
Abstract
The antidepressant desipramine inhibits the reuptake of norepinephrine (NE), leading to activation of both pre- and postsynaptic adrenergic receptors, including α-1, α-2, β-1, and β-2 subtypes. However, it is not clear which adrenergic receptors are involved in mediating its antidepressant effects. Treatment of mice with desipramine (20 mg/kg, i.p.) produced an antidepressant-like effect, as evidenced by decreased immobility in the forced-swim test; this was antagonized by pretreatment with the α-2 adrenergic antagonist idazoxan (0.1–2.5 mg/kg, i.p.). Similarly, idazoxan, administered peripherally (0.5–2.5 mg/kg, i.p.) or centrally (1–10 μg, i.c.v.), antagonized the antidepressant-like effect of desipramine in rats responding under a differential-reinforcement-of-low-rate (DRL) 72-s schedule, ie, decreased response rate and increased reinforcement rate. By contrast, pretreatment with the β-adrenergic antagonists propranolol and CGP-12177 or the α-1 adrenergic antagonist prazosin did not alter the antidepressant-like effect of desipramine on DRL behavior. The lack of involvement of β-adrenergic receptors in mediating the behavioral effects of desipramine was confirmed using knockout lines. In the forced-swim test, the desipramine-induced decrease in immobility was not altered in mice deficient in β-1, β-2, or both β-1 and β-2 adrenergic receptors. In addition, desipramine (3–30 mg/kg) produced an antidepressant-like effect on behavior under a DRL 36-s schedule in mice deficient in both β-1 and β-2 adrenergic receptors. As antagonism of presynaptic α-2 adrenergic receptors facilitates NE release, which potentiates the effects of desipramine, the present results suggest that postsynaptic α-2 adrenergic receptors play an important role in its antidepressant effects.
Similar content being viewed by others


INTRODUCTION
Desipramine, a relatively selective NE reuptake inhibitor, exerts antidepressant-like effects on behavior in rodent models (Poncelet et al, 1986; Wallace-Boone et al, 2007; Wicke et al, 2007). It decreases immobility in the forced-swim and tail-suspension tests in rats and mice (Dableh et al, 2005; Duman et al, 2007; Zhang et al, 2002, 2008) and decreases response rate and increases reinforcement rate in rats responding under a differential-reinforcement-of-low-rate (DRL) schedule (Dekeyne et al, 2002; O'Donnell, 1990). These effects are similar to those of other antidepressant drugs (Cryan et al, 2005; O'Donnell et al, 2005). Although it is not fully understood how desipramine produces antidepressant effects, increased extracellular NE concentrations in the brain caused by desipramine by its inhibition of NE transporters is considered an important mechanism (Frazer, 2000; Mateo et al, 2001; Zhao et al, 2008). NE, by this mechanism, activates all adrenergic receptors, including α-1, α-2, β-1, and β-2 adrenergic receptors that are located pre- and postsynaptically (Molinoff, 1984; Segal et al, 1991). This makes it difficult to know by which receptor subtypes and at which locations the antidepressant-like effects of desipramine are mediated.
Studies to date have shown that α-2 adrenergic receptors contribute to antidepressant-like effects of NE reuptake inhibitors such as desipramine. First, repeated treatment with desipramine decreases the density of α-2 adrenergic receptors to a greater extent than that of α-1 adrenergic receptors in the cerebral cortex (Subhash et al, 2003). This may contribute to desipramine-induced desensitization of α-2 adrenoreceptor-mediated responses in the central nervous system (Esteban et al, 1999; Mongeau et al, 1994); however, these receptors remain responsive to released NE (Lapiz et al, 2007). Second, chronic administration of desipramine markedly enhances the increase in NE release induced by blockade of presynaptic α-2 adrenergic autoreceptors in the brain (Garcia et al, 2004; Invernizzi and Garattini, 2004). Similarly, pretreatment with idazoxan, a selective α-2 adrenergic antagonist that blocks presynaptic α-2 adrenergic autoreceptors, potentiates the increase in extracellular NE induced by intrahippocampal infusions of desipramine (Thomas and Holman, 1991). Third, major depression is accompanied by decreased α-2 adrenergic receptors in platelets and increased α-2 adrenergic receptors in the hippocampus and cerebral cortex (González et al, 1994); these changes are reversed by tricyclic antidepressants (Garcia-Sevilla et al, 1999b; Maes et al, 1999). Therefore, presynaptic α-2 adrenergic receptors appear to be involved in the mediation of the antidepressant activity of desipramine.
However, peripheral or central administration of idazoxan has been reported to block, rather than enhance, the effect of desipramine on forced-swim behavior, ie, decreased immobility and increased climbing (Cervo et al, 1990; Reneric et al, 2001). This cannot be explained by presynaptic mechanisms such as idazoxan-induced NE release and desipramine-induced increases in extracellular NE. Given that central postsynaptic α-2 adrenergic receptors mediate the mydriatic effect of desipramine (Menargues et al, 1990), it is possible that postsynaptic α-2 adrenergic receptors may play a role in the regulation of the antidepressant-like effects of desipramine.
In addition to α-2 adrenergic receptors, β-adrenergic receptors also may be involved in antidepressant activity. Activation of either β-1 or β-2 adrenergic receptors produces antidepressant-like effects on DRL behavior (O'Donnell et al, 1994; Zhang et al, 2001, 2005; Zhang et al, 2003). In addition, desipramine and other NE reuptake inhibitors substitute for the β-adrenergic agonist isoproterenol in a drug discrimination task, indicating that β-adrenergic receptors play a role in mediating the effects of desipramine, as well as other NE reuptake inhibitors (Crissman et al, 2001; Crissman and O'Donnell, 2002). Further, repeated treatment with desipramine downregulates β-1, but not β-2, adrenergic receptors in the brain (Paetsch and Greenshaw, 1993). Consistent with this, downregulation of β-2 adrenergic receptors does not alter the antidepressant-like effect of desipramine (O'Donnell, 1990). Nevertheless, there does appear to be some involvement of β-2 adrenergic receptors in the antidepressant-like effects on behavior (McElroy and O'Donnell, 1988; O'Donnell et al, 1994; Zhang et al, 2005).
To determine the role of adrenergic receptor subtypes in mediating the behavioral effects of desipramine, antagonism experiments were carried out using selective α-1 and α-2 adrenergic antagonists and nonselective β-adrenergic antagonists. Antidepressant-like effects were assessed, in mice and rats, using forced-swim behavior and behavior maintained under DRL schedules. In addition, the role of β-adrenergic receptors were assessed using mice deficient in β-1, β-2, or both β-1 and β-2 adrenergic receptors. Overall, the results of this study support a predominant role for α-2 adrenergic receptors in the mediation of the antidepressant-like effects of desipramine on behavior.
MATERIALS AND METHODS
Animals
Male Sprague–Dawley rats (Harlan, Indianapolis, IN), weighing 280–350 g, were housed individually in clear plastic cages in a temperature-controlled room (22–23°C) and maintained on a 12-h on /12-h off light cycle (lights on at 06:00 hours). Food was freely available, whereas water was restricted to 15–30 ml after daily testing (Sunday–Thursday, 15 ml/day; Friday and Saturday, 30 ml/day) to keep animals at about 85% of free-watering weights.
