Homology Models of Melatonin Receptors: Challenges and Recent Advances

Abstract
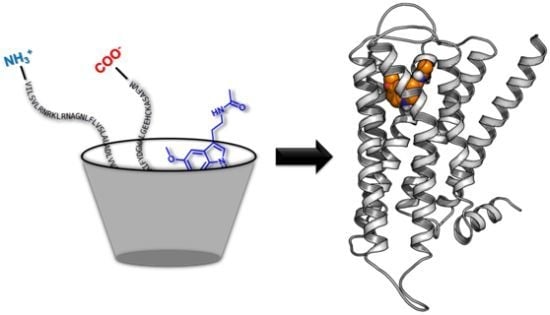
:
{kind=link}
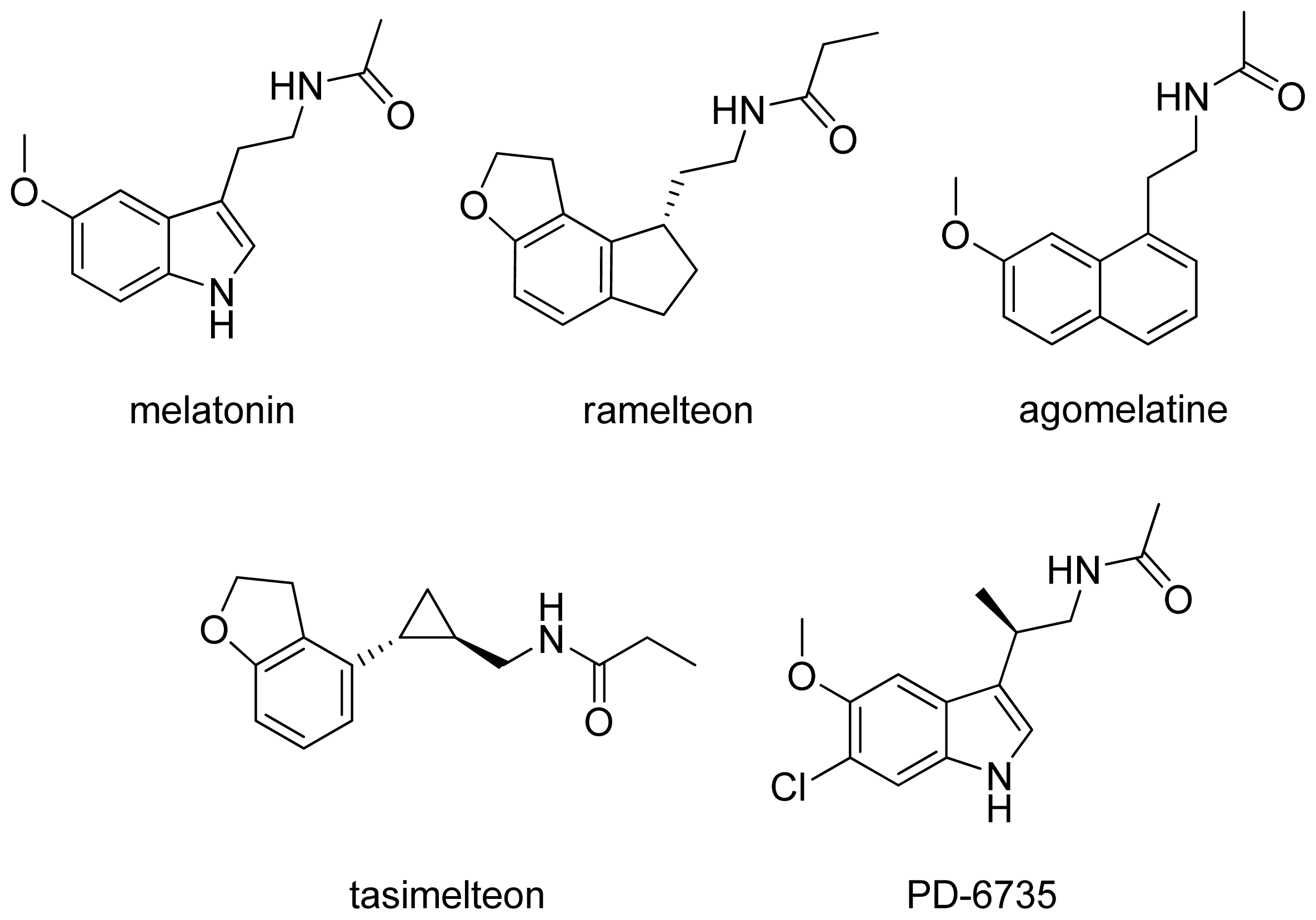{kind=link}
{kind=link}
{kind=link}
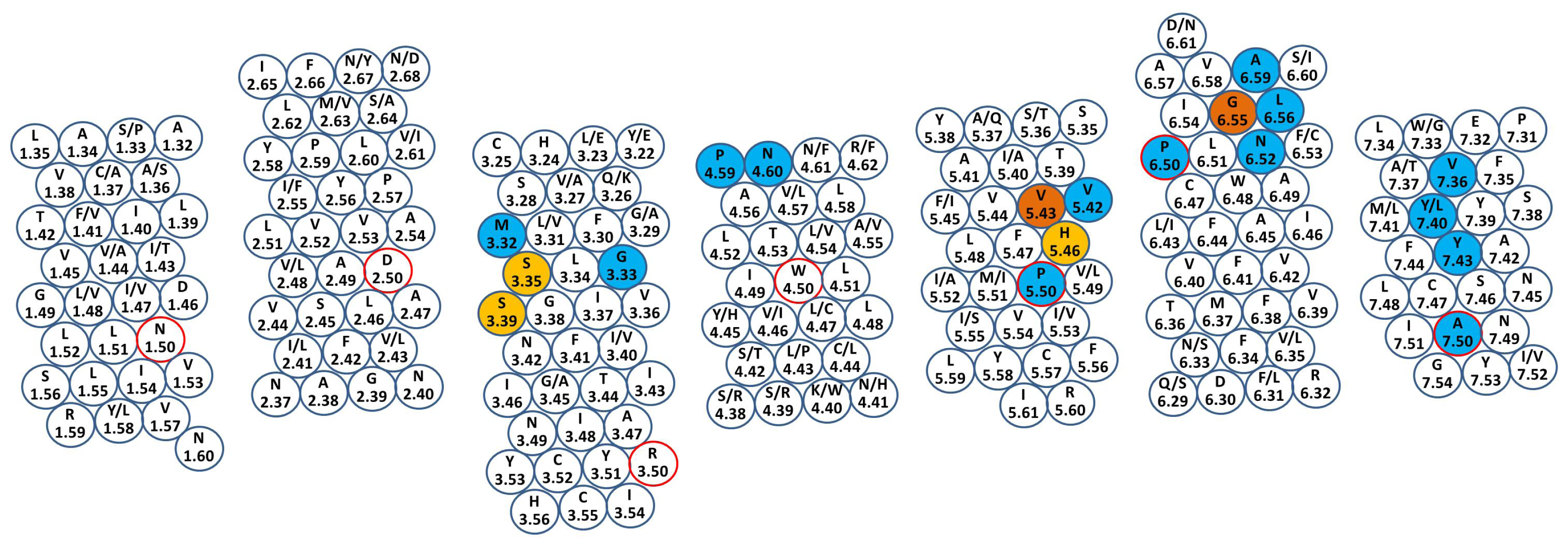{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MT1 and MT2 Melatonin Receptors
2.1. Structure
2.2. Mutagenesis Data
3. MT1 and MT2 Melatonin Receptor Models
3.1. MT1 Receptor Models
3.2. MT2 Receptor Models
4. Conclusions and Perspectives
Conflict of Interest
References
- Lerner, A.B.; Case, J.D.; Heinzelman, R.V. Structure of melatonin. J. Am. Chem. Soc 1959, 81, 6084–6085. [Google Scholar]
- Arendt, J. Melatonin: Characteristics, concerns, and prospects. J. Biol. Rhythms 2005, 20, 291–303. [Google Scholar]
- Pévet, P.; Bothorel, B.; Slotten, H.; Saboureau, M. The chronobiotic properties of melatonin. Cell Tissue Res 2002, 309, 183–191. [Google Scholar]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.M.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol 2008, 85, 335–353. [Google Scholar]
- Pandi-Perumal, S.R.; Srinivasan, V.; Maestroni, G.J.M.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin: Nature’s most versatile biological signal? FEBS J 2006, 273, 2813–2838. [Google Scholar]
- Lyssenko, V.; Nagorny, C.L.F.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spégel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet 2009, 41, 82–88. [Google Scholar]
- Sánchez-Barceló, E.J.; Mediavilla, M.D.; Tan, D.X.; Reiter, R.J. Clinical uses of melatonin: Evaluation of human trials. Curr. Med. Chem 2010, 17, 2070–2095. [Google Scholar]
