responses to p38 inhibitors do not correlate with the kinetics of the CRP response suggests that the mechanism might not truly be “escape” but instead represents an alternative pathway unrelated to signal transduction.
Role of p38 in RA
In the final analysis (and as horrifying as it is to consider), the possibility that p38 does not participate in the pathogenesis of RA should be considered. This seems unlikely given the known functions of p38 and the wealth of preclinical and human synovial data. However, it is possible that p38 is simply not relevant.
And now for something completely different: new small-molecule kinase inhibitors in RA
Increasing the selectivity and potency of p38α inhibitors has not led to more effective treatment in RA. One corollary is that repeated attempts to fine tune p38α inhibitors will probably not circumvent lack of efficacy. A second corollary is that going “downstream”, such as a MAPKAPK2 inhibitor (the key downstream substrate of p38α that regulates cytokines), will probably not improve efficacy. However, we should not despair; biological agents did not succeed with the first attempts. Before the success of TNF blockers, notable failures included anti-CD4, anti-CD5, anti-CD53, IL2-diphtheria toxin fusion protein, interferon γ and others. Even after TNF blockers, success has not been guaranteed, such as IL1 inhibitors (anakinra)53 or B-cell inhibitors (anti-BlyS),54 which showed modest efficacy.
With regard to kinases and signalling, the twin burdens of safety AND efficacy seemed overwhelming. Recent advances, however, demonstrated that kinase inhibitors can clearly improve signs and symptoms of RA. The safety concerns are still significant but can potentially be managed. Some of the lessons learnt from these newer compounds suggest that moving higher in the signalling cascade (and casting a broader penumbra on downstream kinases) and perhaps less selectivity might be more useful than highly selective compounds.55
The most successful small-molecule inhibitors evaluated in RA target protein tyrosine kinases (PTKs). These enzymes are very high upstream in the signalling cascade and catalyse the transfer of phosphate groups to specific tyrosine residues in the substrate. PTKs are categorised in two classes depending on their localisation and function—receptor tyrosine kinases are transmembrane proteins while the non-receptor tyrosine kinases are cytoplasmic. Receptor tyrosine kinases primarily bind growth factors such as epidermal growth factor, non-receptor tyrosine kinases are activated by cytokines like IL6 and IL12 and regulate haematopoiesis and immune response. This class of PTKs has been further divided into nine subfamilies, two of which are the Janus kinases (JAKs) and spleen tyrosine kinases (Syks). Inhibitors of JAK and Syk family members were recently evaluated in phase II trials and showed excellent efficacy in patients with RA for whom MTX and anti-TNF therapies had failed.
Janus kinases (JAKs)
JAKs were named after the Roman god Janus with two faces symbolising beginning and ending.56 The duality refers to the structure of JAKs, which contain a kinase domain adjacent to an enzymatically inactive pseudokinase region with critical regulatory functions.57 JAKs constitutively bind the cytoplasmic region of the transmembrane cytokine receptors. Cytokine–receptor interaction activates combinations of the JAK family members that phosphorylate the tyrosine residues in the receptor. This creates docking sites for one or more signal transducer and activators of phosphorylation (STAT) molecules. JAKs then phosphorylate STATs, which are released from the receptors and function as transcription factors.
In mammals, the JAK family consists of four members—JAK1, JAK2, JAK3 and Tyk2.58 Among these, JAK3 is primarily expressed in haematopoietic cells while the others are ubiquitously expressed. The JAK family members, either individually or in combination, associate with different cytokine receptors and can activate multiple downstream pathways such as MAPKs and NF-κB.59 Mutations that lead to increased activity of JAK2 are seen in haematological malignancies while the loss of JAK3 function results in severe combined immune deficiency characterised by the lack of T cells and NK cells.58
The unique positioning of JAKs in the signalling pathway and their role in cytokine responses make them an attractive target for inflammatory diseases. CP 690550 is a potent JAK-specific inhibitor that was originally identified as a JAK3 blocker but also inhibits JAK1 and JAK2.60 CP 690550 is quite effective in animal models of arthritis.61 The compound has been studied in several phase II RA studies with clear evidence of efficacy. In one randomised, double-blinded, placebo-controlled, 6-month study, dose-dependent improvements in ACR20 were seen as early as 1 week after starting treatment.62 63 By 6 weeks, 70–81% of the patients receiving the active drug achieved ACR20 compared with 29% in the placebo group. About one-quarter of the treated patients fulfilled ACR70 criteria compared with 3% of the placebo group. Headaches, dizziness and nausea were the most common adverse effects. Dose-dependent adverse events such as elevated serum creatinine, hypercholesterolaemia, neutropenia and anaemia were also seen. Some of these are almost certainly mechanism-based, since JAKs are involved with colony stimulating factor and erythropoietin signalling.60
