 Samantha R. Furr Ian Marriott*
Samantha R. Furr Ian Marriott*- Department of Biology, University of North Carolina at Charlotte, Charlotte, NC, USA
Viruses are the major causative agents of central nervous system (CNS) infection worldwide. RNA and DNA viruses trigger broad activation of glial cells including microglia and astrocytes, eliciting the release of an array of mediators that can promote innate and adaptive immune responses. Such responses can limit viral replication and dissemination leading to infection resolution. However, a defining feature of viral CNS infection is the rapid onset of severe neuroinflammation and overzealous glial responses are associated with significant neurological damage or even death. The mechanisms by which microglia and astrocytes perceive neurotropic RNA and DNA viruses are only now becoming apparent with the discovery of a variety of cell surface and cytosolic molecules that serve as sensors for viral components. In this review we discuss the role played by members of the Toll-like family of pattern recognition receptors (PRRs) in the inflammatory responses of glial cells to the principle causative agents of viral encephalitis. Importantly, we also describe the evidence for the involvement of a number of newly described intracellular PRRs, including retinoic acid-inducible gene I and DNA-dependent activator of IFN regulatory factors, that are thought to function as intracellular sensors of RNA and DNA viruses, respectively. Finally, we explore the possibility that cross-talk exists between these disparate viral sensors and their signaling pathways, and describe how glial cytosolic and cell surface/endosomal PRRs could act in a cooperative manner to promote the fulminant inflammation associated with acute neurotropic viral infection.
Microbial and viral infections of the brain and spinal cord result in inflammation that underlies wide ranging symptoms including fever, headache, confusion, and in extreme cases, stroke, seizure, or even death. The resulting disease is classified based on the area of the central nervous system (CNS) affected and can include the membranes that surround the brain (meningitis), spinal cord (myelitis), or inflammation within the brain parenchyma (encephalitis). Of these, encephalitis tends to be associated with more significant alterations in the level of consciousness, cognitive defects, seizures, and focal neurological deficits than meningitis, although it should be noted that clinical presentation of these conditions often overlap. The incidence of encephalitis in the United States has been estimated at approximately 20,000 new cases annually according to the Centers for Disease Control (CDC). While encephalitis and meningitis can be caused by bacterial or fungal pathogens, viruses are the major causative agents of CNS infection worldwide, with an overall incidence of 6.34 per 100,000 for all ages (Jmor et al., 2008). However, it must be noted that this number is likely to be an underestimation given the problems associated with diagnosis, particularly in developing countries (Sejvar, 2006).
Viral CNS infections can be very difficult to treat due to the rapid onset of severe disease symptoms that can occur even in otherwise healthy individuals. Such infections trigger broad activation of glial cells including microglia and astrocytes, and the concomitant release of an array of pro-inflammatory molecules that can elicit both innate and adaptive immune responses. While host responses limit viral replication and dissemination and can lead to infection resolution, overzealous, or sustained inflammation within the CNS can result in significant neurological damage.
One of the major challenges in the study and treatment of viral CNS infections is the wide range of etiological agents (Tunkel et al., 2008). Both RNA and DNA viruses can infect the CNS and cause meningitis or encephalitis, although an etiological diagnosis is established in less than 30% of cases (Stahl et al., 2008). While historically important RNA viruses that can cause encephalitis such as measles, mumps, and rubella have seen steep decreases in incidence as a result of immunization (Koskiniemi et al., 1997), rabies virus (RABV) remains an endemic cause of viral encephalitis in the United States and a serious public health concern. RABV is transmitted from animals to humans and although rare, CNS infection results in uncontrolled virus replication, a prominent pro-inflammatory response, and is 100% fatal once neurological symptoms develop (Theerasurakarn and Ubol, 1998).
In addition, emerging RNA viral pathogens such as West Nile virus (WNV; Gubler, 2008), Japanese encephalitis virus (JEV), Australian bat Lyssavirus (Samaratunga et al., 1998), retroviruses, and Nipah virus (McCormack and Allworth, 2002) have become increasingly important causes of encephalitis. Nipah virus was first identified in 1999 and the mortality rate associated with acute encephalitis caused by this virus has been estimated to be between 40 and 75% (as reviewed in Tyler, 2009). While most survivors make a full recovery, 20% demonstrate neurological sequelae and a small number develop delayed-onset encephalitis.
Worldwide, the flavivirus JEV is the most common cause of arthropod-borne encephalitis with over 50,000 cases reported per year in China, Southeast Asia, and India (Whitley and Kimberlin, 1999). Epidemics due to this arbovirus result in a mortality rate that ranges from 30 to 50% with death usually occurring within the first week (Roos, 1999; Solomon et al., 2003), and the development of sustained neurological deficits in approximately half of the survivors (Ghosh and Basu, 2009). Another flavivirus, WNV, is also transmitted by Culex spp. mosquitoes and can cause fatal encephalitis or long-term neurological sequelae. Once inside the CNS, JEV, and WNV infect neurons (Wang et al., 1997; Shrestha et al., 2003; Koh and Ng, 2005) leading to neuronal apoptosis (Liao et al., 1998; Parquet et al., 2001) and causing severe immunopathology (Licon Luna et al., 2002). WNV has been associated with over 12,000 cases of encephalitis and/or meningitis in the U.S. with a mortality rate of 9.5% as reported by the CDC.
DNA viral agents that can infect the CNS include varicella zoster virus (VZV) and several viruses in the human herpesvirus family that can cause encephalitis, including human herpesvirus 6 (Isaacson et al., 2005), herpes simplex virus 1 (HSV-1), and HSV-2 (Baringer, 2008). VZV is a medically important cause of encephalitis characterized by up to 25% mortality and rates close to 100% in immunocompromised patients (Stahl et al., 2008). However, HSV is considered to be an endemic cause of encephalitis in the United States and is the most frequent cause of this disease in adults (Stahl et al., 2008) while HSV-2 is the most significant etiological agent of severe encephalitis in children (Roos, 1999; Bingham and Saiman, 2000). Neonatal HSV-2 encephalitis, with treatment, has a mortality rate of 14% (compared to 85% without treatment) and severe neurological dysfunction is observed in 50–70% of surviving individuals. HSV-1 encephalitis in older children represents the most common cause of sporadic fatal viral encephalitis (Baringer, 2008). Untreated patients with HSV-1 encephalitis have a 70% mortality rate, while patients who receive early treatment have a 40% chance of recovering without neurological deficits. However, despite appropriate diagnosis and therapy, the mortality rate remains at 30% (Xu et al., 2006; Baringer, 2008). HSV encephalitis may follow primary infection but most cases are the due to the reactivation of latent virus in the olfactory bulb or trigeminal ganglia and subsequent retrograde axonal transport into the CNS (as reviewed in Conrady et al., 2010).
The complexity seen in viral encephalitis due to the array of causative agents, clinical presentations, and severity has made defining the mechanisms that underlie disease pathology challenging. Ideally, host responses following CNS infection should result in rapid neutralization of the invading pathogen with minimal collateral damage to the sensitive and poorly regenerating neural tissue. However, viral CNS infections are often associated with inadequate anti-viral responses and/or the rapid and severe onset of damaging inflammation (as reviewed in Savarin and Bergmann, 2008). While the brain has traditionally been viewed as a “victim organ” of infiltrating leukocytes, it has become apparent that specialized resident glial cells, such as microglia and astrocytes, play an essential role in regulating the permeability of the blood–brain barrier, promoting the recruitment of leukocytes, and the activation of such cells following infiltration.
Glial Cells Play a Critical Role in the Initiation of Virally Induced Neuroinflammation
Microglia and astrocytes are well known to play an important role in homeostasis within the CNS and to support neuronal cell function. However, these cell types are now recognized to be key players in the development of protective immune responses or the progression of damaging inflammation during CNS disease states (Bauer et al., 1995; Stoll and Jander, 1999; Dong and Benveniste, 2001; Fischer and Reichmann, 2001). Microglia are resident myeloid immune cells of the CNS and, similar to other myeloid cells such as macrophages and dendritic cells, these cells are facultative phagocytes and express antigen presenting MHC class II molecules (Hickey and Kimura, 1988). Importantly, microglia produce key pro-inflammatory mediators such as IL-1β (Martin et al., 1993), TNF-α (Streit et al., 1998), IL-6 (Kiefer et al., 1993), and bioactive IL-12 p70 (Aloisi et al., 1997; Suzumura et al., 1998) following activation. Astrocytes are the major glial cell type in the brain and may also function as immune effector cells (Dong and Benveniste, 2001). Stimulated astrocytes can express an array of inflammatory cytokines and chemokines that can initiate leukocyte migration across the blood–brain barrier and promote effector functions in these infiltrating cells (Dong and Benveniste, 2001). As such, these cells are ideally situated to detect and respond to invading pathogens including neurotropic viruses.
