
Abstract
GPR109A has generated expanding interest since its discovery as the receptor for niacin a decade ago, along with deorphanisation as the receptor for endogenous ligand 3-hydroxy-butyrate shortly after. This interest is generated especially because of the continuing exploration of niacin’s “pleiotropic” mechanisms of action and its potential in the “cross-talk” between metabolic and inflammatory pathways. As GPR109A’s primary pharmacological ligand in clinical use, niacin has been used for over 50 years in the treatment of cardiovascular disease, mainly due to its favourable effects on plasma lipoproteins. However, it has become apparent that niacin also possesses lipoprotein-independent effects that influence inflammatory pathways mediated through GPR109A. In addition to its G-protein–mediated effects, recent evidence has emerged to support alternative GPR109A signalling via adaptive protein β-arrestins. In this article, we consider the role of GPR109A and its downstream effects in the context of atherosclerosis and vascular inflammation, along with insights into strategy for future drug development.
Similar content being viewed by others

Introduction
GPR109A belongs to a family of three G-protein–coupled receptors that share significant sequence homology and whose known cognate ligands are metabolites of hydroxycarboxylic acid [1•]. Despite the discovery by Altschul in 1955 of the beneficial effects of nicotinic acid (niacin) on plasma lipoproteins [2], it was not until in 2003 that the G-protein–coupled receptor (GPCR) GPR109A [also known as hydroxy-carboxylic acid (HCA) receptor 2 (HCA2), HM74a, or NIACR1] was found to be a receptor target of niacin [3–5]. Two years later, GPR109A was deorphanised when the endogenous ligand ketone body 3-hydroxy-butyrate was found by Taggart et al. [6]. Since then, many other synthetic agonists of GPR109A had been developed (Acipimox, Acifran, MK-0354, etc.) [1•]. Other members of the HCA receptors family have also been deorphanised: lactate was found as the endogenous ligand for GPR81 (HCA1 receptor) [7], whereas β-oxidation intermediate 3-hydroxyl-octanoic acid was found for GPR109B (also known as HCA3 receptor, HM74, or NIACR2) [8].
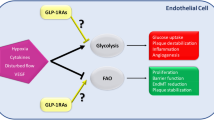
Over the last twenty years, HMG-CoA reductase inhibitors, statins, have become the first-line treatment of dyslipidaemia. Although treatment with these compounds achieves substantial LDL-c reduction, significant residual cardiovascular risk remains [9–11]. There is strong epidemiological evidence of an inverse relationship between HDL-c level and coronary heart disease risk, regardless of the LDL-c level [12, 13], which persists in patients who are treated with statins [10]. Thus, besides targeting LDL-c, recent clinical focus has been expanded to elevate or modify the properties of plasma HDL [14, 15], and to counteract the inflammatory processes associated with atherosclerosis [16–18]. The GPR109A agonist niacin reduces atherogenic lipoproteins LDL-c, VLDL-c, and Lp(a), and is currently the most potent drug available to raise plasma HDL-c (by up to 30 %) [19]. In addition, activation of GPR109A with niacin also antagonises known inflammatory pathways in adipocytes [20] and in some leukocytes [21]. Therefore, with its “pleiotropic” potential, niacin has re-entered the forefront and has fuelled accelerated clinical studies, as well as basic research into the biological role of GPR109A and its downstream signalling pathways (Fig. 1)

Diagram showing the putative GPR109A downstream signalling mechanisms. α, β, γ: G-protein subunits; β-Arr: β-arrestin; numbers refer to respective references. GPR109A activation results in G-protein–coupled receptor kinase (GRK)-mediated self-phosphorylation and recruitment of β-arrestins. In addition to mediating GPR109A receptor internalisation and recycling, β-arrestins also directly mediate downstream signalling independent of the G-protein pathways
.