- Spadoni, G.; Bedini, A.; Rivara, S.; Mor, M. Melatonin receptor agonists: New options for insomnia and depression treatment. CNS Neurosci. Ther. 2011, 17, 733–741. [Google Scholar]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev 2010, 62, 343–380. [Google Scholar]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem 2000, 275, 31311–31317. [Google Scholar]
- Reppert, S.M.; Weaver, D.R.; Ebisawa, T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron 1994, 13, 1177–1185. [Google Scholar]
- Reppert, S.M.; Godson, C.; Mahle, C.D.; Weaver, D.R.; Slaugenhaupt, S.A.; Gusella, J.F. Molecular characterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8734–8738. [Google Scholar]
- Ochoa-Sanchez, R.; Comai, S.; Lacoste, B.; Rodriguez Bambico, F.; Dominguez-Lopez, S.; Spadoni, G.; Rivara, S.; Bedini, A.; Angeloni, D.; Fraschini, F.; et al. Promotion of non-rapid eye movement sleep and activation of reticular thalamic neurons by a novel MT2 melatonin receptor ligand. J. Neurosci 2011, 31, 18439–18452. [Google Scholar]
- Rivara, S.; Mor, M.; Bedini, A.; Spadoni, G.; Tarzia, G. Melatonin receptor agonists: SAR and applications to the treatment of sleep-wake disorders. Curr. Top. Med. Chem 2008, 8, 954–968. [Google Scholar]
- Zlotos, D.P. Recent progress in the development of agonists and antagonists for melatonin receptors. Curr. Med. Chem 2012, 19, 3532–3549. [Google Scholar]
- Michino, M.; Abola, E.; Brooks, C.L., III; Dixon, J.S.; Moult, J.; Stevens, R.C. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat. Rev. Drug Discov. 2009, 8, 455–463. [Google Scholar]