Viral infections in the CNS are known to broadly trigger glial activation and the release of pro-inflammatory molecules. For example, it is known that pro-inflammatory cytokines, IFN-γ and TNF-α, are markedly increased in CNS tissues during HSV-1 infection in the brain and microglia have been shown to respond to HSV-1 by secreting pro-inflammatory and chemotactic molecules such as TNF-α, IL-1β, IL-6, IL-12, CCL7, CCL8, CCL9, CXCL1, CXCL2, CXCL4, and CXCL5 (Lokensgard et al., 2001; Aravalli et al., 2006). Human microglia have also been shown to respond to RNA viruses including WNV by producing cytokines and chemokines (Cheeran et al., 2005). Elevated levels of IL-1β are readily detectable in neural tissue from WNV encephalitis patients and cultured human glia produce this potent inflammatory cytokine in response to WNV challenge (van Marle et al., 2007). We have also documented the ability of primary cultured microglia and astrocytes to respond to several RNA and DNA viruses, including vesicular stomatitis virus (VSV), Sendai virus, murine gammaherpesvirus (MHV)-68, and HSV-1, by producing inflammatory mediators such as IL-6, TNF-α, and IL-1β (Rasley et al., 2004; Chauhan et al., 2010; Furr et al., 2010, 2011). Importantly, we confirmed that in vivo VSV administration results in viral infection of both glial cell types in situ, while earlier studies demonstrated that microglia and astrocytes respond to VSV by proliferating, producing inducible nitric oxide synthase, and increasing the expression of cell surface MHC class II molecules (Bi et al., 1995), responses that are likely to set the stage for subsequent inflammatory damage.
An immune response to a virus in the CNS requires the recognition of this pathogen and the mechanisms by which innate immune cells achieve this have only recently become apparent with the discovery of a variety of cell surface and cytosolic molecules that serve as sensors for viral components. Such pattern recognition receptors (PRRs) are present on and within immune sentinel cells (Furr et al., 2008, 2011; Kielian, 2009) and appear to bind conserved viral structures known as pathogen-associated molecular patterns (PAMPs). These PAMPs have several features that make them ideal targets for innate immune recognition. First, PAMPs are unique to microbes and viruses allowing for “self” versus “non-self” recognition by the innate immune system. Second, PAMPs are often conserved among similar pathogens allowing a relatively small number of PRRs to detect a broad array of challenges. In addition, PAMPs tend to be essential for microbial/viral survival making pathogen evasion by genetic mutation or deletion unlikely (Medzhitov and Janeway, 2002).
Pattern recognition receptor engagement by PAMPs can precipitate the production of anti-viral type I interferons (IFN) and/or the release of chemotactic and inflammatory molecules that can then direct a subsequent adaptive immune response. Three families of PRRs have been defined: Toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid inducible gene (RIG)-I-like receptors (RLRs). As such, these molecules represent important mechanisms by which a protective host response or potentially damaging inflammation is initiated (Kawai and Akira, 2007; Yoneyama and Fujita, 2010).
TLRs Mediate Glial Responses to Extra Cellular or Endosomal Viral Motifs
Toll was first identified as a gene critical for fruit fly embryogenesis (Hashimoto et al., 1988) but its products were subsequently shown to play an important role in Drosophila’s immunity to fungal infections (Lemaitre et al., 1996). Interestingly, similar gene products were found in mammalian cells and these TLRs have since been shown to play an important role in the initiation of innate immune responses to infection (Medzhitov et al., 1997). To date, at least 13 TLRs have been discovered in mammals and members of this family of cell surface receptors can elicit inflammatory and/or anti-viral mediator production and can serve to initiate or modify adaptive immune responses (as reviewed in Kawai and Akira, 2008). Acute virally induced CNS inflammatory responses occur within the first few days following infection and this argues against a central role for activated T cell clones in such responses. Indeed, previous studies have shown that acute viral encephalitis can occur in athymic mice (Bi et al., 1995). That being said, T cell subpopulations, such as Th1 and Th17 cells, are almost certain to play a key role in sustained virally induced neuroinflammation (Kalinke and Prinz, 2012). As such, glial activation via TLRs and other PRRs is likely to facilitate antigen presentation to, and costimulation of, such cells and the role of these receptors in the antigen-specific activation of T cells following viral challenge has been extensively reviewed elsewhere (Kawai and Akira, 2008).
Several TLRs have been demonstrated to specifically recognize viral motifs, including TLRs 2, 3, 7, 8, and 9. While TLR2 is best known to bind a variety of microbial cell wall component including lipoproteins, peptidoglycans, and lipoteichoic acid present in bacterial cell walls and the yeast cell wall component zymosan, this sensor can also recognize as yet unidentified viral motifs. In contrast, endosomal TLRs such as TLR3 recognize viral double-stranded RNA (dsRNA) and its synthetic analog, polyinosine-deoxycytidylic acid (poly(I:C)), while TLR7 and TLR8 mediate responses to GU-rich single-stranded RNA (ssRNA) produced in virus-infected cells. Finally, TLR9 detects bacterial and viral DNA with unmethylated CpG motifs. Studies from our group and others have demonstrated that microglia and astrocytes constitutively express low levels of TLR3 (Jack et al., 2005; Bsibsi et al., 2006), TLR7 (Jack et al., 2005), and TLR9 (Bowman et al., 2003; Jack et al., 2005), but such expression shows marked upregulation following viral infection and/or stimulation with viral components (Bowman et al., 2003; Carpentier et al., 2005; Jack et al., 2005; McKimmie and Fazakerley, 2005; McKimmie et al., 2005; Bsibsi et al., 2006). While these findings suggest that glial cells could become sensitized to the presence of viral pathogens, it is presently unclear whether sustained stimulation via these sensors can alter the pattern of immune mediator production by glial cells to one that favors the resolution of inflammation.
Upon ligand binding, TLRs dimerize and undergo conformational changes precipitating a complex cascade of intracellular signaling events that ultimately result in activation of transcription factors including nuclear factor-κB (NF-κB), and interferon-regulatory factors (IRF) 3 and 7 (as reviewed in Kawai and Akira, 2008; Wilkins and Gale, 2010). NF-κB is a critical transcription factor in the regulation of inflammatory cytokine production, while IRF-3 and IRF-7 activation stimulates expression of anti-viral genes including IFN-β. Hence, activation of cells via these receptors can initiate the repertoire of defense mechanisms used by the innate immune system against viral pathogens.
TLR3 is perhaps the most widely studied PRR in the perception of viral pathogens by glial cells. Isolated murine wild-type microglia are known to respond to the TLR3 ligand poly(I:C) by secreting TNF-α and IL-6 (Melton et al., 2003). In line with these findings, intracerebral administration of poly(I:C) has been shown to elicit microglial (Melton et al., 2003; Town et al., 2006) and astrocyte (Melton et al., 2003) activation associated with neuronal loss, a response that is nearly absent in TLR3 knockout animals (Town et al., 2006). Circumstantial evidence for the involvement of TLR3 in viral encephalitis comes from the finding that the expression of this PRR is upregulated following RABV infection prior to the onset of lethal neuroinflammation (McKimmie and Fazakerley, 2005; McKimmie et al., 2005).
Several studies have implicated TLR3 activation in WNV-associated pathogenicity. dsRNA, a ligand for TLR3, is produced by WNV during infection and has been shown to trigger the release of cytokines and chemokines from human microglia (Cheeran et al., 2005). Seemingly in keeping with these observations, TLR3 deficient mice were found to be resistant to lethal WNV-associated encephalitis. The fulminate CNS inflammatory response seen in wild-type mice, characterized in part by activated microglia, was markedly reduced or absent in knockout animals following intraperitoneal WNV infection (Wang and Fikrig, 2004). However, this reduced CNS disease occurs despite increased peripheral viral loads and reduced systemic production of anti-viral and pro-inflammatory cytokines (Wang et al., 2004). These results suggest that decreased systemic inflammation in the absence of TLR3 expression may maintain blood–brain barrier integrity and prevent WNV access into the CNS (Wang et al., 2004). Such a hypothesis is supported by the demonstration that direct intracerebral administration of WNV leads to identical outcomes in wild-type and TLR3 deficient mice (Wang et al., 2004). However, it must be noted that at least one study appears to contradict this finding in that neuronal WNV replication was increased in TLR3 knockout animals following subcutaneous infection and the reason for this apparent disparity is presently unclear (Daffis et al., 2008).
Another endosome-associated PRR, TLR7, also appears to play an important role in inflammatory and/or protective glial responses to viral pathogens. Intracerebral administration of the TLR7 agonist imiquimod leads to pronounced neuroinflammation, with upregulated expression of TNF-α, IL-1α, IL-1β, IL-6, and IL-12 as well as the chemokines CCL2, CCL3, CXCL1, CXCL9, and CXCL10 (Butchi et al., 2008). Interestingly however, this effect appears to be predominantly due to the activation of astrocytes rather than microglia (Butchi et al., 2008). Upregulation of TLR7 has been demonstrated in the brains of infected mice prior to lethal RABV-induced inflammation (McKimmie et al., 2005). Furthermore, mice lacking the expression of MyD88, a critical downstream effector molecule of TLRs including TLR7, have increased susceptibility to an attenuated RABV strain and TLR7 deficient mice exhibited a phenotype with deficits in both the development of peripheral immunity and RABV clearance from the CNS (Li et al., 2011). As such, these data indicate a possible role for TLR7 in viral clearance from infected brain tissue. In contrast, another report has shown that macrophages isolated from TLR7 deficient animals released less IL-12 and IL-23 in response to in vitro WNV infection and TLR7 knockout mice demonstrated an increased susceptibility to WNV encephalitis (Town et al., 2009).