Adult male mice deficient in β-1 and/or β-2 adrenergic receptors (β-1 KO and β-2 KO, respectively) were generated by Dr Brian K Kobilka, Stanford University, as described previously (Chruscinski et al, 1999; Rohrer et al, 1996, 1999). β-1 or β-2 knockout (KO) mice were on an FVB background, whereas β-1 and β-2 double KO mice (β-1/2 KO) were on a mixed FVB and C57BL/6 background. Age-matched male wild-type FVB and FVB × C57BL/6 mice were used as controls. All the KO and wild-type mice were bred within the laboratory animal facilities of West Virginia University and were housed in groups of 3–4 per cage under the same conditions as rat housing. However, mice tested for DRL behavior were restricted to 2 ml each of water per day after daily training/testing sessions and on weekends, to keep the animals at about 85% of free-watering weights.
Blind observations were used throughout all the experiments, which were carried out from 09:00 hours to 16:30 hours. All experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23, revised 1996). The procedures were approved by the Animal Care and Use Committees of the University of Tennessee Health Science Center and West Virginia University Health Sciences Center.
Drugs and Chemicals
All drugs were purchased from Sigma-Aldrich (St Louis, MO) and dissolved in 0.9% saline (i.p. administration) or artificial cerebrospinal fluid (aCSF; i.c.v. or intrahippocampal infusions) except for prazosin, which was dissolved in a mixture of propylene glycol and distilled water (2 : 1). All doses are expressed in terms of the free bases of the compounds. The injection volumes were 1 or 10 ml/kg body weight (i.p.; rats and mice, respectively), 10 μl (in a 1-min period; i.c.v.), and 1 μl/side (in a 2-min period; intrahippocampal). Central infusions were performed using a syringe pump (CMA/100; CMA Microdialysis Inc., North Chelmsford, MA); the 28-gauge infusion cannulae, which were 1 mm longer than the guide cannulae, were left in place for an additional 1-min (i.c.v.) or 2-min (intrahippocampal infusion) period to permit diffusion. For antagonism experiments, antagonists were given either 15 min before (for i.p. tests) or 20 min after (for i.c.v. tests) desipramine or a β-adrenergic agonist, which were given (i.p.) 30 min before behavioral tests.
Cannula Implantation
Under aseptic conditions, rats previously trained to respond under a DRL 72-s schedule were implanted with guide cannulae. Rats were anesthetized (100 mg/kg ketamine and 6 mg/kg xylazine) and placed in a stereotaxic holder (Stoelting, Wood Dale, IL). Guide cannulae (22-gauge; PlasticOne, Roanoke, VA) were implanted according to the following coordinates: (1) the right lateral ventricle, AP: −0.5 mm from bregma, ML: +1.6 mm from the midline, DV: −3.9 mm from dura (Zhang et al, 2003); (2) bilateral hippocampus, AP: −3.3 mm from bregma, ML: ±2.0 mm from the midline, DV: −2.5 mm from dura (Zhang et al, 2004). The cannulae were cemented to the surface of the skull. About 1 week after surgery, rats resumed daily training sessions under the DRL schedule; drug testing was carried out after performance had stabilized. Following completion of the testing, site of cannula implantation was verified histologically.
Behavioral Procedures
Mouse forced-swim test
The forced-swim test was performed as described previously (Zhang et al, 2002). Each mouse was placed in a plastic cylinder (45 cm high × 20 cm diameter), which was filled to a depth of 28 cm with water (22–23°C). The duration of immobility, which was defined as floating in an upright position without additional activity other than that necessary for the animal to keep its head above water, was recorded for the entire 6-min period.
DRL behavior
Mice and rats were trained to respond under DRL 36- and 72-s schedules, respectively, using operant conditioning chambers (ENV-307A; Med Associates Inc., St. Albans, VT for mice and Model E10-10; Coulbourn Instruments, Allentown, PA for rats), which were enclosed in ventilated sound-attenuating cubicles. Each of the mouse chambers contained two ultra-sensitive, nose-poke holes, which were on each side of a round access port (2.2 cm in diameter) with a dipper (ENV-302M; Med Associates Inc.) submerged in a water reservoir. The holes contained photocells to register nosepokes; only nosepokes in the right opening constituted a response; nosepokes on the left side had no programmed consequence. A house light was located in the center of the wall opposite to the water access port and the nosepoke holes. Each of the rat chambers contained two levers, a water access port, and a house light. A press on the right lever constituted a response; responses on the left lever had no programmed consequence. When the schedule contingencies (correct responses; eg, the interval between nosepokes or lever presses was longer than 36 or 72 s for the DRL 36- and 72-s schedules, respectively) were met, a dipper was raised to the water access port, providing 0.01 ml (for mice) or 0.2 ml (for rats) of water for 4 s, which constituted a reinforcer. Behavioral responses were recorded and scheduled events controlled using a MED Associates interface and operating system (MED Associates Inc.).
The training and testing procedures were performed as described previously, with minor modifications for mice (Pattij et al, 2003; Zhang et al, 2005). Training was conducted during once-daily sessions, 5 days per week (Monday–Friday); unless specified (see below), each session lasted for 60 min before it stopped automatically. Specifically, during the first 2 weeks, mice were trained to poke the front hole using the nose for water reinforcement under a fixed-ratio 1 (FR1) schedule. Next, a DRL 18-s schedule was introduced, followed 3 weeks later by a DRL 36-s schedule. Rats were trained to press the front lever for water reinforcement under an FR1/fixed-interval 1-min schedule for the first 2 weeks. The session automatically stopped once the rat finished 100 responses, which took 15–20 min. A DRL 18-s schedule was then introduced; if the number of responses per session was less than 200, the water ration was reduced. Three weeks later, the DRL 72-s schedule was introduced. Drug tests were started when responses (nosepokes or lever presses) and reinforcers (times of dipper-raising) per session were maintained at a stable baseline for 5 successive days (approximately 2 or 4 weeks later for rats and mice, respectively); these were carried out on Tuesdays and Fridays with Thursdays serving as noninjected control days. We set a performance criterion (response >70 and reinforcers <12) for rats based on our previous studies to prevent ceiling effects and permit antidepressant-like effects on behavior (Zhang et al, 2001, 2003, 2005). For mice, we emphasized on baseline stability because they were more variable in DRL performance.
Statistical Analysis
The DRL data were converted to percentages of control response and reinforcement rates for each individual animal. Control values were the median of response and reinforcement rates for Thursdays (ie, the noninjected control days) during the testing period. These data were analyzed by repeated measures ANOVA followed by Newman–Keuls tests. The forced-swim test data were analyzed by one-way ANOVA followed by Newman–Keuls tests except for those using KO mice (data in Figure 5), which were analyzed by Student's t-tests.
