
Abstract
Prevention is a promising option for the control of cancer. Cellular redox changes have emerged as a pivotal and proximal event in cancer. In this review, we provide a brief background on redox biochemistry, discuss the important distinction between redox signalling and oxidative stress, and outline the ‘multiple biological personalities’ of reactive oxygen and nitrogen species: at low concentrations they protect the cell; at higher concentrations they can damage many biological molecules, such as DNA, proteins, and lipids; and, as we argue here, they may also prevent cancer by initiating the death of the transformed cell. Nitric oxide-donating aspirin is discussed as an instructive example: it generates a state of oxidative stress through which it affects several redox-sensitive signalling pathways, leading ultimately to the elimination of the neoplastic cell via apoptosis or necrosis. As additional examples, we discuss the chemopreventive n–3 polyunsaturated fatty acids and conventional nonsteroidal anti-inflammatory drugs, which induce cell death through redox changes. We conclude that modulation of redox biochemistry represents a fruitful approach to cancer prevention.
Similar content being viewed by others

Main
Prevention has emerged as a promising option for the control of cancer, a defining medical challenge of our time. The ultimate aim of prevention is to accomplish one or more of the following three goals: eliminate the causative agent (e.g., smoking cessation); remove a precancerous lesion (e.g., endoscopic resection of colon adenomas); or counteract pathogenetic pathways (e.g., efforts to prevent cancer using COX-2 inhibitors). Some, although slow, progress has been achieved with the first two approaches, but to date mechanism-driven efforts have rendered no major gains.
Work in recent years has generated enhanced appreciation of the complexity and relevance of pathways altered in the cancer cell. Cellular redox changes, much more explored in cardiology than in oncology (Gutierrez et al, 2006), emerge as a pivotal and proximal event in cancer. Initially thought of as ‘the bad guy’, mainly because oxidative stress promotes DNA damage, redox changes may mediate cancer prevention by natural and pharmaceutical agents. After providing a brief background to redox biochemistry, we focus on the chemopreventive nitric oxide-donating aspirin (NO-ASA) to illustrate the concept that altering the redox status of the cancer cell represents a valid strategy for cancer prevention.
Redox biochemistry: The basics
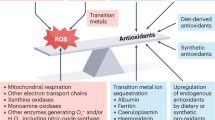
Redox biochemistry is fundamental to life. The energy needs of complex organisms require vast amounts of ATP. The supply of ATP depends heavily on redox chemistry, as it is driven by changes in free energy associated with electron or hydrogen transfers (Frein et al, 2005). Technically, redox, shorthand for reduction/oxidation, describes all chemical reactions in which the oxidation state of atoms changes. In simpler terms, oxidation describes the loss of electrons by a molecule, atom, or ion, with reduction describing the gain of electrons by the same. Redox signalling is the concept that electron-transfer processes play a key messenger role in biological systems. At the heart of redox signalling are the so-called reactive oxygen species (ROS), a term that includes oxygen radicals (e.g., O2•− and OH•) and also nonradical derivatives of O2 (H2O2). The discovery of the reactive nitrogen species expanded this term to reactive oxygen and nitrogen species (RONS) (Table 1). Free radicals contain one or more unpaired electrons. Since all molecules seek to be balanced, that is, to have an equal number of protons and electrons, the unpaired electron spins of these radicals make them highly reactive.
Reactive oxygen and nitrogen species are produced continuously by the mitochondria (O2•−, H2O2, and OH•) of most cells and also by cytochrome P450 (O2•− and H2O2), macrophages (O2•−, H2O2, and NO•), and peroxisomes (H2O2) (Klaunig and Kamendulis, 2004; Genestra, 2007). During mitochondrial oxidative metabolism about 5% of oxygen is converted primarily into O2•−, whereas 95% of it is reduced to water. Given the high reactivity of RONS, it is not surprising that the cell has invested heavily into an antioxidant defence system to contain RONS. This defence system includes (a) classic antioxidant enzymes, such as superoxide dismutase (SOD), catalase, glutathione (GSH) peroxidase, glutaredoxin, and thioredoxin. These enzymes are distributed in mitochondria, peroxisomes, and cytoplasm. (b) Nonclassic antioxidant enzymes, for example, haem oxygenase-1. (c) Phase II detoxifying enzymes, recently shown to be protective, such as GSH reductase, NQO1, and GSH transferase; and (d) nonenzymatic antioxidants, such as vitamins E and C, GSH, and catechins.