Niacin and Its Mechanisms of Action
GPR109A has a high affinity for niacin; however, the effects of niacin on plasma lipoproteins [22] are complex and currently not entirely understood. GPR109A is highly expressed in adipocytes [23], as well as other cell types including neutrophils [24], macrophages [25], keratinocytes [26] and Langerhans cells [26]. In adipocytes, GPR109A activation results in Gi/G0 protein-mediated inhibition of adenylate cyclase, leading to a decreased cAMP response [4, 27]. Suppression of cAMP has also been reported in HM74a-transfected CHO-K1 [4], 293EBNA [3], and HEK293 [27] which is at least partially ascribed to inhibition of adenylate cyclase, mediated by GPR109A. We have also observed a suppression of cAMP in nonstimulated, basal monocytic cells (Chai et al. 2013, unpublished). The reduction in cAMP in adipocytes leads to a reduced activity of protein kinase A and a decrease in hormone sensitive lipase activity resulting in an inhibition of lipolysis. According to one hypothesis, reduced triglyceride hydrolysis and free fatty acid (FFA) release leads to diminished FFA flux to the liver, thus limiting substrate availability for hepatic triglyceride and VLDL-c synthesis [28]. However, this prevailing “FFA hypothesis” was recently refuted by a study using a humanized genetic mouse model, in which niacin lost its efficacy in inhibiting FFA release in animals lacking GPR109A, but retained its effect on plasma HDL and triglycerides.[29••] Treatment in mice with selective GPR109A agonist MK-1903 demonstrated antilipolytic action but showed no effects on plasma triglycerides, LDL-c, as well as HDL-c. The authors went on to further support their findings in separate human clinical trials, in which both MK-1903 and another GPR109A agonist (SCH900271) resulted in reduced FFA lipolysis but neither showed the anticipated effects in plasma lipoproteins. Taken together, this suggests that although niacin exerts its antilipolytic effects via GPR109A, it may have an independent mechanism of action in altering plasma lipoproteins [30].
Niacin has other actions on the liver that are also GPR109A-independent as shown in a study by Jin et al. [31]. Using a human hepatoblastoma (HepG2) cell line, niacin increased intracellular apolipoprotein B degradation and reduced secretion of apolipoprotein B into the culture media. Niacin has also been shown to reduce hepatic reuptake of HDL particles by inhibiting the surface-expressed ATP-synthase β-chains, which facilitate holoparticle HDL endocytosis [32]. In addition, high concentrations of niacin in vitro non-competitively inhibit hepatocyte microsomal diacylglycerol aceltransferase-2 [33], which catalyses the formation of triglyceride from diacylglycerol and fatty acetyl-CoA, a committed step in triglyceride synthesis [34].
The “pleiotropic” Effects of Niacin
Although it remains unclear whether niacin directly affects hepatic HDL-c synthesis via GPR109A, there is intriguing evidence that it does so in adipose tissue. Adipose tissue is the largest free cholesterol reservoir and abundantly expresses ATP-binding cassette transport A1, a key cholesterol transporter for HDL biosynthesis [35]. In this context, niacin was shown to stimulate PPARγ, LXRα and ABCA1 mRNA expression dose-dependently and promote ApoA-I-induced cholesterol efflux in 3T3-L1 adipocytes [36]. In addition, both niacin and its structurally-distinct GPR109A/B agonist acifran were able to induce nuclear expression of PPARγ and enhance PPARγ transcriptional activity [37]. Importantly, the authors showed that these effects were pertussis toxin–sensitive and required phospholipase A2. Furthermore, niacin-mediated PPARγ activity was observed in GPR109A-CHO transfectant cells but not in vector-only control cells, indicating that GPR109A is critical for niacin’s effect on the reverse cholesterol transport (RCT) PPARγ-LXRα-ABCA1 pathway.
Reverse Cholesterol Transport and HDL
The effects of GPR109A-mediated ABCA1 upregulation, the resultant cholesterol efflux, and HDL biosynthesis have potential indirect beneficial effects on vascular inflammation. HDLs have long been shown to exhibit anti-inflammatory properties by virtue of their ability to inhibit monocyte transmigration in response to oxidized LDL [38]. In addition, HDLs inhibit cytokine-induced expression of vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, and E-selectin in human umbilical vein endothelial cells (HUVEC’s) within the physiological range of HDL levels [39–41]. It was shown that HDL’s also inhibit tumour necrosis factor-α (TNF-α) stimulated endothelial cell sphingosine kinase activity, which may also serve to be atheroprotective [42], as there is reduced nuclear translocation of NF-κB, a key step in the inflammatory pathway by which TNF-α stimulates the expression of endothelial adhesion molecules [43]. The ability of HDL’s to alter endothelial cell adhesion protein expression has also been demonstrated in animal models. Infusion of rHDL containing ApoA-I and phosphatidylcholine to apoE-/- mice reduced VCAM-1 expression and monocyte infiltration within 1 week [44]. Interestingly, niacin supplementation (0.6 % and 1.2 %) in the diet of New Zealand white rabbits for 2 weeks was also associated with significantly improved endothelial dysfunction independent of changes in plasma lipids. At 24 hours following periarterial carotid collar implantation, endothelial expression of VCAM-1, ICAM-1 and monocyte chemotactic protein-1 (MCP-1) were reduced in comparison to controls [45]. In human subjects heterozygous for a loss-of-function ABCA1 mutation who have low plasma HDL, their impaired forearm blood flow (a surrogate indicator of impaired endothelial function) was restored to that of normal controls 4 hours after a single infusion of rHDL [46]. Niacin treatment was also found to improve endothelial dysfunction in patients with coronary artery disease and low HDL-c, but not with normal HDL-c [47].