RESULTS
Antagonism by Idazoxan of the Antidepressant-Like Effect of Desipramine on Forced-Swim Behavior in Mice
To determine whether α-2 adrenergic receptors are involved in the behavioral effects of desipramine, the effect of desipramine on forced-swim behavior was examined in wild-type mice in the presence or absence of idazoxan, a selective α-2 adrenergic antagonist. Desipramine, at a dose of 20 mg/kg, decreased immobility in the forced-swim test (p<0.001); this was antagonized by idazoxan (0.1–2.5 mg/kg) in a dose-dependent manner (F4,32=14.1; p<0.0001; Figure 1a). At 0.5 mg/kg, idazoxan produced a complete blockade of the effect of desipramine. Idazoxan alone (0.5 and 2.5 mg/kg) did not alter behavior in the forced-swim test (F2,16=0.12; p=0.89; Figure 1b).

Antagonism by idazoxan of the antidepressant-like effects of desipramine on forced-swim behavior. (a) Blockade by idazoxan (Ida) of the effect of desipramine on immobility in the forced-swim test in wild-type mice. (b) The effect of idazoxan alone on forced-swim behavior in wild-type mice. (c) Blockade by idazoxan of the effect of desipramine on immobility in the forced-swim test in β-1/2 KO mice. (d) The effect of idazoxan alone on forced-swim behavior in β-1/2 KO mice. Ida was injected (i.p.) 30 min before the test or 15 min before desipramine (20 mg/kg, i.p.), which was given 30 min before the test. Data shown represent means±SEM of nine mice per group; *p<0.05, **p<0.01, ***p<0.001 vs vehicle (Veh); ##p<0.01, ###p<0.001 vs desipramine alone.
To examine a potential, secondary role of β-adrenergic receptors in idazoxan's antagonism of the antidepressant-like effect of desipramine, the above tests were repeated in β-1/2 KO mice; deficiency of β-adrenergic receptors was verified by [3H]CGP-12177 binding using cerebral cortical tissues (data not shown). The effect of desipramine in the forced-swim test was antagonized by idazoxan (0.1–2.5 mg/kg) in a dose-dependent manner (F4,32=6.11; p<0.001; Figure 1c). However, the potency of idazoxan appeared to be reduced in the β-1/2 KO mice; only the highest dose (2.5 mg/kg) antagonized the effect of desipramine (p<0.01 vs desipramine alone). Idazoxan (0.5 and 2.5 mg/kg) alone did not alter forced-swim behavior in β-1/2 KO mice (F2,16=1.25; p=0.31; Figure 1d).
Antagonism by Idazoxan of the Antidepressant-Like Effects of Desipramine on DRL Behavior in Rats
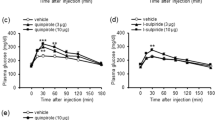
To verify the effects observed above, antagonism by idazoxan of desipramine-induced antidepressant-like behavior was examined in rats maintained under a DRL 72-s schedule, which is sensitive to a wide range of antidepressant drugs (O'Donnell et al, 2005). Desipramine (10 mg/kg) produced an antidepressant-like effect on DRL behavior in rats, ie, decreased response rate (p<0.05) and increased reinforcement rate (p<0.001); this was antagonized by peripheral administration of idazoxan (0.5–2.5 mg/kg) in a dose-dependent manner (F4,24=6.29; p<0.001 for response rate and F4,24=12.73; p<0.0001 for reinforcement rate; Figure 2a). At 0.5 mg/kg, idazoxan attenuated the desipramine-induced decrease in response rate (p<0.05), but did not affect the desipramine-induced increase in reinforcement rate. At doses of 1 and 2.5 mg/kg, idazoxan antagonized the effects of desipramine on both response and reinforcement rate. Similar to the results in the mouse forced-swim test, idazoxan administered alone, at doses as high as 5 mg/kg, did not alter DRL behavior (Figure 2b).

Antagonism by idazoxan of the antidepressant-like effects of desipramine in rats under a DRL 72-s schedule. (a) Blockade of the effects of desipramine on response rates (open symbols) and reinforcement rates (filled symbols) by peripheral administration (i.p.) of idazoxan. (b) The effects of idazoxan alone on DRL behavior. Idazoxan was injected i.p. 30 min before the test (b) or 15 min before desipramine (a), which was injected 30 min before the test. (c) Antagonism of the effects of desipramine on response and reinforcement rates by central administration of idazoxan; desipramine was injected i.p. 20 min before idazoxan, which was infused into the right lateral ventricle (i.c.v.) 10 min before the test. Data shown represent means±SEM of 7–10 rats per group expressed as a percentage of control performance. The control response and reinforcement rates (means±SD) were, respectively: (a) 86.4±6.2 and 9.9±1.7, (b) 96±14.4 and 8.5±1.7, and (c) 96.3±12.7 and 9.8±1.8. *p<0.05, ***p<0.001 vs Veh; #p<0.05, ##p<0.01, ###p<0.001 vs desipramine alone.
To determine whether central α-2 adrenergic receptors were involved its antagonist effect, idazoxan was infused into the lateral cerebral ventricle of rats treated with desipramine. Similar to the effect following peripheral administration, idazoxan (1–10 μg, i.c.v.) also attenuated the desipramine-induced decreases in response rate (F4,32=6.11; p<0.001) and increases in reinforcement rate (F4,32=11.38; p<0.0001) in a dose-dependent manner (Figure 2c).
Effects of Propranolol, CGP-12177, and Prazosin on Antidepressant-Like Behavior Produced by Desipramine in Rats Responding Under the DRL Schedule
To examine the involvement of β- and α-1 adrenergic receptors in the antidepressant-like effect of desipramine in rats, the β-adrenergic antagonists propranolol and CGP-12177 and the α-1 adrenergic antagonist prazosin were tested for their ability to antagonize the effects of desipramine on DRL behavior. The doses were chosen based on their ability to block behavioral effects involving the corresponding receptors (Ge et al, 2005; Itoi et al, 1994; Miao et al, 2003; Zhang et al, 2001; Zhang et al, 2003). Peripheral administration of propranolol (1–5 mg/kg) did not alter the effects of desipramine on response and reinforcement rates (F3,27=2.06; p=0.13 and F3,27=0.60; p=0.62; Figure 3a). Similarly, infusion of the hydrophilic antagonist CGP-12177 into the dorsal hippocampus (3–15 μg/side) did not alter the effect of desipramine on DRL behavior (F3,18=1.17; p=0.35 for response rate and F3,18=2.39; p=0.47 for reinforcement rate; Figure 3b). In addition, central administration of prazosin (5–30 μg, i.c.v.) did not alter the effect of desipramine on DRL behavior (F3,18=4.15; p<0.05 for response rate and F3,18=0.21; p=0.89 for reinforcement rate; Figure 3c).

Effects of (a) propranolol, (b) CGP-12177, and (c) prazosin on the antidepressant-like effects of desipramine in rats under a DRL 72-s schedule. Propranolol (Prop) was injected (i.p.) 15 min before desipramine, which was given (i.p.) 30 min before the test. For central antagonist administration, desipramine was injected (i.p.) 20 min before CGP-12177 or prazosin, which were given by intrahippocampal or i.c.v. infusions, respectively, 10 min before the test. Data shown represent means±SEM of 7–10 rats per group expressed as a percentage of control performance. The control response and reinforcement rates (means±SD) were, respectively: (a) 96±14.4 and 8.5±1.7, (b) 126.2±31.1 and 7.7±2.7, and (c) 86.4±6.2 and 9.9±1.7. *p<0.05, **p<0.01, ***p<0.001 vs Veh.