RONS: Their multiple biological personalities
For years, the intuitive, although perhaps uncritical, assumption has been that all RONS are bad for the cell. The consequent response has been to try to suppress RONS, hoping to prevent or even reverse RONS-related biological damage. Reactive oxygen and nitrogen species, however, have what could be termed ‘multiple biological personalities’: at low concentrations they protect the cell; at higher concentrations they can damage many biological molecules, such as DNA, proteins, and lipids; and yet, as we will argue here, they may also help prevent cancer by initiating the death of the transformed cell.
An important conceptual distinction has become clear in the past several years (even though adequate terminology has not yet been proposed) (Frein et al, 2005; Halliwell, 2007). First, there exists a network of redox-based regulatory mechanisms that are often quite relevant to carcinogenesis. Second, disturbed redox equilibrium is pathophysiological, having been described for years as ‘oxidative stress’. These findings have led to the recent delineation of redox signalling and oxidative stress. Redox signalling is a reversible phase of physiological regulatory reactions occurring over shorter time periods; they concern primarily the main cellular redox systems, for example, GSH, ascorbate, vitamin E, lipoic acid, NADPH, or NADH. The oxidative reactions, leading to posttranslational protein modification, are returned to the resting state by reductive pathways. Such modifications include glutathiolation, S-nitrosylation, methionine sulphoxidation, and oxidations with disulphide formation. In contrast, oxidative stress denotes a persistent (over longer time periods) and often irreversible oxidative shift that characterises a pathophysiological state. It has been defined as an imbalance between oxidants and antioxidants in favour of the former, resulting in increased cellular levels of RONS. Oxidative stress is implicated in the pathogenesis of several diseases such as cancer, cardiovascular and neurodegenerative disorders, sepsis, reperfusion damage, rheumatoid arthritis, and diabetes.
There is significant, although incomplete, evidence for a role of RONS in cancer, including genotoxicity, promotion of transformed cell growth, and angiogenesis as well as regulation of apoptosis. A ‘persistent oxidative stress in cancer’ has been hypothesised, for example, in Toyokuni et al (1995). Such oxidative stress was considered to contribute to oncogene activation, genomic instability, chemotherapy resistance, and even invasion and metastasis. Nuclear factor-κB (NF-κB), MAPK cascades as well as GSH and related antioxidant pathways were hypothesised to be the mediators. Chronic inflammation, widely connected to carcinogenesis, is also a source of RONS and a linkage of inflammation-generated RONS to cancer has also been postulated. The hypoxia-inducible factor-1α (HIF-1α) is linked to cancer through its regulation by RONS (Pouyssegur and Mechta-Grigoriou, 2006); in particular, RONS signalling may account for the high levels of HIF-1α in normoxic areas of tumours. Hypoxia-inducible factor promotes survival in low oxygen conditions, like those encountered in cancer, by upregulating an array of hypoxia-induced genes, including the vascular endothelial factor, which promotes angiogenesis. Finally, RONS have been associated with the induction of apoptotic and necrotic cell death, the specific outcome depending, among others, on the cellular levels of RONS.
NO-ASA: a case of rons-mediated cancer prevention
Nitric oxide-donating aspirin consists of conventional aspirin to which the NO-releasing moiety –ONO2 has been attached via a chemical linker as reviewed in Rigas and Kashfi (2004) (Figure 1). Studies using animal tumour models have established the strong chemopreventive effect of NO-ASA against colon and pancreatic cancer. The remarkably potent inhibition by NO-ASA of the growth of various human cell lines, for example, colon, prostate, lung, pancreas, tonsil, breast cancer, and leukaemia, suggests that a similar chemopreventive effect on these types of malignancies is also possible.