- Kufareva, I.; Rueda, M.; Katritch, V.; Stevens, R.C.; Abagyan, R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19, 1108–1126. [Google Scholar]
- Beuming, T.; Sherman, W. Current assessment of docking into GPCR crystal structures and homology models: Successes, challenges, and guidelines. J. Chem. Inf. Model 2012, 52, 3263–3277. [Google Scholar]
- Forrest, L.R.; Tang, C.L.; Honig, B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J 2006, 91, 508–517. [Google Scholar]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar]
- Kolakowski, L.F., Jr. GCRDb: A G-protein-coupled receptor database. Recept. Channels 1994, 2, 1–7. [Google Scholar]
- Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Rosser, E.M.; Pin, J.-P.; Davenport, A.P.; Spedding, M.; Harmar, A.J. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev 2005, 57, 279–288. [Google Scholar]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 2003, 63, 1256–1272. [Google Scholar]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-dimensional Models and Computational Probing of Structure-function Relations in G Protein-coupled Receptors. In Methods in Neurosciences; Sealfon, S.C., Conn, P.M., Eds.; Academic Press: San Diego, CA USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Mirzadegan, T.; Benkö, G.; Filipek, S.; Palczewski, K. Sequence analyses of G-protein-coupled receptors: Similarities to rhodopsin. Biochemistry 2003, 42, 2759–2767. [Google Scholar]
- GMOS Web Interface. Available online: http://lmc.uab.cat/gmos/ (accessed on 4 February 2013).