Spleen tyrosine kinase (Syk)
Syk is a non-receptor protein tyrosine kinase predominantly expressed in bone marrow derived cells as well as synoviocytes and vascular endothelial cells.64 65 Syk binds to the cytoplasmic region of receptors that contain the immune-receptor tyrosine-based activation motif (ITAM).66 This motif is located in the cytoplasmic portion of FcγR, FcεR, Igα (B cells), CD3ζ (T cells) and integrins.67 Immune complexes or antigens that bind these receptors phosphorylate ITAMs, which in turn activate Syk. Subsequently, Syk activates multiple pathways that regulate inflammation, including MAPKs, phosphoinositide-3-kinase (PI3K) and phospholipase C γ. Proinflammatory cytokines such as TNF and IL1 can also activate Syk in synoviocytes, leading to JNK activation and expression of MMPs and IL6.68 69 R788 (fostamatinib disodium), a prodrug of R406, inhibits proinflammatory cytokines and joint destruction in animal models of arthritis.70
Fostamatinib was recently evaluated in a randomised, placebo-controlled, phase II trial in patients with RA who were resistant to MTX. Clinical improvement was seen as early as 1 week after the start of treatment.71 At 12 weeks, approximately 72% of the patients in the highest-dose group achieved at least an ACR20 response, versus 38% in the placebo group. ACR70 response was seen in 19% of patients treated with the highest dose compared with 4% in the placebo group. CRP levels decreased rapidly and remained suppressed; serum MMP3 and IL6 were also suppressed. The most common adverse events were diarrhoea, increased blood pressure and neutropenia. A recent phase IIb study suggested that fostamatinib might not be effective in patients for whom anti-TNF biological agents have failed, providing possible insight into the drug's mechanism of action.72
Where to next?
While the safety of signal transduction inhibitors needs to be carefully scrutinised, these drugs represent attractive alternatives for patients with RA for whom conventional treatments have failed or who are unable to take biological agents. The success of the JAK and Syk inhibitors provides much-needed confidence to researchers focused on targeting kinases in autoimmune diseases. One distinguishing characteristic between the successful agents and p38 inhibitors is that the former act at a higher point in the signal transduction cascade. Upstream targets might have fewer opportunities for escape or for bypassing a downstream kinase through redundant pathways. Downstream targets, such as MAPKAPK2, would be less interesting in this scenario.
How can these lessons be applied to the p38 pathway? One possible approach is to target upstream kinases that regulate p38, such as the MKK3 and MKK6. Both of these are expressed and activated in RA synovium.73 MKK3 activates p38α, β, δ isoforms while MKK6 activates all four isoforms.8 Unlike p38α deficiency, which is lethal, MKK3−/− mice are viable and healthy.45 Moreover, MKK3−/− mice are protected from passive K/BxN arthritis and appear to have normal in vivo responses to LPS. Therefore, MKK3 inhibition could selectively suppress inflammation while sparing some aspects of host defences. MKK3 deficiency also inhibits TNF-mediated NF-κB activation, which might be one “escape” route if p38 is blocked. MKK6 deficiency also decreases arthritis,34 but only MKK3 has effects on pain processing and analgesia.32
Another potential target in the p38 pathway that is even further upstream is transforming growth factor activated kinase 1 (TAK1). TAK1 is a ubiquitous MAP3K that activates JNK, p38 and NF-κB pathways in response to TNF and IL1.46 TAK1 deficiency decreases proinflammatory cytokine and MMP production in cultured synoviocytes.74 Because it intersects several signalling pathways, TAK1 has therapeutic potential in RA.
Other possible targets reside at the apex of the signalling cascade. The PI3 kinases, including the relatively leukocyte-specific γ and δ isoforms, could suppress chemokine-dependent cell recruitment as well as T- and B-cell activation.75 76 I kappa B kinase β (IKKβ) is a critical convergence point for NF-κB activation in synoviocytes as well as other cell lineages and could suppress an array of proinflammatory cytokines.77 Sphingosine 1-phosphate kinase (SK1) also regulates lipid mediators that control numerous processes involved with inflammation and SK1 inhibitors are effective in arthritis models.78 Other signalling molecules, like IL1 receptor-associated kinase 4 (IRAK4), MyD88, IKK-related kinases, or ρ kinase are also sufficiently upstream that they might be useful.79
An alternative strategy is to decrease the selectivity of inhibitors. The general trend for greater and greater specificity might be counterproductive owing to the redundancy of signalling networks. Perhaps a combination JNK–p38 inhibitor or one that targets multiple MAP kinase kinases would overcome this problem.
The hurdles for developing new treatments for RA in the era of biological agents are daunting. However, the seemingly endless series of failures has ended with a better and improved understanding of the kinome. There is still much work to be done, especially in evaluating the safety risks. Nevertheless, the opportunities for success appear to be closer than ever.
REFERENCES
Footnotes
Competing interests None.
Provenance and Peer review Not commissioned; externally peer reviewed.