It has recently been shown that TLR2 contributes to detrimental inflammatory cytokine production in mice following intracranial administration with HSV-1, with the absence of TLR2 expression leading to decreased peripheral (Kurt-Jones et al., 2004) and CNS (Aravalli et al., 2006) inflammatory response sand increased survival. In addition, multiple reports have shown that maximal in vitro microglial and astrocyte responses to HSV-1 are dependent on the presence of TLR2 (Lokensgard et al., 2001; Aravalli et al., 2006; Schachtele et al., 2010; Wang et al., 2012) and HSV-1-infected microglia derived from TLR2 deficient mice have been demonstrated to elicit less neuronal oxidative damage and death in mixed neural cell cultures in comparison to infected wild-type microglia (Schachtele et al., 2010). However, it should be noted that these studies differ in their assessments of the relative importance of this PRR in such responses, and marked elevations in serum IL-6 concentrations have been found in TLR2 deficient mice following HSV-1 infection (Kurt-Jones et al., 2004). Together, these data suggest while TLR2 may play a major role in HSV-induced pathology and mortality, additional mechanism(s) are likely to be involved.
In addition to the detection of HSV virion motifs by TLR2, genomic HSV DNA can be recognized by TLR9 and at least one study suggests that this sensor might play a more significant role in lethal HSV-1 encephalitis than TLR2 (Lima et al., 2010). In contrast, others have shown that the absence of TLR9 alone does not impact survival, type I IFN levels, or viral replication in the brain following HSV infection (Wang et al., 2012). Since TLR2/9 double knockout mice show significantly greater susceptibility to HSV-1 infection than mice deficient in either TLR2 or TLR9 alone (Lima et al., 2010), it appears likely that these sensors act in a cooperative or synergistic manner.
Taken together, this body of work suggests that TLR activation is a significant contributor to sustained glial activation, inflammatory mediator production, and neuronal loss during viral CNS infections. However, it is important to note that TLRs can also mediate beneficial effects. For example, the activation of glial cells via TLR2, 3, or 9 has been shown to initiate the production of type I IFNs (Zhou et al., 2009; Conrady et al., 2010) which limit viral replication through the induction of proteins such as RNase L and IFN-induced dsRNA-activated protein kinase, resulting in mRNA degradation and cessation of translation, respectively (Samuel, 2001). Furthermore, two recent studies (Zhang et al., 2007; Guo et al., 2011) indicate that a genetic deficiency in TLR3 expression can predispose children to HSV-1 encephalitis and implicate a key role for this PRR in protective CNS immune responses against this, but not other, pathogens. However, the mechanisms that keep the balance between damaging and protective glial responses have not been identified and are likely to include a multitude of variables including the number of infectious organisms, the kinetics of glial activation and immune mediator production, the specific identity of the responsive glial cell, and the direct influence of the pathogen itself on the cellular response. Moreover, evidence is accumulating that TLR expression is not limited to glial cells and that neurons can also express such PRRs under certain conditions (as reviewed in Carty and Bowie, 2011), although, the functional relevance of TLR expression in neurons has not been resolved.
While cell-surface receptors like TLRs appear to play an important role in glial responses to extracellular pathogens or those present in intracellular compartments, the ability of such cells to respond to pathogens in the absence of TLR expression suggests that additional mechanisms are present. Furthermore, our recent studies employing the model neurotropic RNA virus, VSV, demonstrate that viral replication is required to elicit robust immune responses by infected microglia and astrocytes, and indicate that mechanisms exist to sense the presence of replicative viral products within infected cells. We have shown that heat inactivated wild-type VSV or a host-range limited VSV mutant (Grdzelishvili et al., 2005) elicit glial immune responses that are an order of magnitude smaller than those induced by wild-type viral particles (Chauhan et al., 2010). These results suggest that VSV-induced glial responses are not predominantly mediated by cell surface and/or endosomal PRRs and indicate that active viral replication is a critical requirement for such responses.
The recent demonstration that microglia and astrocytes express members of the newly described RLR family of PRRs may provide a mechanism underlying the replication-dependent nature of glial responses (Furr et al., 2008). RLRs such as RIG-I and melanoma differentiation-associated gene 5 (MDA5) have been shown to serve as intracellular PRRs for viral nucleic acid motifs (Kato et al., 2006; Takeuchi and Akira, 2008), and their involvement in inflammatory and anti-viral responses in other cell types raises the intriguing possibility that these cytosolic sensors could play an important role in glial responses to viral infection.
RIG-Like Receptors can Mediate Protective Immune Responses and Damaging CNS Inflammation Following RNA Virus Infection
In contrast to cell-surface TLRs, RLRs detect viral RNA motifs or processed self-RNA in the cytoplasm to trigger innate immunity and inflammation (Takeuchi and Akira, 2010). These intracellular sensors are responsible for the production of type I IFNs in most cell types in response to RNA virus infection (Sumpter et al., 2005; Kato et al., 2006; Saito et al., 2007; Lui et al., 2008). In addition to such anti-viral responses, RLR engagement also elicits robust inflammatory cytokine production (Takeuchi and Akira, 2010). As shown in Figure 1, helicase interaction with viral RNA induces the recruitment of downstream effector molecules including interferon promoter stimulator (IPS)-1 (also known as VISA, Cardif, or MAVS), TNF receptor associated factor (TRAF) 3 (Takeuchi and Akira, 2010), and perhaps stimulator of interferon genes (STING; Ishikawa and Barber, 2008), leading to the activation of mitogen activated protein (MAP) kinases, and the IκB kinase (IKK)-related kinases, TRAF family member-associated NF-κB activator (TANK) binding kinase 1 and IKK-I. These kinases activate the transcription factors NF-κB and IRF-3, which translocate into the nucleus and activate transcription of IFN stimulated genes and inflammatory cytokines. RLRs are comprised of three domains: N-terminal caspase activation and recruitment domains (CARDs), a helicase domain, and a C-terminal domain (CTD). Although RIG-I and MDA5 show structural homology (Yoneyama et al., 2004) and share signaling features (Yoneyama et al., 2005), it is now known that the two helicases discriminate between differing ligands to trigger innate immune responses to RNA viruses.
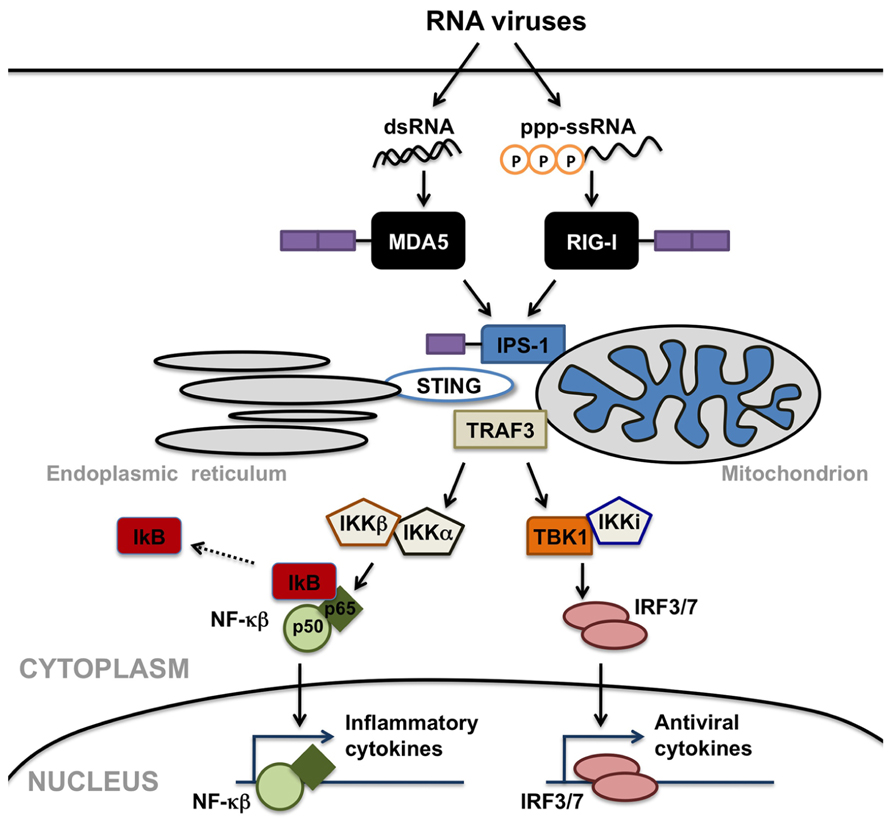
FIGURE 1. Retinoic acid inducible gene (RIG)-I-like receptors serve as cytosolic sensors for viral RNA motifs and can initiate anti-viral and inflammatory cell responses. Melanoma differentiation-associated gene 5 (MDA5) recognizes double-stranded RNA moieties (dsRNA), while RIG-I perceives short single or double-stranded RNAs with 5’ triphosphate ends (ppp-ssRNA). Helicase interaction with these viral RNAs induces the recruitment of downstream effector molecules including mitochondrial- or peroxisomal-associated interferon promoter stimulator (IPS)-1, TNF receptor associated factor (TRAF) 3, and/or stimulator of interferon genes (STING), leading to the activation of IκB kinase (IKK)-related kinases, TRAF family member-associated NF-κB activator binding kinase 1 (TBK1), and IKK-I. These kinases activate the transcription factors NF-κB and interferon-regulatory factors (IRF) 3, which translocate into the nucleus and activate transcription of anti-viral and inflammatory cytokines.