- Klco, J.M.; Nikiforovich, G.V.; Baranski, T.J. Genetic analysis of the first and third extracellular loops of the C5a receptor reveals an essential WXFG motif in the first loop. J. Biol. Chem 2006, 281, 12010–12019. [Google Scholar]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci 2012, 33, 17–27. [Google Scholar]
- Duvernay, M.T.; Dong, C.; Zhang, X.; Robitaille, M.; Hébert, T.E.; Wu, G. A single conserved leucine residue on the first intracellular loop regulates ER export of G protein-coupled receptors. Traffic 2009, 10, 552–566. [Google Scholar]
- Conway, S.; Canning, S.J.; Barrett, P.; Guardiola-Lemaitre, B.; Delagrange, P.; Morgan, P.J. The roles of valine 208 and histidine 211 in ligand binding and receptor function of the ovine Mel1aβ melatonin receptor. Biochem. Biophys. Res. Commun 1997, 239, 418–423. [Google Scholar]
- Kokkola, T.; Foord, S.M.; Watson, M.-A.; Vakkuri, O.; Laitinen, J.T. Important amino acids for the function of the human MT1 melatonin receptor. Biochem. Pharmacol 2003, 65, 1463–1471. [Google Scholar]
- Gerdin, M.J.; Mseeh, F.; Dubocovich, M.L. Mutagenesis studies of the human MT2 melatonin receptor. Biochem. Pharmacol 2003, 66, 315–320. [Google Scholar]
- Almaula, N.; Ebersole, B.J.; Ballesteros, J.A.; Weinstein, H.; Sealfon, S.C. Contribution of a helix 5 locus to selectivity of hallucinogenic and nonhallucinogenic ligands for the human 5-hydroxytryptamine2A and 5-hydroxytryptamine2C receptors: Direct and indirect effects on ligand affinity mediated by the same locus. Mol. Pharmacol 1996, 50, 34–42. [Google Scholar]
- Wetzel, J.M.; Salon, J.A.; Tamm, J.A.; Forray, C.; Craig, D.; Nakanishi, H.; Cui, W.; Vaysse, P.J.; Chiu, G.; Weinshank, R.L.; et al. Modeling and mutagenesis of the human alpha 1a-adrenoceptor: Orientation and function of transmembrane helix V sidechains. Recept. Channels 1996, 4, 165–177. [Google Scholar]
- Song, Z.H.; Slowey, C.A.; Hurst, D.P.; Reggio, P.H. The difference between the CB1 and CB2 cannabinoid receptors at position 5.46 is crucial for the selectivity of WIN55212-2 for CB2. Mol. Pharmacol 1999, 56, 834–840. [Google Scholar]
- Conway, S.; Mowat, E.S.; Drew, J.E.; Barrett, P.; Delagrange, P.; Morgan, P.J. Serine residues 110 and 114 are required for agonist binding but not antagonist binding to the melatonin MT1 receptor. Biochem. Biophys. Res. Commun 2001, 282, 1229–1236. [Google Scholar]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Ijzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar]
- Gao, Z.G.; Ijzerman, A.P. Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodium ions. Biochem. Pharmacol 2000, 60, 669–676. [Google Scholar]
- Zuscik, M.J.; Porter, J.E.; Gaivin, R.; Perez, D.M. Identification of a conserved switch residue responsible for selective constitutive activation of the β2-adrenergic receptor. J. Biol. Chem 1998, 273, 3401–3407. [Google Scholar]
- Kam, K.W.L.; New, D.C.; Wong, Y.H. Constitutive activation of the opioid receptor-like (ORL1) receptor by mutation of Asn133 to tryptophan in the third transmembrane region. J. Neurochem 2002, 83, 1461–1470. [Google Scholar]