Nitric oxide-donating aspirin, RONS, and cancer prevention. Nitric oxide-donating aspirin induces cell death by altering the redox balance. It achieves it by reducing the levels of cellular antioxidants, such as GSH, and/or by generating RONS, such as superoxide anion, nitric oxide, peroxynitrite, and hydrogen peroxide. The net effect is time and concentration dependently increased RONS levels. Moderately elevated levels of RONS initiate RONS signalling, such as through MAPKs (p38, JNK), NF-κB, COX-2, NOS, Wnt, and Keap1-Nrf2, which can lead to apoptotic cell death. Excessive RONS levels constitute oxidative stress, which drives the cell directly to apoptosis or necrosis. The redox-mediated cytokinetic effect of NO-ASA, which eliminates neoplastic cells, constitutes perhaps its major chemopreventive action against cancer.
The growth inhibitory effect of NO-ASA is due to a profound cytokinetic effect consisting of reduced cell proliferation, enhanced cell death, and inhibition of cell cycle phase transitions. We have studied signalling pathways that potentially underlie this effect. NO-ASA (a) inhibited the activation of NF-κB in various cancer cell lines and in vivo (Williams et al, 2003); (b) inhibited the transcriptional activity of β-catenin by diminishing its physical association with TCF (Nath et al, 2003); (c) increased the phosphorylation of p38 and JNK, representing two out of the three main MAPK cascades; its effect on the ERK1/2 pathway was minimal (Hundley and Rigas, 2006); (d) inhibited both the catalytic activity and expression of iNOS, the inducible isoform of nitric oxide synthase (NOS) (Spiegel et al, 2005); (e) had mixed effects on the COX pathway: it induced the expression of COX-2 (no effect on COX-1) in various cell lines, whereas in animals it inhibited COX activity and the levels of various prostaglandins (Williams et al, 2003; Rao et al, 2006); and (f) induced the translocation of Nrf2 into the nucleus, promoting its dissociation from its cytoplasmic anchor Keap1 (Gao et al, 2006).
A shared characteristic of these pathways is that they are redox sensitive or involved in the regulation of the redox status of the cell. This prompted us to examine whether NO-ASA increased ROS levels. Both O2•− and a series of RONS collectively assayed by the DCFDA probe were elevated in response to NO-ASA (Gao et al, 2005); subsequent work has revealed increased intracellular levels of H2O2, peroxynitrite, and NO (Sun Y, Rigas B, unpublished data). The oxidative stress that was thus induced led to apoptosis by activating the intrinsic apoptosis pathway. Pretreatment of the cells with N-acetylcysteine abrogated cell death and restored cell growth to control levels, clearly indicating that generating a state of oxidative stress in the target cells was a critical proximal event in the action of NO-ASA in cancer. Nitric oxide-donating aspirin also induced apoptosis in small intestinal cells of Min mice that bear a somatic mutation predisposing them to intestinal cancer (Williams et al, 2004). A derivative of NO-ASA in which the NO-donating moiety was substituted by diethylphosphate also induced oxidative stress (Kashfi and Rigas, 2007; Sun Y, Rigas B, unpublished data). Interestingly, this compound, provisionally termed ‘phosphoaspirin’ is quite effective in preventing colon cancer in a xenograft animal model (Rigas and Kozoni, 2008). This finding emphasises that the oxidative stress induced by NO-ASA is not due exclusively to the NO that it releases.
The specifics of this effect are currently unclear but its mechanistic significance is unmistakable. It is quite likely that many of the changes in the signalling pathways mentioned above are derivative events, generated by redox changes that either lead to reversible redox signalling or irreversible oxidative stress that culminates in cell death and thus elimination of the neoplastic cell (Figure 1).