Signaling by RIG-I is triggered during infection by a number of RNA viruses and by the presence of synthetic RNA transcribed in vitro (Sumpter et al., 2005; Kato et al., 2006; Saito et al., 2007; Lui et al., 2008; Rehwinkel et al., 2010). It appears that RIG-I recognizes ssRNA or dsRNA moieties with 5′ triphosphate ends (5′ppp-ssRNA and 5′ppp-dsRNA, respectively; Hornung et al., 2006; Pichlmair et al., 2006) or RNAs that assume complex secondary structures (Sumpter et al., 2005; Saito et al., 2007). More recent studies have elucidated the structural basis underlying RIG-I activation. Inactive RIG-I has an open conformation in which the CARD domains are sequestered by a helical domain inserted between the helicase moieties. When activated, ATP and RNA binding induce a major rearrangement in RIG-I to a closed conformation in which the helicase and the CTD bind to the blunt end 5′ppp-dsRNA with perfect complementarity (Kowalinski et al., 2011).
In contrast, MDA5 has been reported to be more selective and is triggered during picornavirus infections or in the presence of poly(I:C) (Gitlin et al., 2006; Kato et al., 2006; Loo et al., 2008). The CTD domains of MDA5 and RIG-I involved in RNA binding have been suggested to be responsible for the differences in ligand affinity seen between these RLRs. Each have a similar fold and basic surface but distinct RNA binding loops exist within the CTD for RIG-I and MDA5, and the conformation of this RNA binding loop seems to be responsible for sensitivity to dsRNA or 5′ppp-ssRNA. Such a conclusion is supported by RLR structural analysis and studies featuring mutation of the basic surface and the RNA binding loop (Li et al., 2009; Takahasi et al., 2009).
We have recently demonstrated that expression of both RIG-I and MDA5 is discernable in uninfected mouse brain tissue (Furr et al., 2008). Importantly, we have shown that isolated murine microglia and astrocytes constitutively express RIG-I and MDA5 transcripts and protein, as well as the critical downstream effector molecule, IPS-1 (Furr et al., 2008). Furthermore, the expression of these novel viral sensors has been confirmed in human glial cells (Yoshida et al., 2007; Furr et al., 2010). Circumstantial evidence of a role for these RLRs in RNA virus infections of the CNS comes from our observation that RIG-I and MDA5 expression is elevated in the brains of mice following intranasal administration of VSV (Furr et al., 2008). VSV is a neurotropic negative-sense ssRNA virus that closely resembles RABV. In mice, intranasal VSV infection results in a severe encephalitis with rapid activation and proliferation of microglia and astrocytes (Huneycutt et al., 1993; Bi et al., 1995) and our in vitro studies show that this virus elicits marked increases in RIG-I expression by murine and human glial cells (Furr et al., 2008, 2010).
More direct evidence for the functional nature of RIG-I expression in glia comes from our observation that this molecule associates with IPS-1 following VSV infection and from the finding that the specific RIG-I ligand, 5′ppp-ssRNA, elicits human astrocyte immune responses (Furr et al., 2010). Importantly, we also showed that RIG-I knockdown significantly reduces inflammatory cytokine production by VSV-infected human astrocytes and inhibits the production of soluble neurotoxic mediators by virally challenged cells (Furr et al., 2010). These findings directly implicate RIG-I in the initiation of glial inflammatory immune responses and suggest a potential mechanism underlying the neuronal cell death associated with acute viral CNS infections.
In contrast to the proposed detrimental role for RLR activation following challenge with VSV, other work suggests that IPS-1 is essential for triggering protective innate and adaptive immunity against WNV in the CNS. In these studies, mice deficient in the RLR adaptor molecule IPS-1 exhibited increased susceptibility to WNV infection, characterized by enhanced inflammation, viral replication, and rapid dissemination into the CNS (Suthar et al., 2010). However, it is important to note that glial cells were not the focus of this study and IPS-1 dependent IFN responses and limiting of viral replication was attributed to effects on systemically generated immune cells and cortical neurons, perhaps via changes in the numbers and/or function of regulatory T cells (Suthar et al., 2010). As such, these data suggest an innate/adaptive immune interface mediated through RLR signaling that regulates the balance of the immune response to WNV infection. Together, these findings raise the exciting possibility that RLR molecules play important roles in the detection of viral CNS pathogens and, depending upon the host cell type and viral pathogen, the initiation of protective immune responses or damaging inflammation within the brain.
Newly Discovered Cytosolic Viral Sensors may Mediate Glial Responses to DNA Viruses
Recently, novel cytosolic DNA sensors have been discovered that include DNA-dependent activator of interferon-regulatory factors (DAI; also known as Z-DNA binding protein 1; Takaoka et al., 2007) and a DNA-sensing inflammasome consisting of the HIN200 protein, absent in melanoma 2 (AIM2; Schroder and Tschopp, 2010). Acting in concert with TLRs, these receptors may provide a diverse repertoire of mechanisms to alert the cell to viral and microbial DNA, leading to the activation of the innate immune system in a similar manner to that seen with RLR-mediated responses to viral RNA. DAI reportedly senses cytosolic DNA using two N-terminal Z-DNA binding domains (ZBDs) and a third putative DNA binding domain located next to the second ZBD. Analysis of the DAI/Z-DNA complex structure reveals that DAI adopts an unusual binding mode for Z-DNA recognition (Ha et al., 2008). The DAI ZBDs bind DNA and both must be bound for full B-to-Z conversion, and so it is conceivable that the binding of two DAI proteins to each cytoplasmic dsDNA elicits DAI dimerization and subsequent innate immune activation (Ha et al., 2008; Wang et al., 2008).
Importantly, DAI has been shown to recognize double-stranded DNA in its canonical B helical form (B-DNA; Takaoka et al., 2007) and elicit type I IFN and inflammatory cytokine production in a TLR9-independent manner (Ishii et al., 2006; Stetson and Medzhitov, 2006) in other cells types. As shown in Figure 2, DAI elicits such immune functions via activation of the IRF and NF-κB transcription factors. DAI-induced IRF activation is dependent upon TANK-binding kinase 1 (TBK1). In contrast, DAI-induced NF-κB activation is mediated through interactions between two receptor-interacting protein (RIP) homotypic interaction motifs (RHIMs) in the DAI protein and RHIM-containing kinases, RIP1 and RIP3 (Kaiser et al., 2008; Rebsamen et al., 2009), as shown by the demonstration that knockdown of either RIP1 or RIP3 inhibits DAI-induced NF-κB activation. Both of these activation pathways appear to be induced via the recruitment of the adaptor molecules IPS-1, TRAF3, and STING. The importance of these adaptor molecules in host immune responses to DNA viruses is exemplified by the demonstration that the loss of STING renders mice susceptible to lethal HSV-1 infection (Ishikawa and Barber, 2008).