- Braden, M.R.; Nichols, D.E. Assessment of the roles of serines 5.43(239) and 5.46(242) for binding and potency of agonist ligands at the human serotonin 5-HT2A receptor. Mol. Pharmacol 2007, 72, 1200–1209. [Google Scholar]
- Pollock, N.J.; Manelli, A.M.; Hutchins, C.W.; Steffey, M.E.; MacKenzie, R.G.; Frail, D.E. Serine mutations in transmembrane V of the dopamine D1 receptor affect ligand interactions and receptor activation. J. Biol. Chem 1992, 267, 17780–17786. [Google Scholar]
- Gubitz, A.K.; Reppert, S.M. Chimeric and point-mutated receptors reveal that a single glycine residue in transmembrane domain 6 is critical for high affinity melatonin binding. Endocrinology 2000, 141, 1236–1244. [Google Scholar]
- Conway, S.; Drew, J.E.; Mowat, E.S.; Barrett, P.; Delagrange, P.; Morgan, P.J. Chimeric melatonin mt1 and melatonin-related receptors. J. Biol. Chem 2000, 275, 20602–20609. [Google Scholar]
- Shim, J.-Y.; Bertalovitz, A.C.; Kendall, D.A. Identification of essential cannabinoid-binding domains: Structural insights into early dynamic events in receptor activation. J. Biol. Chem 2011, 286, 33422–33435. [Google Scholar]
- Manivet, P.; Schneider, B.; Smith, J.C.; Choi, D.-S.; Maroteaux, L.; Kellermann, O.; Launay, J.-M. The serotonin binding site of human and murine 5-HT2B receptors: Molecular modeling and site-directed mutagenesis. J. Biol. Chem 2002, 277, 17170–17178. [Google Scholar]
- Mialet, J.; Dahmoune, Y.; Lezoualc’h, F.; Berque-Bestel, I.; Eftekhari, P.; Hoebeke, J.; Sicsic, S.; Langlois, M.; Fischmeister, R. Exploration of the ligand binding site of the human 5-HT4 receptor by site-directed mutagenesis and molecular modeling. Br. J. Pharmacol 2000, 130, 527–538. [Google Scholar]
- Mazna, P.; Berka, K.; Jelinkova, I.; Balik, A.; Svoboda, P.; Obsilova, V.; Obsil, T.; Teisinger, J. Ligand binding to the human MT2 melatonin receptor: The role of residues in transmembrane domains 3, 6, and 7. Biochem. Biophys. Res. Commun 2005, 332, 726–734. [Google Scholar]
- Mazna, P.; Obsilova, V.; Jelinkova, I.; Balik, A.; Berka, K.; Sovova, Z.; Ettrich, R.; Svoboda, P.; Obsil, T.; Teisinger, J. Molecular modeling of human MT2 melatonin receptor: The role of Val204, Leu272 and Tyr298 in ligand binding. J. Neurochem 2004, 91, 836–842. [Google Scholar]
- Kim, J.; Wess, J.; van Rhee, A.M.; Schöneberg, T.; Jacobson, K.A. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem 1995, 270, 13987–13997. [Google Scholar]
- Huang, X.P.; Williams, F.E.; Peseckis, S.M.; Messer, W.S., Jr. Differential modulation of agonist potency and receptor coupling by mutations of Ser388Tyr and Thr389Pro at the junction of transmembrane domain VI and the third extracellular loop of human M1 muscarinic acetylcholine receptors. Mol. Pharmacol 1999, 56, 775–783. [Google Scholar]
- Sautel, M.; Rudolf, K.; Wittneben, H.; Herzog, H.; Martinez, R.; Munoz, M.; Eberlein, W.; Engel, W.; Walker, P.; Beck-Sickinger, A.G. Neuropeptide Y and the nonpeptide antagonist BIBP 3226 share an overlapping binding site at the human Y1 receptor. Mol. Pharmacol 1996, 50, 285–292. [Google Scholar]