Additional examples of RONS-mediated cancer prevention: N–3 PUFA and conventional NSAIDS
Chapkin's team, studying the impact of n–3 polyunsaturated fatty acids (PUFA) (dietary fish oil) on colon carcinogenesis, showed that n–3 PUFA inhibited carcinogenesis through modulation of ROS levels. Several mechanistic findings supported this notion – and underscored the complexity of the relevant phenomena. Fish oil reduced oxidative DNA damage, lowered cell proliferation, and increased apoptosis in rats, preventing colon cancer (Hong et al, 2005). Interestingly, n–3 PUFA induced apoptosis either alone or synergistically when combined with butyrate (a product of fermentable fibre). Apoptosis was associated with increased ROS production and inhibition of antioxidant enzymes, such as SOD, catalase, and GSH peroxidase (Sanders et al, 2004). Membrane phospholipid hydroperoxides, one of the ROS (Sanders et al, 2004; Ng et al, 2005), disrupted the mitochondrial permeability transition pore and released cytochrome c into cytosol, triggering apoptosis. n–3 PUFA induced oxidative damage not only of the mitochondrial membrane but also of other membrane structures, such as lipid rafts in plasma membrane, which play an important role in the modulation and integration of cell signalling (Switzer et al, 2004). Reactive oxygen species generation by these chemopreventive interventions was the critical biochemical change that appeared to precede a host of relevant signalling events.
Conventional NSAIDs provide another example of how RONS may mediate cancer prevention. Epidemiological and interventional studies have established that NSAIDs reduce the risk of colon cancer and regress colorectal adenomas in patients with familial adenomatous polyposis (Baron, 2003). Besides their classical inhibitory effect on cyclooxygenase, NSAIDs exert pleiotropic non-COX effects (Hanif et al, 1996), including the generation of ROS. Although at times methodologically limited, the relevant literature provides a consensus that treatment of various cancer cell lines with NSAIDs does indeed generate increased ROS levels.
Giardina and colleagues determined the effect of the NSAIDs indomethacin and salicylic acid, and the short-chain fatty acids butyrate and propionate on ROS metabolism in the HT-29 human colorectal carcinoma cell line. All these agents increased cellular peroxide generation. Interestingly, (a) these agents sensitised the normally resistant HT-29 cells to TNF-α-induced apoptosis, and (b) arachidonic acid, which also increased ROS, synergised with indomethacin in this reaction (Giardina and Inan, 1998; Giardina et al, 1999). The NSAID sulindac, and its derivatives sulindac sulphide and sulindac sulphone increased RONS levels in DLD-1 colon cancer cells, assayed with a probe that reacts with several such species (Oh et al, 2003; Adachi et al, 2007). Of note, these three compounds had broader effects on redox homeostasis, as they increased transiently the activity of catalase, SOD, and GSH peroxidase. An antioxidant effect was also suggested by in vitro studies assessing scavenging activity of sulindac and its derivatives against various RONS (Fernandes et al, 2003; Costa et al, 2005). It is, however, the balance of these antithetic events that determines the final redox status and all evidence indicates it to be pro-oxidant. A study by Chung et al (2003) demonstrated that sodium salicylate mediates ROS production followed by decreased mitochondrial membrane potential, release of cytochrome c, and activation of caspase-9 and -3. Reactive oxygen species scavengers and an inhibitor of NADPH oxidase as well as expression of a dominant-negative form of Rac1 blocked ROS production and the consequent apoptotic cell death.
Chemotherapeutic agents are also known to induce the production of RONS. For example, the topoisomerase inhibitor etoposide (Oh et al, 2007), arsenic trioxide (Nakagawa et al, 2002), and cisplatin (Berndtsson et al, 2007) all induced cell death in various cancers through oxidative stress.
Conclusions
Our data and those reported by others make it clear that an array of agents controlling cancer either through prevention or treatment have as their common denominator the production of RONS. An additional point is equally clear: a significant fraction of cell signalling pathways modulated by these agents is RONS dependent or RONS responsive; activation of such pathways culminates to the death of the relevant cell through either apoptosis or its variants or through necrosis (Heck et al, 2005).