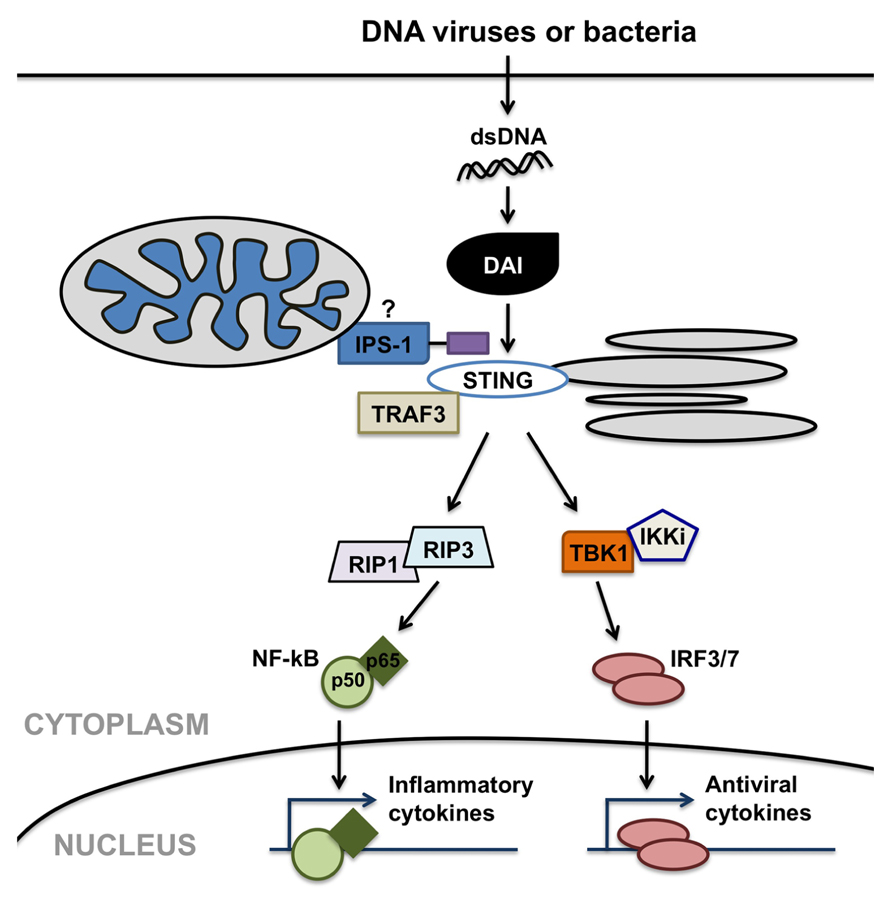
FIGURE 2. DNA-dependent activator of IFN regulatory factors (DAI) can serve as a cytosolic sensor for viral or microbial double-stranded DNA (dsDNA) motifs and initiate anti-viral and inflammatory cell responses. DAI elicits such immune functions via possible interactions with mitochondrial- or peroxisomal-associated interferon promoter stimulator (IPS)-1, and/or stimulator of interferon genes (STING), facilitating the activation of the interferon-regulatory factors (IRF) and NF-κB transcription factors. DAI-induced IRF activation is dependent on TANK-binding kinase 1 (TBK1). In contrast, DAI-induced NF-κB activation is mediated through interactions involving receptor-interacting protein (RIP) 1 and RIP3 kinases.
Overexpression of DAI in mouse fibroblasts enhances DNA-mediated type I IFN production while DAI knockdown attenuates responses to viral DNA (Takaoka et al., 2007). In an additional study, DNA-mediated activation of NF-κB and the induction of NF-κB-dependent genes and their products were also significantly inhibited in cells following DAI knockdown (Rebsamen et al., 2009). As such, these data are suggestive of a significant role for DAI in DNA-mediated activation of the NF-κB pathway and viral recognition. However, it is interesting to note that DAI-mediated NF-κB activation appears to facilitate HIV-1 replication although the mechanisms underlying this effect remain unclear (Hayashi et al., 2010).
DAI is reported to be expressed in the lymph nodes and spleen constitutively, but detectable levels are also found in circulating leukocytes and other tissues including bone marrow and small intestines (Rothenburg et al., 2002). In the CNS, we have shown that microglia and astrocytes constitutively express DAI and its effector molecules RIP3 and STING, and show that such expression is upregulated following DNA virus challenge (Furr et al., 2011). In vivo HSV-1 infection elicited an upregulation in microglial DAI expression by up to 23.8-fold over that seen in resting cells, and despite strong constitutive expression, HSV-1 exposure was also able to further increase DAI expression in astrocytes with a maximal increase of 2.2-fold over that seen in unstimulated cells. Interestingly, the ability of viral challenge to augment DAI expression by primary glial cells was not limited to this neurotropic alpha herpesvirus, as the lymphotropic gammaherpesvirus, MHV-68, was also capable of eliciting robust increases in microglial DAI expression and caused modest increases in the expression of this molecule in astrocytes.
Importantly, we showed that transfection with the DAI specific ligand B-DNA elicits inflammatory cytokine production by isolated glial cells, with induced production of the inflammatory cytokines TNF-α and IL-6 as rapidly as 6 h post-transfection at levels that matched or exceeded those elicited following a 24-h HSV-1 challenge (Furr et al., 2011). To confirm the functional status of DAI in glial cells and to begin to determine the relative importance of this innate immune sensor in their responses to neurotropic DNA viruses, we also assessed the effect of DAI knockdown on inflammatory cytokine production by HSV-1 challenged microglia and astrocytes (Furr et al., 2011). We found that siRNA directed against DAI significantly attenuated HSV-1 induced TNF-α and IL-6 production by murine microglia. Such an approach also markedly reduced IL-6 release by HSV-1 infected astrocytes but was not as effective in reducing TNF-α expression by these cells, where a statistically significant reduction was only observed at the highest viral MOI used (Furr et al., 2011). Finally, we demonstrated that HSV-1 challenged microglia and astrocytes release neurotoxic mediators and showed that such production is significantly attenuated following DAI knockdown (Furr et al., 2011).
As such, the functional expression of DAI by microglia and astrocytes may represent an important innate immune mechanism underlying the rapid and potentially lethal inflammation associated with neurotropic DNA virus infection. However, the involvement of DNA sensor(s) other than DAI cannot be excluded. For example, a new DNA sensor, AIM2, has been identified which binds to DNA (as reviewed in Ranjan et al., 2009). Upon DNA ligation, AIM2 associates with the apoptosis-associated speck-like protein containing a CARD (ASC) leading to NF-κB and caspase-1 activation and inflammatory cytokine production. AIM2 knockdown has been shown to reduce such inflammasome formation and caspase-1 activation in response to dsDNA administration and infection with vaccinia virus (Hornung et al., 2009) and stable transfection of cell-lines with AIM2 has been demonstrated to confer responsiveness to cytoplasmic DNA (Fernandes-Alnemri et al., 2009). To date, the role of AIM2 in innate immune responses to DNA viruses in the CNS has not been explored.
PRRs Act in a Cooperative Manner to Induce Glial Responses to Viral Challenges
While the preceding sections have focused on the roles of specific individual PRRs in the detection of viral pathogens by CNS cells, it is highly likely that glial responses represent the sum of the inputs from disparate cell surface, endosomal, and cytosolic viral sensors (as shown in Figure 3). We, and others, have demonstrated that the level of expression of a particular TLR, NLR, or RLR by microglia and astrocytes can be regulated by ligands for dissimilar PRRs (Bowman et al., 2003; Jack et al., 2005; Carpentier et al., 2008). Furthermore, mechanisms of positive and negative regulation of RLR signaling have been identified that include signaling cross-talk between RLR, NLR and DNA-sensing pathways, and caspase networks. For example, it has been shown that mice deficient in the expression of the DAI downstream effector molecule STING are highly susceptible to infection by negative-stranded viruses including VSV suggesting a role for this signaling molecule in both DAI and RIG-I mediated cell activation (Ishikawa and Barber, 2008). Immunoprecipitation studies indicate STING associates with RIG-I complexes in close proximity to endoplasmic reticulum associated mitochondria (Ishikawa et al., 2009; Dixit et al., 2010). Interestingly, a number of RNA viruses including yellow fever virus, dengue virus, and hepatitis C virus, encode proteins with significant structural homology to STING and at least one of these proteins produced by the dengue virus has been shown to inhibit STING function (Ishikawa et al., 2009). As such, these observations suggest a role for STING in anti-viral host responses to both DNA and RNA viruses.
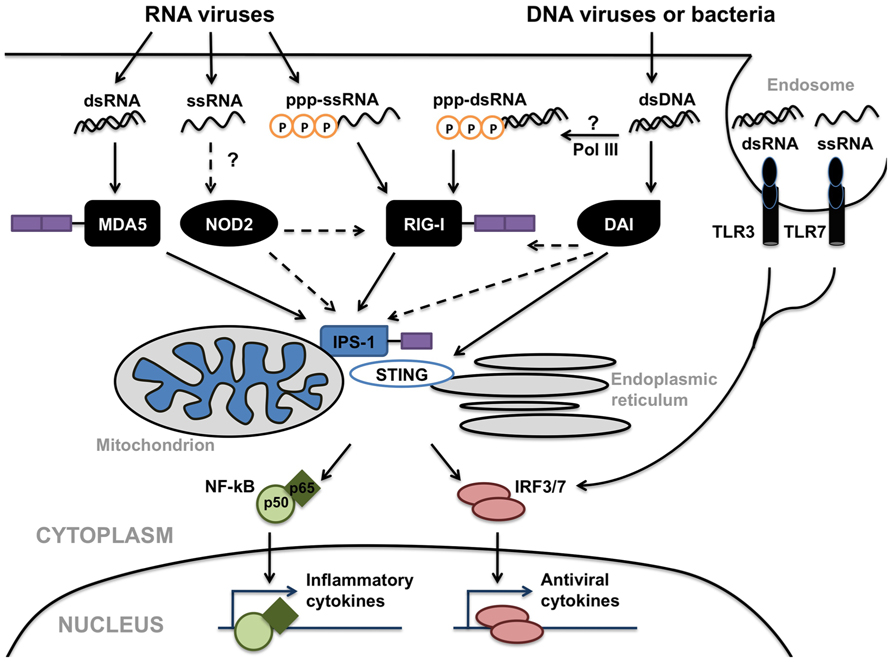
FIGURE 3. Possible interactions between Toll-like receptors (TLRs; e.g., TLR3 and TLR7/8), nucleotide oligomerization domain (NOD)-like receptors (NLRs; e.g., NOD2), retinoic acid inducible gene (RIG)-I-like receptors (RLRs; e.g., RIG-I and MDA5) and DNA-dependent activator of IFN regulatory factors (DAI) in the detection of viruses and the initiation of glial immune responses. These disparate cell surface, endosomal, and cytosolic sensors share common signaling components including interferon promoter stimulator (IPS)-1 and stimulator of interferon genes (STING), leading to the activation of the transcription factors NF-κB and interferon-regulatory factor (IRF) 3, which translocate into the nucleus and initiate anti-viral and inflammatory cytokine production. In addition, the activity of RNA polymerase III (Pol III) could provide an additional mechanism for the perception of DNA viruses via RLRs.