Direct Anti-inflammatory Effects
In addition to the indirect anti-inflammatory effects mediated by HDLs, more recently, the relevance of niacin and GPR109A activation with its direct anti-inflammatory potential in the vasculature has begun to emerge. A number of laboratories have reported nonlipoprotein–mediated effects of niacin that may have a bearing on atherosclerosis progression and risk [48••, 49]. In addition to the potentially favourable lipoprotein modulating effects of niacin, study of the pharmacology and mechanisms of action of niacin have revealed anti-inflammatory effects in monocytes/macrophages [50, 51], adipose tissue [20], and vascular endothelium [49, 52]. In an elegant study by Lukasova et al., using LDL-receptor knockout mice, niacin reduced the progression of atherosclerosis. Importantly, this was lipoprotein independent, as there were no changes to LDL-c, VLDL-c and HDL-c levels. Moreover, these beneficial effects were abrogated in Ldlr -/- & GPR109A -/- ‘double knockout’ mice [50]. Through bone marrow transplantation, mediation of antiatherosclerotic mechanisms was shown to be via GPR109A in marrow-derived cells, which was further supported by the inhibition of MCP-1 induced recruitment of macrophages into the peritoneal cavity and impaired macrophage recruitment to atherosclerotic plaques. This study also reported a reduction in the expression of adhesion molecules in atherosclerotic vessels of nicotinic acid–treated Ldlr –/– mice. These data suggest novel GPR109A receptor mediated antiatherosclerotic effects of niacin, which are not dependent on alterations in lipoproteins.
In support of this finding, we have recently reported potent anti-inflammatory effects of niacin in both human adipocytes [20] and monocytes [21], which are mediated via GPR109A-dependent mechanisms. In adipocytes, niacin inhibits TNF-α stimulated expression and secretion of inflammatory cytokines MCP-1, regulated and normal T cell expressed and secreted (RANTES) and fractalkine [20]. Under conditions of inflammation associated with cardiovascular disease, increased secretion of proatherogenic, proinflammatory cytokines and chemokines contribute significantly to the recruitment of inflammatory T-cells and macrophages into atherosclerotic lesions [53–55]. Adipose tissue has the potential to contribute to processes involved in both systemic and local (perivascular) inflammation in the context of atherosclerosis, both of which may be influenced by the actions of niacin. More importantly, niacin also inhibited toll-like receptor (TLR)-4 and TLR-2 induced expression and secretion of pro-inflammatory cytokines TNF-α, interleukin-6 (IL-6), and MCP-1 in human monocytes [21]. Furthermore, this anti-inflammatory effect of niacin was contingent upon the expression of GPR109A as siRNA knockdown of GPR109A abrogated the effect. Niacin profoundly inhibited TLR-4 induced nuclear accumulation of activated NF-κB in human monocytes and that this inhibition was independent of the prostaglandin and PPAR-γ pathways. Interestingly, the effect of niacin on cell adhesion and chemotaxis was very rapid. Treatment of monocytes with niacin for just one hour potently inhibited monocyte adhesion to activated HUVEC, and to VCAM, mediated by the integrin, very late antigen 4 (VLA-4). Monocyte chemotaxis was also significantly reduced. Since monocyte recruitment to activated vascular endothelium and chemotaxis within the sub-endothelial space are key events in vascular inflammation and atherosclerosis, this suggests that niacin can potentially exert very rapid effect in modulating events of early vascular inflammation.
Niacin promotes the expression of ABC transporter proteins in adipocytes [36], and importantly, in macrophages [48••], mediated by GPR109A. Recently, the strong relationship between ABC transporters and vascular inflammation is emerging. ABCA1 primarily interacts with apoA-I to efflux cholesterol to form nascent HDL particles. The interaction between apoA-I and ABCA1 was shown to activate signalling molecules such as Janus Kinase 2 [56]. This ABCA1-mediated Janus Kinase signalling activates STAT3 independently of the lipid transport function of ABCA1. ABCA1 was also shown to be responsible in regulating macrophage responsiveness to TLR agonists by modulation of lipid raft cholesterol and MyD88-dependent TLR mobilization to lipid rafts [57]. Similar findings have also been reported for another ABC transporter protein ABCG1, where ABCG1-deficient macrophages showed increased cholesterol accumulation and enhanced TLR signalling in response to TLR-4 stimulation [58]. In animal models deficient in ABCA1 and ABCG1 there is expansion of Lin – Sca-1 + Kit + (LSK) haematopoietic progenitor cells leading to leukocytosis, and accelerated atherosclerosis in mice, suggesting a role of ABC transporters in haematopoietic stem cell proliferation [58]. Moreover, transplantation of Abca1 –/– Abcg1 –/– bone marrow into apoA-1 transgenic mice with elevated levels of HDL suppressed the LSK population, reduced leukocytosis, reversed the myeloproliferative disorder, and accelerated atherosclerosis. This implies that cholesterol efflux mechanisms, such as ABCA1, ABCG1, and HDL, may directly regulate the proliferation of myeloid progenitor populations, which are intricately linked to leukocyte flux and accelerated atherosclerosis [59].