That the chemopreventive agents discussed here work by increasing RONS levels suggests that modulation of redox biochemistry represents a fruitful approach to cancer prevention. Conceptualising this underappreciated target for cancer control should guide efforts to develop novel agents or combinations thereof, in which RONS levels are the guiding read-out. This approach illustrates how mechanistic understanding of the pharmacological mode of action of effective agents can be translated into an effective chemoprevention strategy.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adachi M, Sakamoto H, Kawamura R, Wang W, Imai K, Shinomura Y (2007) Nonsteroidal anti-inflammatory drugs and oxidative stress in cancer cells. Histol Histopathol 22: 437–442
Baron JA (2003) Epidemiology of non-steroidal anti-inflammatory drugs and cancer. Prog Exp Tumor Res 37: 1–24
Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S (2007) Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer 120: 175–180
Chung YM, Bae YS, Lee SY (2003) Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic Biol Med 34: 434–442
Costa D, Gomes A, Reis S, Lima JL, Fernandes E (2005) Hydrogen peroxide scavenging activity by non-steroidal anti-inflammatory drugs. Life Sci 76: 2841–2848
Fernandes E, Toste SA, Lima JL, Reis S (2003) The metabolism of sulindac enhances its scavenging activity against reactive oxygen and nitrogen species. Free Radic Biol Med 35: 1008–1017
Frein D, Schildknecht S, Bachschmid M, Ullrich V (2005) Redox regulation: a new challenge for pharmacology. Biochem Pharmacol 70: 811–823
Gao J, Kashfi K, Liu X, Rigas B (2006) NO-donating aspirin induces phase II enzymes in vitro and in vivo. Carcinogenesis 27: 803–810
Gao J, Liu X, Rigas B (2005) Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc Natl Acad Sci USA 102: 17207–17212
Genestra M (2007) Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal 19: 1807–1819
Giardina C, Boulares H, Inan MS (1999) NSAIDs and butyrate sensitize a human colorectal cancer cell line to TNF-alpha and Fas ligation: the role of reactive oxygen species. Biochim Biophys Acta 1448: 425–438
Giardina C, Inan MS (1998) Nonsteroidal anti-inflammatory drugs, short-chain fatty acids, and reactive oxygen metabolism in human colorectal cancer cells. Biochim Biophys Acta 1401: 277–288
Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A (2006) Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res 99: 924–932
Halliwell B (2007) Oxidative stress and cancer: have we moved forward? Biochem J 401: 1–11
Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B (1996) Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 52: 237–245
Heck DE, Kagan VE, Shvedova AA, Laskin JD (2005) An epigrammatic (abridged) recounting of the myriad tales of astonishing deeds and dire consequences pertaining to nitric oxide and reactive oxygen species in mitochondria with an ancillary missive concerning the origins of apoptosis. Toxicology 208: 259–271
Hong MY, Bancroft LK, Turner ND, Davidson LA, Murphy ME, Carroll RJ, Chapkin RS, Lupton JR (2005) Fish oil decreases oxidative DNA damage by enhancing apoptosis in rat colon. Nutr Cancer 52: 166–175
Hundley TR, Rigas B (2006) Nitric oxide-donating aspirin inhibits colon cancer cell growth via mitogen-activated protein kinase activation. J Pharmacol Exp Ther 316: 25–34
Kashfi K, Rigas B (2007) The mechanism of action of nitric oxide-donating aspirin. Biochem Biophys Res Commun 358: 1096–1101
Klaunig JE, Kamendulis LM (2004) The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol 44: 239–267
Nakagawa Y, Akao Y, Morikawa H, Hirata I, Katsu K, Naoe T, Ohishi N, Yagi K (2002) Arsenic trioxide-induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sci 70: 2253–2269