Antidepressant-Like Effects of Desipramine on Forced-Swim Behavior in Mice Deficient in β-Adrenergic Receptors
To verify the lack of involvement of β-adrenergic receptors in the behavioral effects of desipramine, its effects on forced-swim behavior were examined in β-1 KO, β-2 KO, β-1/2 KO mice, and their wild-type controls. Although FVB × C57BL/6 mice, which were the wild-type controls of β-1/2 KO mice, exhibited a high baseline of immobility, FVB mice, which were the wild-type controls of β-1 or β-2 KO mice, initially exhibited a relatively low baseline of immobility in the forced-swim test. The latter was consistent with previous findings (Lucki et al, 2001). However, 2–3 sessions of pre-test training increased the baseline of immobility of FVB mice up to fourfold; this was stable for months (data not shown). Desipramine (20 mg/kg) decreased immobility in the forced-swim test relative to the corresponding vehicle control in both strains of wild-type mice (p<0.001; Figure 4). Consistent with the pharmacological tests using propranolol and CGP-12177, desipramine significantly decreased immobility in mice deficient in individual β-1 or β-2 adrenergic receptors (p<0.01; Figure 4a), as well as in mice deficient in both β-1 and β-2 adrenergic receptors (p<0.01; Figure 4b), suggesting that β-1 and/or β-2 adrenergic receptors are not essential for the mediation of the effect of desipramine on forced-swim behavior.

Deficiency of β-adrenergic receptors did not alter the antidepressant-like effects of desipramine on forced-swim behavior. (a) Effects of desipramine on immobility in mice deficient in β-1 (β-1 KO) or β-2 (β-2 KO) adrenergic receptors and their wild-type controls (ie, FVB mice). (b) Effects of desipramine on immobility in mice deficient in both β-1 and β-2 adrenergic receptors (β-1/2 KO) and their wild-type controls (ie, FVB × C57BL/6 mice). Desipramine (20 mg/kg, i.p.) was injected (i.p.) 30 min before the test. Data shown represent means±SEM of 9–10 mice per group; **p<0.05, ***p<0.001 vs the corresponding vehicle control in the same genotype; ##p<0.01 vs the wild-type control with the same treatment.
The Effect of Propranolol on Desipramine-Induced Antidepressant-Like Behavior in the Forced-Swim Test in Mice
To confirm the above observations using the KO mice, the effects of desipramine, the β-1 adrenergic agonist dobutamine, and the β-2 adrenergic agonist clenbuterol on forced-swim behavior were examined in FVB mice in the absence or presence of propranolol. Peripheral administration (i.p.) of dobutamine (30 mg/kg), clenbuterol (0.5 mg/kg), or desipramine (20 mg/kg) produced an antidepressant-like effect in the forced-swim test, as evidenced by decreased immobility relative to the vehicle control (p<0.01, p<0.05, and p<0.001, respectively; Figure 5). The effects of dobutamine and clentuberol were blocked by pretreatment with 2.5 mg/kg propranolol (p<0.05), a dose that did not alter immobility when administered alone. By contrast, the desipramine-induced decrease in immobility was not blocked by propranolol, indicating that β-adrenergic receptors may not be involved to a large degree in the antidepressant-like effect of desipramine on forced-swim behavior.

Propranolol (Prop) antagonized the antidepressant-like effects of clenbuterol (Clen) and dobutamine (Dob), but not that of desipramine (Des), in the forced-swim test in wild-type mice. Propranolol (2.5 mg/kg, i.p.) was given 15 min before clenbuterol (0.5 mg/kg, i.p.), dobutamine (30 mg/kg, i.p.), or desipramine (20 mg/kg, i.p.), which were injected 30 min before the test. Data shown represent means±SEM of 10 mice per group; *p<0.05, **p<0.01, ***p<0.001 vs vehicle (Veh); #p<0.05 vs dobutamine alone.
Antidepressant-Like Behavior of Desipramine in β-1/2 KO Mice Responding Under a DRL Schedule
To verify the results observed in the forced-swim test, the effects of desipramine on behavior under a DRL 36-s schedule were examined in β-1/2 KO mice and their wild-type controls. Similar to the effects observed in rats, desipramine (3–30 mg/kg) produced antidepressant-like effects on DRL behavior in the wild-type mice, ie, decreased response rate (F3,27=5.60, p<0.01) and increased reinforcement rate (F3,27=4.00, p<0.05) in a dose-dependent manner (Figure 6a). Post hoc comparisons revealed that desipramine at 30 mg/kg significantly increased reinforcement rate (p<0.05) and decreased response rates at all doses (p<0.05 for 3 mg/kg; p<0.01 for 10 and 30 mg/kg). In the β-1/2 KO mice, desipramine also decreased response rate (F3,24=3.66, p<0.05) and increased reinforcement rate (F3,24=3.77, p<0.05), similar to its effects in wild-type controls (Figure 6b). Although the minimum effective dose (30 mg/kg) of desipramine for decreasing response rate in the β-1/2 KO mice was higher than that in the wild-type controls (3 mg/kg), the minimum effective doses for increasing reinforcement rate were the same in both genotypes. At a higher dose (50 mg/kg), desipramine produced a sedative effect on DRL behavior, ie, decreased response rate and reinforcement rate (data not shown).

Desipramine produced an antidepressant-like effect in β-1/2 KO mice and their wild-type controls under a DRL 36-s schedule. Effects of desipramine on response rates (open symbols) and reinforcement rates (filled symbols) in (a) wild-type mice and (b) β-1/2 KO mice. Desipramine (20 mg/kg, i.p.) was given 30 min before the DRL test. Data shown represent means±SEM of 9–10 mice per group expressed as a percentage of control performance. The control response and reinforcement rates (means±SD) were, respectively: (a) 278±31 and 18±3, (b) 187±66 and 29±10. *p<0.05, **p<0.01 vs corresponding vehicle (Veh) treatment.
DISCUSSION
Although it is well established that desipramine produces antidepressant-like effects by its inhibition of NE reuptake and a subsequent increase in extracellular NE concentrations (Frazer, 2000; Gillman, 2007; Zhao et al, 2008), the role of downstream mechanisms remains poorly understood. In this study, we found that the antidepressant-like effects of desipramine on mouse forced-swim and rat DRL behavior were antagonized by the α-2 adrenergic antagonist idazoxan, but not the α-1 adrenergic antagonist prazosin nor the β-adrenergic antagonists propranolol and CGP-12177. The lack of involvement of β-adrenergic receptors was verified in the forced-swim and DRL tasks using mice deficient in β-1, β-2, or both β-1 and β-2 adrenergic receptors. Desipramine remained effective in mice lacking one or both β-adrenergic receptor subtypes. These results suggest that α-2 adrenergic receptors play an important and distinct role in the mediation of the antidepressant-like effect of desipramine.