- Grånäs, C.; Larhammar, D. Identification of an amino acid residue important for binding of methiothepin and sumatriptan to the human 5-HT1B receptor. Eur. J. Pharmacol 1999, 380, 171–181. [Google Scholar]
- Yan, F.; Mosier, P.D.; Westkaemper, R.B.; Stewart, J.; Zjawiony, J.K.; Vortherms, T.A.; Sheffler, D.J.; Roth, B.L. Identification of the molecular mechanisms by which the diterpenoid salvinorin A binds to κ-opioid receptors. Biochemistry 2005, 44, 8643–8651. [Google Scholar]
- Roth, B.L.; Shoham, M.; Choudhary, M.S.; Khan, N. Identification of conserved aromatic residues essential for agonist binding and second messenger production at 5-hydroxytryptamine2A receptors. Mol. Pharmacol 1997, 52, 259–266. [Google Scholar]
- Mansour, A.; Taylor, L.P.; Fine, J.L.; Thompson, R.C.; Hoversten, M.T.; Mosberg, H.I.; Watson, S.J.; Akil, H. Key residues defining the μ-opioid receptor binding pocket: A site-directed mutagenesis study. J. Neurochem 1997, 68, 344–353. [Google Scholar]
- Befort, K.; Tabbara, L.; Kling, D.; Maigret, B.; Kieffer, B.L. Role of aromatic transmembrane residues of the δ-opioid receptor in ligand recognition. J. Biol. Chem 1996, 271, 10161–10168. [Google Scholar]
- Uchikawa, O.; Fukatsu, K.; Tokunoh, R.; Kawada, M.; Matsumoto, K.; Imai, Y.; Hinuma, S.; Kato, K.; Nishikawa, H.; Hirai, K.; et al. Synthesis of a novel series of tricyclic indan derivatives as melatonin receptor agonists. J. Med. Chem 2002, 45, 4222–4239. [Google Scholar]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar]
- Ivanov, A.A.; Voronkov, A.E.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. The study of the mechanism of binding of human ML1A melatonin receptor ligands using molecular modeling. Dokl. Biochem. Biophys 2004, 394, 49–52. [Google Scholar]
- Okada, T.; Fujiyoshi, Y.; Silow, M.; Navarro, J.; Landau, E.M.; Shichida, Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 5982–5987. [Google Scholar]
- Chugunov, A.O.; Farce, A.; Chavatte, P.; Efremov, R.G. Differences in binding sites of two melatonin receptors help to explain their selectivity to some melatonin analogs: A molecular modeling study. J. Biomol. Struct. Dyn 2006, 24, 91–107. [Google Scholar]
- Farce, A.; Chugunov, A.O.; Logé, C.; Sabaouni, A.; Yous, S.; Dilly, S.; Renault, N.; Vergoten, G.; Efremov, R.G.; Lesieur, D.; et al. Homology modeling of MT1 and MT2 receptors. Eur. J. Med. Chem 2008, 43, 1926–1944. [Google Scholar]
- Kokkola, T.; Watson, M.A.; White, J.; Dowell, S.; Foord, S.M.; Laitinen, J.T. Mutagenesis of human Mel1a melatonin receptor expressed in yeast reveals domains important for receptor function. Biochem. Biophys. Res. Commun 1998, 249, 531–536. [Google Scholar]
- Rivara, S.; Pala, D.; Lodola, A.; Mor, M.; Lucini, V.; Dugnani, S.; Scaglione, F.; Bedini, A.; Lucarini, S.; Tarzia, G.; et al. MT1-selective melatonin receptor ligands: Synthesis, pharmacological evaluation, and molecular dynamics investigation of N-{[(3-O-substituted)anilino]alkyl}amides. ChemMedChem 2012, 7, 1954–1964. [Google Scholar]