Another example of cross-talk between viral sensing pathways is the recent demonstration that RIG-I may be important in the production of type I IFNs by cells in response to DNA viral challenge in a DNA-dependent RNA polymerase III manner (Ablasser et al., 2009; Chiu et al., 2009). In the proposed model, AT-rich double-stranded DNA serves as a template for RNA polymerase III and is transcribed into RNA containing a 5′-triphosphate, the ligand for RIG-I. Subsequent activation of RIG-I by this ligand has been shown to induce type I IFN production and to activate NF-κB, and studies employing RNA polymerase III knockdown or pharmacological inhibitors of this enzyme have shown that this pathway is important in cell responses to the gammaherpesvirus Epstein-Barr virus (Ablasser et al., 2009; Chiu et al., 2009). Together, these studies suggest that the transcription of cytosolic viral DNA by RNA polymerase III and subsequent RNA recognition by RIG-I could provide an additional mechanism by which glial cells perceive DNA viruses.
Interestingly, there has been a recent report of a role for the well-characterized bacterial sensor, NOD2, in the recognition of viral infections and the activation of IRF-3 via IPS-1 (Sabbah et al., 2009). While NOD2 is known to recognize the bacterial cell wall component muramyl dipeptide and trigger NF-κB activation via the adaptor molecule RIP2, Sabbah et al. (2009) have provided evidence that this sensor is also important in the detection of ssRNA derived from respiratory syncytial virus. However, it is currently unknown if NOD2 is directly activated by this viral ligand, or is activated secondarily following association with other PRRs or RNA binding factors. Furthermore, it remains to be determined whether such interactions between NLRs and RIG-I-associated signaling components occur in glial cells or whether they play a significant role during viral CNS infections.
Concluding Remarks
The importance of innate immunity in host responses to viral CNS infection is evident. Microglia and astrocytes express a diverse array of cell surface, endosomal, and cytosolic molecules that can serve as sensors for RNA and DNA viruses, either constitutively or following activation. It is also clear that resident CNS cells can respond to a diverse array of ligands and that exquisite control of the associated signaling pathways exists leading to the observed complexity in the interactions between the innate and adaptive arms of the host immune response. However, it is obvious that we still do not fully understand the precise nature and structure of the ligands for many of these PRRs or their mode of generation during infection. Furthermore, the available data suggest that models that ascribe discrete PRRs to each particular pathogen are overly simplistic. Rather, it appears that TLRs, RLRs, NLRs, and DAI function as PRRs and/or adaptor molecules that act in a cooperative/synergistic manner to promote glial responses to viral pathogens.
In addition to these caveats, it is almost certain that additional PRRs await discovery in glial cells and the nature of the interactions between these viral sensors and cellular activation pathways have yet to be determined. Finally, it is unclear whether and to what degree viral CNS pathogens evade detection or manipulate immune responses to their advantage. Answering these questions will lead to greater understanding of the fundamental mechanisms underlying glial activation by neurotropic viruses and will contribute significantly to our understanding of the events that underlie the development of either protective host responses or life threatening neuroinflammation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ablasser, A., Bauernfeind, F., Hartmann, G., Latz, E., Fitzgerald, K. A., and Hornung, V. (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072.
Aloisi, F., Penna, G., Cerase, J., Menendez Iglesias, B., and Adorini, L. (1997). IL-12 production by central nervous system microglia is inhibited by astrocytes. J. Immunol. 159, 1604–1612.
Aravalli, R. N., Hu, S., Rowen, T. N., Gekker, G., and Lokensgard, J. R. (2006). Differential apoptotic signaling in primary glial cells infected with herpes simplex virus 1. J. Neurovirol. 12, 501–510.
Bauer, J., Huitinga, I., Zhao, W., Lassmann, H., Hickey, W. F., and Dijkstra, C. D. (1995). The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia 15, 437–446.
Bi, Z., Barna, M., Komatsu, T., and Reiss, C. S. (1995). Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J. Virol. 69, 6466–6472.
Bingham, A. C., and Saiman, L. (2000). Short course viral encephalitis. Office Emerg. Pediatr. 13, 184–189.
Bowman, C. C., Rasley, A., Tranguch, S. L., and Marriott, I. (2003). Cultured astrocytes express Toll-like receptors for bacterial products. Glia 43, 281–291.
Bsibsi, M., Persoon-Deen, C., Verwer, R. W., Meeuwsen, S., Ravid, R., and Van Noort, J. M. (2006). Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53, 688–695.
Butchi, N. B., Pourciau, S., Du, M., Morgan, T. W., and Peterson, K. E. (2008). Analysis of the neuroinflammatory response to TLR7 stimulation in the brain: comparison of multiple TLR7 and/or TLR8 agonists. J. Immunol. 11, 7604–7612.
Carpentier, P. A., Begolka, W. S., Olson, J. K., Elhofy, A., Karpus, W. J., and Miller, S. D. (2005). Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49, 360–374.
Carpentier, P. A., Duncan, D. S., and Miller, S. D. (2008). Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav. Immun. 22, 140–147.
Carty, M., and Bowie, A. G. (2011). Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem. Pharmacol. 81, 825–837.
Chauhan, V. S., Furr, S. R., Sterka, D. G. Jr., Nelson, D. A., Moerdyk-Schauwecker, M., Marriott, I., and Grdzelishvili, V. Z. (2010). Vesicular stomatitis virus infects resident cells of the central nervous system and induces replication-dependent inflammatory responses. Virology 10, 187–196.
Cheeran, M. C., Hu, S., Sheng, W. S., Rashid, A., Peterson, P. K., and Lokensgard, J. R. (2005). Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 11, 512–524.
Chiu, Y. H., Macmillan, J. B., and Chen, Z. J. (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591.
Conrady, C. D., Drevets, D. A., and Carr, D. J. (2010). Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J. Neuroimmunol. 220, 1–9.
Daffis, S., Samuel, M. A., Suthar, M. S., Gale, M. Jr., and Diamond, M. S. (2008). Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 82, 10349–10358.
Dixit, E., Boulant, S., Zhang, Y., Lee, A. S., Odendall, C., Shum, B., Hacohen, N., Chen, Z. J., Whelan, S. P., Fransen, M., Nibert, M. L., Superti-Furga, G., and Kagan, J. C. (2010). Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681.
Fernandes-Alnemri, T., Yu, J., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513.
Fischer, H. G., and Reichmann, G. (2001). Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J. Immunol. 166, 2717–2726.
Furr, S. R., Chauhan, V. S., Moerdyk-Schauwecker, M. J., and Marriott, I. (2011). A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflammation 8, 99.
Furr, S. R., Chauhan, V. S., Sterka, D. Jr., Grdzelishvili, V., and Marriott, I. (2008). Characterization of retinoic acid-inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus. J. Neurovirol. 14, 503–513.
Furr, S. R., Moerdyk-Schauwecker, M., Grdzelishvili, V. Z., and Marriott, I. (2010). RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia 58, 1620–1629.
Ghosh, D., and Basu, A. (2009). Japanese encephalitis – a pathological and clinical perspective. PLoS Negl. Trop. Dis. 3, e437. doi: 10.1371/journal.pntd.0000437
Gitlin, L., Barchet, W., Gilfillan, S., Cella, M., Beutler, B., Flavell, R. A., Diamond, M. S., and Colonna, M. (2006). Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. U.S.A. 30, 8459–8464.
Grdzelishvili, V. Z., Smallwood, S., Tower, D., Hall, R. L., Hunt, D. M., and Moyer, S. A. (2005). A single amino acid change in the L-polymerase protein of vesicular stomatitis virus completely abolishes viral mRNA cap methylation. J. Virol. 79, 7327–7337.
Gubler, D. J. (2008). The continuing spread of west Nile virus in the Western hemisphere. Clin. Infect. Dis. 45, 1039–1046.