Both peripheral (i.p.) and central (i.c.v.) administration of idazoxan blocked the antidepressant-like behavioral effects of desipramine. This antagonism is consistent with the results from two previous studies that used the forced-swim test in rats (Cervo et al, 1990; Reneric et al, 2001); these earlier studies, in contrast to the present work, examined the effects of chronic desipramine treatment. Involvement of α-2 adrenergic receptor in the mediation of the effects of desipramine also is supported by electrophysiological evidence, which shows that the desipramine-induced decrease in locus coeruleus activity is reversed by idazoxan (Curet et al, 1992). Overall, the convergence of data indicate the importance of α-2 adrenergic receptors in the mediation of the antidepressant effects of desipramine, regardless of the route and duration of desipramine administration or the paradigms used for testing its actions.
Peripheral administration of desipramine increases extracellular NE levels 2- to 4-fold in the brain (Invernizzi and Garattini, 2004). Released NE activates all adrenergic receptors, including α-1, α-2, β-1, and β-2 subtypes, both pre- and postsynaptically, although it has a low affinity for β-2 adrenergic receptors (Molinoff, 1984; Segal et al, 1991). Presynaptic α-2 adrenergic receptors are inhibitory autoreceptors that regulate NE release; their antagonism increases NE release (Dennis et al, 1987). If presynaptic α-2 adrenergic receptors are important for the antidepressant-like effect of desipramine, antagonism by idazoxan would be expected to potentiate, rather than block, its effects on behavior. In addition, idazoxan alone would be expected to result in antidepressant-like behavior. The present results, which are opposite to these predictions, suggest that postsynaptic, not presynaptic, α-2 adrenergic receptors play the major role in the mediation of antidepressant effects of desipramine.
α-2 adrenergic receptors, which are classified into three subtypes (α-2A, 2B, and 2C; Bylund et al, 1994; Hieble, 2007), are involved in the mediation of both pro- and antidepressant actions. Studies using postmortem specimens from depressed suicide victims show an increase in α-2A receptors in the prefrontal cortex (Garcia-Sevilla et al, 1999b), which suggests that α-2 adrenergic antagonists may produce antidepressant effects. Consistent with this, the antidepressant effects of fluoxetine are potentiated by coadministration of yohimbine, another α-2 adrenergic antagonist (Sanacora et al, 2004); further, neuronal responsiveness to the antidepressant imipramine is enhanced by idazoxan (Linner et al, 1999). In addition, chronic treatment with the α-2 adrenergic antagonist dexefaroxan increases hippocampal neurogenesis (Rizk et al, 2006), which is associated with antidepressant activity (Santarelli et al, 2003). On the other hand, in the rat forced-swim test, α-2 adrenergic antagonists such as idazoxan block the antidepressant-like effect of desipramine (Cervo et al, 1990; Reneric et al, 2001). This is consistent with α-2 adrenergic antagonist-induced increases in immediate early genes in the brain, which are responsive to stress stimuli (Shen and Gundlach, 2000). Other studies have shown that α-2 receptor subtypes may play different roles in the regulation of antidepressant activity. Mice deficient in α-2A adrenergic receptors display depressive-like behavior in the forced-swim test and are not sensitive to the tricyclic antidepressant imipramine (Schramm et al, 2001). Consistent with this, chronic administration of desipramine selectively upregulates α-2A receptors in rats (Callado et al, 1999). By contrast, mice deficient in the α-2C subtype exhibit antidepressant-like behavior; consistently, over-expression of α-2C adrenergic receptors produces depressive-like effects in the forced-swim test (Sallinen et al, 1999). The insensitivity of α-2A KO mice to imipramine in the forced-swim test is consistent with the antagonism by idazoxan of the antidepressant-like effect of desipramine, suggesting that the α-2A subtype may be the primary α-2 adrenergic receptors involved in the mediation of antidepressant activity of desipramine. Idazoxan alone did not produce any effect on either forced-swim or DRL behavior most likely due to the balance of antagonism of both α-2A and α-2C adrenergic receptors, which may lead to depressive- and antidepressant-like effects, respectively.
Idazoxan also antagonizes imidazoline I2 receptors, which are associated with depression and antidepressant activity (Garcia-Sevilla et al, 1999a; Finn et al, 2003). However, selective imidazoline I2 antagonists do not alter forced-swim behavior nor block the antidepressant-like effects of imipramine (O'Neill et al, 2001), suggesting that idazoxan blocks the effect of desipramine via α-2 adrenergic receptors. It is reasonable to believe that other α-2 adrenergic antagonists also could block the antidepressant-like effect of desipramine and that idazoxan could block the effect of other desipramine-like antidepressants. Further studies are needed to verify this.
Presynaptic β-2 adrenergic receptors can serve a facilitatory autoreceptor function. Activation of presynaptic β-2 autoreceptors increases NE release (Murugaiah and O'Donnell, 1995); this has been suggested to contribute in the regulation of antidepressant actions of desipramine. In addition, β-1 adrenergic receptors may also contribute to the antidepressant-like effect of desipramine, given that repeated treatment with desipramine reduces the density of this subtype in the brain (Daws et al, 1998; Paetsch and Greenshaw, 1993). Consistent with this, it has been found that desipramine and related drugs produce β-1 adrenergic receptor-mediated discriminative stimulus effects (Crissman et al, 2001; Crissman and O'Donnell, 2002). Dobutamine and clenbuterol, β-1 and β-2 adrenergic agonists, respectively, produced antidepressant-like effects on forced-swim behavior. This is consistent with our previous findings that both agonists produce antidepressant-like effects on DRL behavior (Zhang et al, 2003, 2005). Propranolol blocked the effects of these two β-adrenergic agonists; this is in agreement with antagonism by propranolol of the antidepressant-like effect of clenbuterol on DRL behavior (O'Donnell, 1987). By contrast, propranolol at the same dose did not alter the antidepressant-like effect of desipramine on forced-swim behavior in mice. This was confirmed in rats responding under a DRL 72-s schedule. These results are not consistent with some previous studies, in which propranolol blocks the effect of desipramine on DRL behavior in rats (O'Donnell, 1987) and behavior produced by chronic mild stress in mice (Yalcin et al, 2007). To further verify the results in the present studies, we directly infused CGP-12177, a nonselective, hydrophilic β-adrenergic antagonist, bilaterally into the dorsal hippocampi of rats, because β-adrenergic receptors in the dorsal hippocampus play an important role in the mediation of DRL behavior (Zhang et al, 2001). CGP-12177 is much more potent than the lipophilic antagonist propranolol when administered intrahippocampally (Zhang et al, 2003). In addition, the effects of desipramine on both forced-swim and DRL behavior also were examined in mice deficient in β-1, β-2, or both β-1 and β-2 adrenergic receptors. It was found that the antidepressant-like effects of desipramine were not altered in any of these tests, suggesting that β-adrenergic receptors are not essential for the antidepressant-like effects of desipramine. This is consistent with the failure of propranolol to block the antidepressant-like effect of desipramine in the forced-swim test in rats (Pulvirenti and Samanin, 1986).