- Rasmussen, S.G.F.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; DeVree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar]
- Rivara, S.; Diamantini, G.; di Giacomo, B.; Lamba, D.; Gatti, G.; Lucini, V.; Pannacci, M.; Mor, M.; Spadoni, G.; Tarzia, G. Reassessing the melatonin pharmacophore—Enantiomeric resolution, pharmacological activity, structure analysis, and molecular modeling of a constrained chiral melatonin analogue. Bioorg. Med. Chem 2006, 14, 3383–3391. [Google Scholar]
- Jaakola, V.-P.; Lane, J.R.; Lin, J.Y.; Katritch, V.; Ijzerman, A.P.; Stevens, R.C. Ligand binding and subtype selectivity of the human A2A adenosine receptor. J. Biol. Chem 2010, 285, 13032–13044. [Google Scholar]
- Conner, M.; Hawtin, S.R.; Simms, J.; Wootten, D.; Lawson, Z.; Conner, A.C.; Parslow, R.A.; Wheatley, M. Systematic analysis of the entire second extracellular loop of the V1a vasopressin receptor. J. Biol. Chem 2007, 282, 17405–17412. [Google Scholar]
- Cavalli, A.; Fanelli, F.; Taddei, C.; de Benedetti, P.G.; Cotecchia, S. Amino acids of the α1B-adrenergic receptor involved in agonist binding: Differences in docking catecholamines to receptor subtypes. FEBS Lett 1996, 399, 9–13. [Google Scholar]
- Grol, C.J.; Jansen, J.M. The high affinity melatonin binding site probed with conformationally restricted ligands II. Homology modeling of the receptor. Bioorg. Med. Chem 1996, 4, 1333–1339. [Google Scholar]
- Rivara, S.; Lorenzi, S.; Mor, M.; Plazzi, P.V.; Spadoni, G.; Bedini, A.; Tarzia, G. Analysis of structure-activity relationships for MT2 selective antagonists by melatonin MT1 and MT2 receptor models. J. Med. Chem 2005, 48, 4049–4060. [Google Scholar]
- Okada, T.; Sugihara, M.; Bondar, A.-N.; Elstner, M.; Entel, P.; Buss, V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J. Mol. Biol 2004, 342, 571–583. [Google Scholar]
- Ballesteros, J.A.; Shi, L.; Javitch, J.A. Structural mimicry in G protein-coupled receptors: Implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol. Pharmacol 2001, 60, 1–19. [Google Scholar]
- Durieux, S.; Chanu, A.; Bochu, C.; Audinot, V.; Coumailleau, S.; Boutin, J.A.; Delagrange, P.; Caignard, D.H.; Bennejean, C.; Renard, P.; et al. Design and synthesis of 3-phenyltetrahydronaphthalenic derivatives as new selective MT2 melatoninergic ligands. Part II. Bioorg. Med. Chem 2009, 17, 2963–2974. [Google Scholar]
- Karageorge, G.N.; Bertenshaw, S.; Iben, L.; Xu, C.; Sarbin, N.; Gentile, A.; Dubowchik, G.M. Tetrahydroisoquinoline derivatives as melatonin MT2 receptor antagonists. Bioorg. Med. Chem. Lett 2004, 14, 5881–5884. [Google Scholar]
- Voronkov, A.E.; Ivanov, A.A.; Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Molecular modeling study of the mechanism of ligand binding to human melatonin receptors. Dokl. Biochem. Biophys 2005, 403, 284–288. [Google Scholar]
- Teh, M.T.; Sugden, D. Comparison of the structure-activity relationships of melatonin receptor agonists and antagonists: Lengthening the N-acyl side-chain has differing effects on potency on Xenopus melanophores. Naunyn Schmiedebergs Arch. Pharmacol 1998, 358, 522–528. [Google Scholar]