Guo, Y., Audry, M., Ciancanelli, M., Alsina, L., Azevedo, J., Herman, M., Anguiano, E., Sancho-Shimizu, V., Lorenzo, L., Pauwels, E., Philippe, P. B., Pérez de Diego, R., Cardon, A., Vogt, G., Picard, C., Andrianirina, Z. Z., Rozenberg, F., Lebon, P., Plancoulaine, S., Tardieu, M., Doireau, V., Jouanguy, E., Chaussabel, D., Geissmann, F., Abel, L., Casanova, J. L., and Zhang, S. Y. (2011). Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 208, 2083–2098.
Ha, S. C., Kim, D., Hwang, H. Y., Rich, A., Kim, Y. G., and Kim, K. K. (2008). The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc. Natl. Acad. Sci. U.S.A. 105, 20671–20676.
Hashimoto, C., Hudson, K. L., and Anderson, K. V. (1988). The Toll gene of Drosophila, required for dorsal–ventral embryonic polarity, appears to encode a transmembrane protein. Cell 52, 269–279.
Hayashi, T., Nishitsuji, H., Takamori, A., Hasegawa, A., Masuda, T., and Kannagi, M. (2010). DNA-dependent activator of IFN-regulatory factors enhances the transcription of HIV-1 through NF-κB. Microbes Infect. 12, 937–947.
Hickey, W. F., and Kimura, H. (1988). Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239, 290–292.
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald KA. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518.
Hornung, V., Ellegast, J., Kim, S., Brzozka, K., Jung, A., Kato, H., Poeck, H., Akira, S., Conzelmann, K. K., Schlee, M., Endres, S., and Hartmann, G. (2006). 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997.
Huneycutt, B. S., Bi, Z., Aoki, C. J., and Reiss, C. S. (1993). Central neuropathogenesis of vesicular stomatitis virus infection of immunodeficient mice. J. Virol. 67, 6698–6706.
Isaacson, E., Glaser, C. A., Forghani, B., Amad, Z., Wallace, M., Armstrong, R. W., Exner, M. M., and Schmid, S. (2005). Evidence of human herpesvirus 6 infection in 4 immunocompetent patients with encephalitis. Clin. Infect. Dis. 40, 890–893.
Ishii, K. J., Coban, C., Kato, H., Takahashi, K., Torii, Y., and Takeshita, F. (2006). A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48.
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. Erratum in Nature 2008; 456, 274.
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792.
Jack, C. S., Arbour, N., Manusow, J., Montgrain, V., Blain, M., McCrea, E., Shapiro, A., and Antel, J. P. (2005). TLR signaling tailors innate immune responses in human microglia and astrocytes. J. Immunol. 175, 4320–4330.
Jmor, F., Emsley, H. C., Fischer, M., Solomon, T., and Lewthwaite, P. (2008). The incidence of acute encephalitis syndrome in Western industrialised and tropical countries. Virol. J. 30, 134.
Kaiser, W. J., Upton, J. W., and Mocarski, E. S. (2008). Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 181, 6427–6434.
Kalinke, U., and Prinz, M. (2012). Endogenous, or therapeutically induced, type I interferon responses differentially modulate Th1/Th17-mediated autoimmunity in the CNS. Immunol. Cell Biol. 90, 505–509.
Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K. J., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C. S., Reis e Sousa, C., Matsuura, Y., Fujita, T., and Akira, S. (2006). Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105.
Kawai, T., and Akira, S. (2007). Antiviral signaling through pattern recognition receptors. J. Biochem. 141, 137–145.
Kawai, T., and Akira, S. (2008). Toll-like receptor and RIG-I-like receptor signaling. Ann. N.Y. Acad. Sci. 1143, 1–20.
Kiefer, R., Lindholm, D., and Kreutzberg, G. W. (1993). Interleukin-6 and transforming growth factor-beta 1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur. J. Neurosci. 5, 775–781.
Kielian, T. (2009). Overview of toll-like receptors in the CNS. Curr. Top. Microbiol. Immunol. 336, 1–14.
Koh, W. L., and Ng, M. L. (2005). Molecular mechanisms of West Nile virus pathogenesis in brain cell. Emerg. Infect. Dis. 11, 629–632.
Koskiniemi, M., Korppi, M., Mustonen, K., Rantala, H., Muttilainen, M., Herrgård, E., Ukkonen, P., and Vaheri, A. (1997). Epidemiology of encephalitis in children: a prospective multicentre study. Eur. J. Pediatr. 156, 541–545.
Kowalinski, E., Lunardi, T., McCarthy, A. A., Louber, J., Brunel, J., Grigorov, B., Gerlier, D., and Cusack, S. (2011). Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147, 423–435.
Kurt-Jones, E. A., Chan, M., Zhou, S., Wang, J., Reed, G., Bronson, R., Arnold, M. M., Knipe, D. M., and Finberg, R. W. (2004). Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U.S.A. 101, 1315–1320.
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983.
Li, J., Faber, M., Dietzschold, B., and Hooper, D. C. (2011). The role of toll-like receptors in the induction of immune responses during rabies virus infection. Adv. Virus Res. 79, 115–126.
Li, X., Lu, C., Stewart, M., Xu, H., Strong, R. K., Igumenova, T., and Li, P. (2009). Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch. Biochem. Biophys. 488, 23–33.
Liao, C. L., Lin, Y. L., Shen, S. C., Shen, J. Y., Su, H. L., Huang, Y. L., Ma, S. H., Sun, Y. C., Chen, K. P., and Chen, L. K. (1998). Antiapoptotic but not antiviral function of human bcl-2 assists establishment of Japanese encephalitis virus persistence in cultured cells. J. Virol. 72, 9844–9854.
Licon Luna, R. M., Lee, E., Müllbacher, A., Blanden, R. V., Langman, R., and Lobigs, M. (2002). Lack of both Fas ligand and perforin protects from flavivirus-mediated encephalitis in mice. J. Virol. 76, 3202–3211.
Lima, G. K., Zolini, G. P., Mansur, D. S., Freire Lima, B. H., Wischhoff, U., Astigarraga, R. G., Dias, M. F., das Graças Almeida Silva, M., Béla, S. R., do Valle Antonelli, L. R., Arantes, R. M., Gazzinelli, R. T., Báfica, A., Kroon, E. G., and Campos, M. A. (2010). Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am. J. Pathol. 177, 2433–2445.
Lokensgard, J. R., Hu, S., Sheng, W., vanOijen, M., Cox, D., Cheeran, M. C., and Peterson, P. K. (2001). Robust expression of TNF-alpha, IL-1beta, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 7, 208.
Loo, Y. M., Fornek, J., Crochet, N., Bajwa, G., Perwitasari, O., Martinez-Sobrido, L., Akira, S., Gill, M. A., García-Sastre, A., Katze, M. G., and Gale, M. Jr. (2008). Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82, 335–345.
Lui, P., Li, K., Garofalo, R. P., and Brasier, A. R. (2008). Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I NF- kappa B-inducing kinase signaling pathway. J. Biol. Chem. 283, 23169–23178.
Martin, F. C., Anton, P. A., Gornbein, J. A., Shanahan, F., and Merrill, J. E. (1993). Production of interleukin-1 by microglia in response to substance P: role for a non-classical NK-1 receptor. J. Neuroimmunol. 42, 53–60.
McCormack, J. G., and Allworth, A. M. (2002). Emerging viral infections in Australia. Med. J. Aust. 177, 45–49.
McKimmie, C. S., Johnson, N., Fooks, A. R., and Fazakerley, J. K. (2005). Viruses selectively upregulate Toll-like receptors in the central nervous system. Biochem. Biophys. Res. Commun. 28, 925–933.
McKimmie, C. S., and Fazakerley, J. K. (2005). In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J. Neuroimmunol. 169, 116–125.
Medzhitov, R., and Janeway, C. A. (2002). Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300.
Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397.
Melton, L. M., Keith, A. B., Davis, S., Oakley, A. E., Edwardson, J. A., and Morris, C. M. (2003). Chronic glial activation, neurodegeneration, and APP immunoreactive deposits following acute administration of double-stranded RNA. Glia 44, 1–12.
Parquet, M. C., Kumatori, A., Hasebe, F., Morita, K., and Igarashi, A. (2001). West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 500, 17–24.
Pichlmair, A., Schulz, O., Tan, C. P., Näslund, T. I., Liljeström, P., Weber, F., and Reis e Sousa, C. (2006). RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001.
Ranjan, P., Bowzard, J. B., Schwerzmann, J. W., Jeisy-Scott, V., Fujita, T., and Sambhara, S. (2009). Cytoplasmic nucleic acid sensors in antiviral immunity. Trends Mol. Med. 15, 359–368.
Rasley, A., Bost, K. L., and Marriott, I. (2004). Murine gammaherpesvirus-68 elicits robust levels of interleukin-12 p40, but not interleukin-12 p70 production, by murine microglia and astrocytes. J. Neurovirol. 10, 171–180.