The basis of the discrepant results is not clear. Based on the studies to date, this might be attributed at least partially to changes in density of β-adrenergic receptors induced by repeated treatment with desipramine or propranolol. For instance, Yalcin et al (2007) treated animals with desipramine and propranlol for 4 weeks before behavioral tests. Antagonism of the effect of desipramine by propranolol may occur at lower levels of receptor expression because chronic desipramine downregulates β-adrenergic receptors (Daws et al, 1998; Paetsch and Greenshaw, 1993) whereas chronic exposure to β-adrenergic antagonists such as propranolol upregulates β-adrenergic receptors (Brodde et al, 1990; Ohkuma et al, 2006; Warner et al, 1992).
It was noted that the potency of idazoxan was reduced in mice deficient in β-adrenergic receptors, as evidenced by the increased minimum effective dose of idazoxan required to block the antidepressant-like effect of desipramine on FST behavior in mice deficient in both β-1 and β-2 adrenergic receptors relative to the wild-type controls (Figure 1). This appears to be due to the increased sensitivity of β-1/2 KO mice to desipramine (Figure 4). Although there is no evidence for alterations of α-adrenergic receptors in the brain of β-1/2 KO mice, studies have shown that chronic treatment with the β-adrenergic antagonist propranolol upregulates α-1 adrenergic receptors in the heart (Mügge et al, 1985; Steinkraus et al, 1989), indicating that α-1 and also likely α-2 adrenergic receptors might be increased in the brains of β-1/2 KO mice. This may cause the decreased potency of idazoxan against the antidepressant-like effects of desipramine in β-1/2 KO mice. This idea appears to be consistent with desipramine-induced increases in α-2 adrenergic receptors in the hippocampus. An alternative interpretation may include inhibition of the positive motivational brain network resulting from deficiency of β-adrenergic receptors, in particular the β-1 subtype (Zhang et al, 2001), and disinhibition of the stress network (for review see Stone et al, 2008) due to upregulation of α-2 adrenergic receptors. The imbalance of these factors not only leads to depressive-like behavior, but also suppresses the antagonist effect of idazoxan on the antidepressant effect of desipramine. Further studies are needed to clarify this point.
Although the rat DRL task has been well established for evaluating the effects of antidepressant drugs (Dekeyne et al, 2002; O'Donnell et al, 2005; Sokolowski and Seiden, 1999; Zhang et al, 2006), little is known about the use of this task in mice. Desipramine produced effects on DRL behavior in mice similar to those in rats, ie, decreased response and increased reinforcement rates, indicating that the mouse DRL task is also sensitive to antidepressant drugs such as desipramine. Mice deficient in both β-1 and β-2 adrenergic receptors and their wild-type controls displayed similar responsiveness to desipramine. However, the former appeared to be less responsive to idazoxan, as evidenced by the increased minimum dose of idazoxan required to block the desipramine-induced decreases in immobility in the forced-swim test in β-1/2 KO mice. This result is consistent with antagonism by propranolol of idazoxan-induced hyperactivity in rats (Haller et al, 1997), suggesting that postsynaptic β-adrenergic receptors may be involved in the effects of idazoxan on desipramine-induced antidepressant-like behavior. Further studies need to be done to examine this issue.
In conclusion, the antidepressant-like effects of desipramine on forced-swim and DRL behavior were antagonized by either peripheral or central administration of the α-2 adrenergic antagonist idazoxan, but were not affected by α-1 or β-adrenergic antagonists. In addition, desipramine remained effective in mice deficient in β-1 and/or β-2 adrenergic receptors. Therefore, the antidepressant-like effect of desipramine appears to be mediated primarily by postsynaptic α-2 adrenergic receptors in the brain. This needs to be taken into account when attempts are made to enhance the therapeutic effect of norepinephrine reuptake inhibitors by combination of α-2 adrenergic antagonists (Invernizzi and Garattini, 2004).
References
Brodde OE, Daul A, Michel MC (1990). Subtype-selective modulation of human beta 1- and beta 2-adrenoceptor function by beta-adrenoceptor agonists and antagonists. Clin Physiol Biochem 8: 11–17.
Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP et al (1994). International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46: 121–136.
Callado LF, Gabilondo AM, Meana JJ (1999). Differential modulation of alpha2-adrenoceptor subtypes in rat kidney by chronic desipramine treatment. Life Sci 64: 2327–2339.
Cervo L, Grignaschi G, Samanin R (1990). Alpha 2-adrenoceptor blockade prevents the effect of desipramine in the forced swimming test. Eur J Pharmacol 175: 301–307.
Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK (1999). Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem 274: 16694–16700.
Crissman AM, Makhay MM, O'Donnell JM (2001). Discriminative stimulus effects of centrally administered isoproterenol in rats: mediation by beta-1 adrenergic receptors. Psychopharmacology (Berl) 154: 70–75.
Crissman AM, O'Donnell JM (2002). Effects of antidepressants in rats trained to discriminate centrally administered isoproterenol. J Pharmacol Exp Ther 302: 606–611.
Cryan JF, Valentino RJ, Lucki I (2005). Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev 29: 547–569.
Curet O, De Montigny C, Blier P (1992). Effect of desipramine and amphetamine on noradrenergic neurotransmission: electrophysiological studies in the rat brain. Eur J Pharmacol 221: 59–70.
Dableh LJ, Yashpal K, Rochford J, Henry JL (2005). Antidepressant-like effects of neurokinin receptor antagonists in the forced swim test in the rat. Eur J Pharmacol 507: 99–105.
Daws LC, Lopez R, Frazer A (1998). Effects of antidepressant treatment on inhibitory avoidance behavior and amygdaloid beta-adrenoceptors in rats. Neuropsychopharmacology 19: 300–313.
Dekeyne A, Gobert A, Auclair A, Girardon S, Millan MJ (2002). Differential modulation of efficiency in a food-rewarded ‘differential reinforcement of low-rate’ 72-s schedule in rats by norepinephrine and serotonin reuptake inhibitors. Psychopharmacology (Berl) 162: 156–167.
Dennis T, L'Heureux R, Carter C, Scatton B (1987). Presynaptic alpha-2 adrenoceptors play a major role in the effects of idazoxan on cortical noradrenaline release (as measured by in vivo dialysis) in the rat. J Pharmacol Exp Ther 241: 642–649.
Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS (2007). A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry 61: 661–670.
Esteban S, Llado J, Sastre-Coll A, Garcia-Sevilla JA (1999). Activation and desensitization by cyclic antidepressant drugs of alpha2-autoreceptors, alpha2-heteroreceptors and 5-HT1A-autoreceptors regulating monamine synthesis in the rat brain in vivo. Naunyn Schmiedebergs Arch Pharmacol 360: 135–143.
Finn DP, Martí O, Harbuz MS, Vallès A, Belda X, Márquez C et al (2003). Behavioral, neuroendocrine and neurochemical effects of the imidazoline I2 receptor selective ligand BU224 in naive rats and rats exposed to the stress of the forced swim test. Psychopharmacology (Berl) 167: 195–202.
Frazer A (2000). Norepinephrine involvement in antidepressant action. J Clin Psychiatry 61: 25–30.
Garcia AS, Barrera G, Burke TF, Ma S, Hensler JG, Morilak DA (2004). Autoreceptor-mediated inhibition of norepinephrine release in rat medial prefrontal cortex is maintained after chronic desipramine treatment. J Neurochem 91: 683–693.