- Lira-Rocha, A.; Espejo-González, O.; Naranjo-Rodríguez, E.B. Receptor-binding studies of 1-N-substituted melatonin analogues. Eur. J. Med. Chem 2002, 37, 945–951. [Google Scholar]
- Garratt, P.J.; Travard, S.; Vonhoff, S.; Tsotinis, A.; Sugden, D. Mapping the melatonin receptor. 4. Comparison of the binding affinities of a series of substituted phenylalkyl amides. J. Med. Chem 1996, 39, 1797–1805. [Google Scholar]
- Mazna, P.; Grycova, L.; Balik, A.; Zemkova, H.; Friedlova, E.; Obsilova, V.; Obsil, T.; Teisinger, J. The role of proline residues in the structure and function of human MT2 melatonin receptor. J. Pineal Res 2008, 45, 361–372. [Google Scholar]
- Scheerer, P.; Park, J.H.; Hildebrand, P.W.; Kim, Y.J.; Krauss, N.; Choe, H.-W.; Hofmann, K.P.; Ernst, O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008, 455, 497–502. [Google Scholar]
- Zefirova, O.N.; Baranova, T.Y.; Ivanova, A.A.; Ivanov, A.A.; Zefirov, N.S. Application of the bridgehead fragments for the design of conformationally restricted melatonin analogues. Bioorg. Chem 2011, 39, 67–72. [Google Scholar]
- Ivanov, A.A.; Barak, D.; Jacobson, K.A. Evaluation of homology modeling of G-protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J. Med. Chem 2009, 52, 3284–3292. [Google Scholar]
- Kneissl, B.; Leonhardt, B.; Hildebrandt, A.; Tautermann, C.S. Revisiting automated G-protein coupled receptor modeling: The benefit of additional template structures for a neurokinin-1 receptor model. J. Med. Chem 2009, 52, 3166–3173. [Google Scholar]
- Evers, A.; Klebe, G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J. Med. Chem 2004, 47, 5381–5392. [Google Scholar]
- Johnston, J.M.; Filizola, M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Curr. Opin. Struct. Biol 2011, 21, 552–558. [Google Scholar]
- Goldfeld, D.A.; Zhu, K.; Beuming, T.; Friesner, R.A. Successful prediction of the intra- and extracellular loops of four G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 8275–8280. [Google Scholar]
- Goldfeld, D.A.; Zhu, K.; Beuming, T.; Friesner, R.A. Loop prediction for a GPCR homology model: Algorithms and results. Proteins 2013, 81, 214–228. [Google Scholar]











© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pala, D.; Lodola, A.; Bedini, A.; Spadoni, G.; Rivara, S. Homology Models of Melatonin Receptors: Challenges and Recent Advances. Int. J. Mol. Sci. 2013, 14, 8093-8121. https://doi.org/10.3390/ijms14048093
Pala D, Lodola A, Bedini A, Spadoni G, Rivara S. Homology Models of Melatonin Receptors: Challenges and Recent Advances. International Journal of Molecular Sciences. 2013; 14(4):8093-8121. https://doi.org/10.3390/ijms14048093
Chicago/Turabian StylePala, Daniele, Alessio Lodola, Annalida Bedini, Gilberto Spadoni, and Silvia Rivara. 2013. "Homology Models of Melatonin Receptors: Challenges and Recent Advances" International Journal of Molecular Sciences 14, no. 4: 8093-8121. https://doi.org/10.3390/ijms14048093