Rebsamen, M., Heinz, L. X., Meylan, E., Michallet, M. C., Schroder, K., Hofmann, K., Vazquez, J., Benedict, C. A., and Tschopp, J. (2009). DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 10, 916–922.
Rehwinkel, J., Tan, C. P., Goubau, D., Schulz, O., Pichlmair, A., Bier, K., Robb, N., Vreede, F., Barclay, W., Fodor, E., and Reis e Sousa, C. (2010). RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140, 397–408.
Rothenburg, S., Schwartz, T., Koch-Nolte, F., and Haag, F. (2002). Complex regulation of the human gene for the Z-DNA binding protein DLM-1. Nucleic Acids Res. 30, 993–1000.
Sabbah, A., Chang, T. H., Harnack, R., Frohlich, V., Tominaga, K., Dube, P. H., Xiang, Y., and Bose, S. (2009). Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 10, 1073–1080.
Saito, T., Hirai, R., Loo, Y. M., Owen, D., Johnson, C. L., Sinha, S. C., Akira, S., Fujita, T., and Gale, M. Jr. (2007). Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. U.S.A. 104, 582–587.
Samaratunga, H., Searle, J. W., and Hudson, N. (1998). Non-rabies lyssavirus human encephalitis from fruit bats: Australian bat lyssavirus (pteropid lyssavirus) infection. Neuropathol. Appl. Neurobiol. 24, 331–335.
Savarin, C., and Bergmann, C. C. (2008). Neuroimmunology of central nervous system viral infections: the cells, molecules and mechanisms involved. Curr. Opin. Pharmacol. 8, 472–479.
Schachtele, S. J., Hu, S., Little, M. R., and Lokensgard, J. R. (2010). Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J. Neuroinflammation 28, 7–35
Sejvar, J. J. (2006). The evolving epidemiology of viral encephalitis. Curr. Opin. Neurol. 19, 350–357.
Shrestha, B., Gottlieb, D., and Diamond, M. S. (2003). Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 77, 13203–13213.
Solomon, T., Ni, H., Beasley, D. W., Ekkelenkamp, M., Cardosa, M. J., and Barrett, A. D. (2003). Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 77, 3091–3098.
Stahl, J. P., Mailles, A., Vaillant, V., Floret, D., and Encephalitis Protocol Group. (2008). Acute infectious encephalitis and pathogens coming from animals. Medicina (Kaunas) 44, 821–826.
Stetson, D. B., and Medzhitov, R. (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103.
Stoll, G., and Jander, S. (1999). The role of microglia and macrophages in the pathophysiology of the CNS. Prog. Neurobiol. 58, 233–247.
Streit, W. J., Semple-Rowland, S. L., Hurley, S. D., Miller, R. C., Popovich, P. G., and Stokes, B. T. (1998). Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp. Neurol. 152, 74–87.
Sumpter, R. Jr., Loo, Y. M., Foy, E., Li, K., Yoneyama, M., Fujita, T., Lemon, S. M., and Gale, M. Jr. (2005). Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79, 2689–2699.
Suthar, M. S., Ma, D. Y., Thomas, S., Lund, J. M., Zhang, N., Daffis, S., Rudensky, A. Y., Bevan, M. J., Clark, E. A., Kaja, M. K., Diamond, M. S., and Gale, M. Jr. (2010). IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6, e1000757. doi: 10.1371/journal.ppat.1000757
Suzumura, A., Sawada, M., and Takayanagi, T. (1998). Production of interleukin-12 and expression of its receptors by murine microglia. Brain Res. 787, 139–142.
Takahasi, K., Kumeta, H., Tsuduki, N., Narita, R., Shigemoto, T., Hirai, R., Yoneyama, M., Horiuchi, M., Ogura, K., Fujita, T., and Inagaki, F. (2009). Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J. Biol. Chem. 284, 17465–17474.
Takaoka, A., Wang, Z., Choi, M., Yanai, H., Negishi, H., Ban, T., Lu, Y., Miyagishi, M., Kodama, T., Hondra, K., Ohba, Y., and Taniguchi, T. (2007). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–506.
Takeuchi, O., and Akira, S. (2008). MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 20, 17–22.
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820.
Theerasurakarn, S., and Ubol, S. (1998). Apoptosis induction in brain during the fixed strain of rabies virus infection correlates with onset and severity of illness. J. Neurovirol. 4, 407–414.
Town, T., Jeng, D., Alexopoulou, L., Tan, J., and Flavell, R. A. (2006). Microglia recognize double-stranded RNA via TLR3. J. Immunol. 176, 3804–3812.
Town, T., Bai, F., Wang, T., Kaplan, A. T., Qian, F., Montgomery, R. R., Anderson, J. F., Flavell, R. A., and Fikrig, E. (2009). Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 30, 242–253.
Tunkel, A. R., Glaser, C. A., Bloch, K. C., Sejvar, J. J., Marra, C. M., Roos, K. L., Hartman, B. J., Kaplan, S. L., Scheld, W. M., and Whitley, R. J. (2008). The management of encephalitis: clinical practice guidelines by the Infectious Disease Society of America. Clin. Infect. Dis. 47, 303–327.
Tyler, K. L. (2009). Emerging viral infections of the central nervous system: part 2. Arch. Neurol. 66, 1065–1074.
van Marle, G., Antony, J., Ostermann, H., Dunham, C., Hunt, T., Halliday, W., Maingat, F., Urbanowski, M. D., Hobman, T., Peeling, J., and Power, C. (2007). West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 81, 10933–10949.
Wang, J. J., Liao, C. L., Chiou, Y. W., Chiou, C. T., Huang, Y. L., and Chen, L. K. (1997). Ultrastructure and localization of E proteins in cultured neuron cells infected with Japanese encephalitis virus. Virology 238, 30–39.
Wang, J. P., Bowen, G. N., Zhou, S., Cerny, A., Zacharia, A., Knipe, D. M., Finberg, R. W., and Kurt-Jones, E. A. (2012). Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 86, 2273–2281.
Wang, T., Town, T., Alexopoulou, L., Anderson, J. F., Fikrig, E., and Flavell, R. A. (2004). Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10, 1366–1373.
Wang, Z., Choi, M. K., Ban, T., Yanai, H., Negishi, H., Lu, Y., Tamura, T., Takaoka, A., Nishikura, K., and Taniguchi, T. (2008). Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. U.S.A. 105, 5477–5482.
Wilkins, C., and Gale, M. Jr. (2010). Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22, 41–47.
Xu, F., Sternberg, M. R., Kottiri, B. J., McQuillan, G. M., Lee, F. K., Nahmias, A. J., Berman, S. M., and Markowitz, L. E. (2006). Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296, 964–973.
Yoneyama, M., and Fujita, T. (2010). Recognition of viral nucleic acids in innate immunity. Rev. Med. Virol. 20, 4–22.
Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M.,. Taira, K., Foy, E., Loo, Y. M., Gale, M. Jr., Akira, S., Yonehara, S., Kato, A., and Fujita, T. (2005). Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851–2858.
Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S., and Fujita, T. (2004). The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 7, 730–737.
Yoshida, H., Imaizumi, T., Lee, S. J., Tanji, K., Sakaki, H., Matsumiya, T., Ishikawa, A., Taima, K., Yuzawa, E., Mori, F., Wakabayashi, K., Kimura, H., and Satoh, K. (2007). Retinoic acid-inducible gene-I mediates RANTES/CCL5 expression in U373MG human astrocytoma cells stimulated with double-stranded RNA. Neurosci. Res. 58, 199–206.
Zhang, S. Y., Jouanguy, E., Ugolini, S., Smahi, A., Elain, G., Romero, P., Segal, D., Sancho-Shimizu, V., Lorenzo, L., Puel, A., Picard, C., Chapgier, A., Plancoulaine, S., Titeux, M., Cognet, C., von Bernuth, H., Ku, C. L., Casrouge, A., Zhang, X. X., Barreiro, L., Leonard, J., Hamilton, C., Lebon, P., Héron, B., Vallée, L., Quintana-Murci, L., Hovnanian, A., Rozenberg, F., Vivier, E., Geissmann, F., Tardieu, M., Abel, L., and Casanova, J. L. (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527.
Keywords: microglia, astrocytes, viral encephalitis, neuroinflammation, pattern recognition receptors, RLR, TLR
Citation: Furr SR and Marriott I (2012) Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front. Microbio. 3:201. doi:10.3389/fmicb.2012.00201
Received: 19 March 2012; Paper pending published: 04 April 2012;
Accepted: 15 May 2012; Published online: 20 June 2012.
Edited by:
Amy Rasley, Lawrence Livermore National Laboratory, USAReviewed by:
Monica Borucki, Lawrence Livermore National Laboratory, USAJune Round, University of Utah, USA
Copyright: © 2012 Furr and Marriott. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ian Marriott, Department of Biology, University of North Carolina at Charlotte, 9201 University City Boulevard, Charlotte, NC 28223, USA. e-mail: imarriot@uncc.edu