Garcia-Sevilla JA, Escriba PV, Guimon J (1999a). Imidazoline receptors and human brain disorders. Ann NY Acad Sci 881: 392–409.
Garcia-Sevilla JA, Escriba PV, Ozaita A, La Harpe R, Walzer C, Eytan A et al (1999b). Up-regulation of immunolabeled alpha2A-adrenoceptors, Gi coupling proteins, and regulatory receptor kinases in the prefrontal cortex of depressed suicides. J Neurochem 72: 282–291.
Ge ZJ, Zeng YM, Tan YF (2005). Effects of intrathecal 6-hydroxydopamine, alpha1 and alpha2 adrenergic receptor antagonists on antinociception of propofol in mice. Acta Pharmacol Sin 26: 186–191.
Gillman PK (2007). Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol 151: 737–748.
González AM, Pascual J, Meana JJ, Barturen F, del Arco C, Pazos A et al (1994). Autoradiographic demonstration of increased alpha 2-adrenoceptor agonist binding sites in the hippocampus and frontal cortex of depressed suicide victims. J Neurochem 63: 256–265.
Haller J, Makara GB, Pintér I, Gyertyán I, Egyed A (1997). The mechanism of action of alpha 2 adrenoceptor blockers as revealed by effects on open field locomotion and escape reactions in the shuttle-box. Psychopharmacology (Berl) 134: 107–114.
Hieble JP (2007). Subclassification and nomenclature of alpha- and beta-adrenoceptors. Curr Top Med Chem 7: 129–134.
Invernizzi RW, Garattini S (2004). Role of presynaptic alpha2-adrenoceptors in antidepressant action: recent findings from microdialysis studies. Prog Neuropsychopharmacol Biol Psychiatry 28: 819–827.
Itoi K, Suda T, Tozawa F, Dobashi I, Ohmori N, Sakai Y et al (1994). Microinjection of norepinephrine into the paraventricular nucleus of the hypothalamus stimulates corticotropin-releasing factor gene expression in conscious rats. Endocrinology 135: 2177–2182.
Lapiz MD, Zhao Z, Bondi CO, O'Donnell JM, Morilak DA (2007). Blockade of autoreceptor-mediated inhibition of norepinephrine release by atipamezole is maintained after chronic reuptake inhibition. Int J Neuropsychopharmacol 10: 827–833.
Linner L, Arborelius L, Nomikos GG, Bertilsson L, Svensson TH (1999). Locus coeruleus neuronal activity and noradrenaline availability in the frontal cortex of rats chronically treated with imipramine: effect of alpha 2-adrenoceptor blockade. Biol Psychiatry 46: 766–774.
Lucki I, Dalvi A, Mayorga AJ (2001). Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology (Berl) 155: 315–322.
Maes M, Van Gastel A, Delmeire L, Meltzer HY (1999). Decreased platelet alpha-2 adrenoceptor density in major depression: effects of tricyclic antidepressants and fluoxetine. Biol Psychiatry 45: 278–284.
Mateo Y, Fernandez-Pastor B, Meana JJ (2001). Acute and chronic effects of desipramine and clorgyline on alpha(2)-adrenoceptors regulating noradrenergic transmission in the rat brain: a dual-probe microdialysis study. Br J Pharmacol 133: 1362–1370.
McElroy JF, O'Donnell JM (1988). Discriminative stimulus properties of clenbuterol: evidence for beta adrenergic involvement. J Pharmacol Exp Ther 245: 155–163.
Menargues A, Obach R, García-Sevilla JA (1990). Modulation by antidepressant drugs of CNS postsynaptic alpha 2-adrenoceptors mediating mydriasis in the rat. Naunyn Schmiedebergs Arch Pharmacol 341: 101–107.
Miao CY, Xie HH, Yu H, Chu ZX, Su DF (2003). Ketanserin stabilizes blood pressure in conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 30: 189–193.
Molinoff PB (1984). Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs 28: 1–15.
Mongeau R, de Montigny C, Blier P (1994). Electrophysiologic evidence for desensitization of alpha 2-adrenoceptors on serotonin terminals following long-term treatment with drugs increasing norepinephrine synaptic concentration. Neuropsychopharmacology 10: 41–51.
Mügge A, Reupcke C, Scholz H (1985). Increased myocardial alpha 1 adrenoceptor density in rats chronically treated with propranolol. Eur J Pharmacol 112: 249–252.
Murugaiah KD, O'Donnell JM (1995). Facilitation of noradrenaline release from rat brain slices by beta-adrenoceptors. Naunyn Schmiedebergs Arch Pharmacol 351: 483–490.
O'Donnell JM (1987). Effects of clenbuterol and prenalterol on performance during differential reinforcement of low response rate in the rat. J Pharmacol Exp Ther 241: 68–75.
O'Donnell JM (1990). Behavioral effects of beta adrenergic agonists and antidepressant drugs after down-regulation of beta-2 adrenergic receptors by clenbuterol. J Pharmacol Exp Ther 254: 147–157.
O'Donnell JM, Frith S, Wilkins J (1994). Involvement of beta-1 and beta-2 adrenergic receptors in the antidepressant-like effects of centrally administered isoproterenol. J Pharmacol Exp Ther 271: 246–254.
O'Donnell JM, Marek GJ, Seiden LS (2005). Antidepressant effects assessed using behavior maintained under a differential-reinforcement-of-low-rate (DRL) operant schedule. Neurosci Biobehav Rev 29: 785–798.
Ohkuma S, Katsura M, Shibasaki M, Tsujimura A, Hirouchi M (2006). Expression of beta-adrenergic receptor up-regulation is mediated by two different processes. Brain Res 1112: 114–125.
O'Neill MF, Osborne DJ, Woodhouse SM, Conway MW (2001). Selective imidazoline I2 ligands do not show antidepressant-like activity in the forced swim test in mice. J Psychopharmacol 15: 18–22.
Paetsch PR, Greenshaw AJ (1993). Effects of chronic antidepressant treatment on beta-adrenoceptor subtype binding in the rat cerebral cortex and cerebellum. Mol Chem Neuropathol 20: 21–31.
Pattij T, Broersen LM, van der Linde J, Groenink L, van der Gugten J, Maes RA et al (2003). Operant learning and differential-reinforcement-of-low-rate 36-s responding in 5-HT1A and 5-HT1B receptor knockout mice. Behav Brain Res 141: 137–145.
Poncelet M, Gaudel G, Danti S, Soubrié P, Simon P (1986). Acute versus repeated administration of desipramine in rats and mice: relationships between brain concentrations and reduction of immobility in the swimming test. Psychopharmacology (Berl) 90: 139–141.
Pulvirenti L, Samanin R (1986). Antagonism by dopamine, but not noradrenaline receptor blockers of the anti-immobility activity of desipramine after different treatment schedules in the rat. Pharmacol Res Commun 18: 73–80.
Reneric JP, Bouvard M, Stinus L (2001). Idazoxan and 8-OH-DPAT modify the behavioral effects induced by either NA, or 5-HT, or dual NA/5-HT reuptake inhibition in the rat forced swimming test. Neuropsychopharmacology 24: 379–390.