Nath N, Kashfi K, Chen J, Rigas B (2003) Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci USA 100: 12584–12589
Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, Lupton JR, Chapkin RS (2005) The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis 26: 1914–1921
Oh KW, Qian T, Brenner DA, Lemasters JJ (2003) Salicylate enhances necrosis and apoptosis mediated by the mitochondrial permeability transition. Toxicol Sci 73: 44–52
Oh SY, Sohn YW, Park JW, Park HJ, Jeon HM, Kim TK, Lee JS, Jung JE, Jin X, Chung YG, Choi YK, You S, Lee JB, Kim H (2007) Selective cell death of oncogenic Akt-transduced brain cancer cells by etoposide through reactive oxygen species mediated damage. Mol Cancer Ther 6: 2178–2187
Pouyssegur J, Mechta-Grigoriou F (2006) Redox regulation of the hypoxia-inducible factor. Biol Chem 387: 1337–1346
Rao CV, Reddy BS, Steele VE, Wang CX, Liu X, Ouyang N, Patlolla JM, Simi B, Kopelovich L, Rigas B (2006) Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Mol Cancer Ther 5: 1530–1538
Rigas B, Kashfi K (2004) Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol Med 10: 324–330
Rigas B, Kozoni V (2008) The novel phenylester anticancer compounds: study of a derivative of aspirin (‘phosphoaspirin’). Int J Oncol 32: 97–100
Sanders LM, Henderson CE, Hong MY, Barhoumi R, Burghardt RC, Wang N, Spinka CM, Carroll RJ, Turner ND, Chapkin RS, Lupton JR (2004) An increase in reactive oxygen species by dietary fish oil coupled with the attenuation of antioxidant defenses by dietary pectin enhances rat colonocyte apoptosis. J Nutr 134: 3233–3238
Spiegel A, Hundley TR, Chen J, Gao J, Ouyang N, Liu X, Go MF, Tsioulias GJ, Kashfi K, Rigas B (2005) NO-donating aspirin inhibits both the expression and catalytic activity of inducible nitric oxide synthase in HT-29 human colon cancer cells. Biochem Pharmacol 70: 993–1000
Switzer KC, McMurray DN, Chapkin RS (2004) Effects of dietary n–3 polyunsaturated fatty acids on T-cell membrane composition and function. Lipids 39: 1163–1170
Toyokuni S, Okamoto K, Yodoi J, Hiai H (1995) Persistent oxidative stress in cancer. FEBS Lett 358: 1–3
Williams JL, Kashfi K, Ouyang N, del Soldato P, Kopelovich L, Rigas B (2004) NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem Biophys Res Commun 313: 784–788
Williams JL, Nath N, Chen J, Hundley TR, Gao J, Kopelovich L, Kashfi K, Rigas B (2003) Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: implications for chemoprevention. Cancer Res 63: 7613–7618
Acknowledgements
This work was supported by Grant NIH 2R01 CA92423.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rigas, B., Sun, Y. Induction of oxidative stress as a mechanism of action of chemopreventive agents against cancer. Br J Cancer 98, 1157–1160 (2008). https://doi.org/10.1038/sj.bjc.6604225
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604225
Keywords
This article is cited by
-
The protective effect of Sophora japonica on prostatic hypertrophy and inflammation in rat
Inflammopharmacology (2020)
-
Transcriptomic analysis of the effect of (E)-3-(3,5-dimethoxyphenyl)-1-(2-methoxyphenyl) prop-2-en-1-one (DPP23) on reactive oxygen species generation in MIA PaCa-2 pancreatic cancer cells
Genes & Genomics (2020)
-
In Vitro Anti/Pro-oxidant Activities of R. ferruginea Extract and Its Effect on Glioma Cell Viability: Correlation with Phenolic Compound Content and Effects on Membrane Dynamics
The Journal of Membrane Biology (2018)
-
Cytotoxic, antioxidative, genotoxic and antigenotoxic effects of Horchata, beverage of South Ecuador
BMC Complementary and Alternative Medicine (2017)
-
Aqueous extract of Polygonum bistorta modulates proteostasis by ROS-induced ER stress in human hepatoma cells
Scientific Reports (2017)