Rizk P, Salazar J, Raisman-Vozari R, Marien M, Ruberg M, Colpaert F et al (2006). The alpha2-adrenoceptor antagonist dexefaroxan enhances hippocampal neurogenesis by increasing the survival and differentiation of new granule cells. Neuropsychopharmacology 31: 1146–1157.
Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK (1999). Cardiovascular and metabolic alterations in mice lacking both beta1- and beta2-adrenergic receptors. J Biol Chem 274: 16701–16708.
Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula Jr DP, Barsh GS et al (1996). Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci USA 93: 7375–7380.
Sallinen J, Haapalinna A, MacDonald E, Viitamaa T, Lahdesmaki J, Rybnikova E et al (1999). Genetic alteration of the alpha2-adrenoceptor subtype c in mice affects the development of behavioral despair and stress-induced increases in plasma corticosterone levels. Mol Psychiatry 4: 443–452.
Sanacora G, Berman RM, Cappiello A, Oren DA, Kugaya A, Liu N et al (2004). Addition of the alpha2-antagonist yohimbine to fluoxetine: effects on rate of antidepressant response. Neuropsychopharmacology 29: 1166–1171.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301: 805–809.
Schramm NL, McDonald MP, Limbird LE (2001). The alpha(2a)-adrenergic receptor plays a protective role in mouse behavioral models of depression and anxiety. J Neurosci 21: 4875–4882.
Segal M, Markram H, Richter-Levin G (1991). Actions of norepinephrine in the rat hippocampus. Prog Brain Res 88: 323–330.
Shen PJ, Gundlach AL (2000). Differential modulatory effects of alpha- and beta-adrenoceptor agonists and antagonists on cortical immediate-early gene expression following focal cerebrocortical lesion-induced spreading depression. Brain Res Mol Brain Res 83: 133–144.
Sokolowski JD, Seiden LS (1999). The behavioral effects of sertraline, fluoxetine, and paroxetine differ on the differential-reinforcement-of-low-rate 72-second operant schedule in the rat. Psychopharmacology (Berl) 147: 153–161.
Steinkraus V, Nose M, Scholz H, Thormählen K (1989). Time course and extent of alpha 1-adrenoceptor density changes in rat heart after beta-adrenoceptor blockade. Br J Pharmacol 96: 441–449.
Stone EA, Lin Y, Quartermain D (2008). A final common pathway for depression? Progress toward a general conceptual framework. Neurosci Biobehav Rev 32: 508–524.
Subhash MN, Nagaraja MR, Sharada S, Vinod KY (2003). Cortical alpha-adrenoceptor downregulation by tricyclic antidepressants in the rat brain. Neurochem Int 43: 603–609.
Thomas DN, Holman RB (1991). A microdialysis study of the regulation of endogenous noradrenaline release in the rat hippocampus. J Neurochem 56: 1741–1746.
Wallace-Boone TL, Newton AE, Wright RN, Lodge NJ, McElroy JF (2007). Behavioral and pharmacological validation of the gerbil forced-swim test: effects of neurokinin-1 receptor antagonists. Neuropsychopharmacology 33: 1919–1928.
Warner AL, Bellah KL, Raya TE, Roeske WR, Goldman S (1992). Effects of beta-adrenergic blockade on papillary muscle function and the beta-adrenergic receptor system in noninfarcted myocardium in compensated ischemic left ventricular dysfunction. Circulation 86: 1584–1595.
Wicke KM, Rex A, Jongen-Relo A, Groth I, Gross G (2007). The guinea pig forced swim test as a new behavioral despair model to characterize potential antidepressants. Psychopharmacology (Berl) 195: 95–102.
Yalcin I, Aksu F, Bodard S, Chalon S, Belzung C (2007). Antidepressant-like effect of tramadol in the unpredictable chronic mild stress procedure: possible involvement of the noradrenergic system. Behav Pharmacol 18: 623–631.
Zhang HT, Frith SA, Wilkins J, O'Donnell JM (2001). Comparison of the effects of isoproterenol administered into the hippocampus, frontal cortex, or amygdala on behavior of rats maintained by differential reinforcement of low response rate. Psychopharmacology (Berl) 159: 89–97.
Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M et al (2002). Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology 27: 587–595.
Zhang HT, Huang Y, Masood A, Stolinski LR, Li YF, Zhang L et al (2008). Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33: 1611–1623.
Zhang HT, Huang Y, Mishler K, Roerig SC, O'Donnell JM (2005). Interaction between the antidepressant-like behavioral effects of beta adrenergic agonists and the cyclic AMP PDE inhibitor rolipram. Psychopharmacology (Berl) 182: 104–115.
Zhang HT, Huang Y, O'Donnell JM (2003). Antagonism of the antidepressant-like effects of clenbuterol by central administration of beta-adrenergic antagonists in rats. Psychopharmacology (Berl) 170: 102–107.
Zhang HT, Zhao Y, Huang Y, Deng C, Hopper AT, De Vivo M et al (2006). Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl) 186: 209–217.
Zhang HT, Zhao Y, Huang Y, Dorairaj NR, Chandler LJ, O'Donnell JM (2004). Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology 29: 1432–1439.
Zhao Z, Baros AM, Zhang HT, Lapiz MD, Bondi CO, Morilak DA et al (2008). Norepinephrine transporter regulation mediates the long-term behavioral effects of the antidepressant desipramine. Neuropsychopharmacology, in press (April 16 2008, Epub ahead of print).
Acknowledgements
This work was supported by research grants (MH040694, MH051175, MH072646) from the National Institute of Mental Health. We thank Dr Brian K Kobilka of Stanford University School of Medicine for providing the β-adrenergic receptor KO mice and Mr Ajay K Venkatesan for his technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
James M O'Donnell is on the Scientific Advisory Board of Fission Pharmaceuticals (unpaid). Han-Ting Zhang and James M O'Donnell have received financial support for their research from Memory Pharmaceuticals, Lundbeck Pharmaceuticals, and Wyeth Pharmaceuticals.
Rights and permissions
About this article
Cite this article
Zhang, HT., Whisler, L., Huang, Y. et al. Postsynaptic α-2 Adrenergic Receptors are Critical for the Antidepressant-Like Effects of Desipramine on Behavior. Neuropsychopharmacol 34, 1067–1077 (2009). https://doi.org/10.1038/npp.2008.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.184
Keywords
This article is cited by
-
G protein-coupled receptors in neurodegenerative diseases and psychiatric disorders
Signal Transduction and Targeted Therapy (2023)
-
Echinacoside exhibits antidepressant-like effects through AMPAR–Akt/ERK–mTOR pathway stimulation and BDNF expression in mice
Chinese Medicine (2022)
-
Toward a Quantification of Anhedonia: Unified Matching Law and Signal Detection for Clinical Assessment and Drug Development
Perspectives on Behavior Science (2021)
-
Circuit-specific hippocampal ΔFosB underlies resilience to stress-induced social avoidance
Nature Communications (2020)
-
Modulation of Monoaminergic Systems by Antidepressants in the Frontal Cortex of Rats After Chronic Mild Stress Exposure
Molecular Neurobiology (2019)