The observation that niacin plays a role in atherosclerosis regression in both mice [48••] and humans [60], and that, via GPR109a, it has the capacity to act directly on monocyte/macrophage function by upregulating proteins that are involved in cellular cholesterol efflux, raises the important possibility that niacin may exert GPR109A–mediated effects in human macrophage-derived foam cells to induce cholesterol efflux leading to plaque regression. We have recently found expression of GPR109A in ex vivo human carotid atherosclerotic plaques and in non–foam cell plaque macrophages but not in lipid-laden “foam cell” macrophages. We have also found in vitro evidence that foam cell transformation downregulates GPR109A mRNA and protein expression, and that niacin loses its effect on ABC transporter protein upregulation, and importantly, functional cholesterol efflux promotion in transformed “foam cells” compared to basal human macrophages (Chai et al. 2013, unpublished). Although the mechanism of GPR109A downregulation in “foam cells” remains to be elucidated, involvement of the adaptor proteins, β-arrestins, which recently emerged to be responsible to some downstream GPR109A effects, is thought to be probable.
Arrestins
As a member of 7-transmembrane receptor (7TMR) GPCR, GPR109A expression is regulated by agonist-induced internalisation and recycling mediated by arrestins [61], which are partly responsible for the desensitisation characteristic of GPCR signalling. In recent years, new evidence has shed light into how this pathway may not only act as a negative feedback to regulate GPR109A expression but may also transduce cellular signals independent of the G-protein pathway. The arrestin family consists of four isoforms, two expressed only in the visual system (visual and cone arrestin) and two that are ubiquitously expressed, β-arrestins 1 and 2 [61–63]. Both β-arrestin 1 and 2 are expressed in 3T3-L1 adipocytes, differentiated THP-1 macrophages, and Langerhans cells, and are thought to mediate the pharmacological effects of GPR109A activation.[64•]
Using GPR109A-expressing stable cells transfected with yellow-fluorescent protein (mYFP) tagged β-arrestin 1 or β-arrestin 2, Walters et al. found that GPR109A activation by niacin rapidly recruited β-arrestin 1 from the cytosolic compartment to the cell membrane.[64•] This interaction with the activated GPR109A was shown to cause β-arrestin to undergo receptor activation–dependent conformational changes using an intramolecular bioluminescence resonance energy transfer–based (BRET-based) biosensor. Both β-arrestin 1 and 2 were shown to interact with IκBα, which prevented its phosphorylation and degradation [65, 66]. As a result, they effectively inhibited NF-κB activity and modulated NF-κB mediated gene activation in the classic inflammatory pathway. Furthermore, recent evidence has suggested a direct involvement of β-arrestin 1 in specific gene activation in cell nuclei. It was observed that GPCR activation leads to nuclear translocation of β-arrestin 1 [67], which is selectively enriched at specific promoters such as that of p27 and c-fos, where it facilitates the recruitment of histone acetyltransferase p300, resulting in enhanced local histone H4 acetylation and transcription of these genes. Collectively, these studies have expanded our knowledge of the role of β-arrestins in cell signalling, and revealed how they can act as key scaffolding proteins to guide receptor signals from cell membrane to various target cascades to activate different cellular pathways.
One important consequence of the divergent signalling of GPR109A, independently via both G-proteins pathways and β-arrestins, is to challenge our previous thinking about the linearity of GRCR signalling. Walters et al. elegantly dissected the signalling pathways mediating niacin’s pharmacological action and side effects and found that while niacin’s anti-lipolytic effect was mediated by G-proteins, its side effect of cutaneous flushing, caused by activation of cytosolic phospholipase A2 and subsequent release of prostaglandin D2 in cutaneous Langerhans’ cells (and keratinocytes), was mediated by β-arrestin 1.[64•] β-arrestin 1–deficient mice displayed reduced cutaneous flushing in response to niacin, although the improvement in serum free fatty acid levels was similar to that observed in wild-type mice. This “biased” or preferential agonism opens the opportunity to dissociate side-effects from a drug’s therapeutic actions through the development of “biased” ligands. In this regard, recently developed GPR109A partial agonists, such as MK-0354, have been shown to decrease serum FFAs, but do not inducing cutaneous flushing [68–70].
Endogenous Ligand 3-HB and Other Inflammatory Pathologies
Besides its potential pharmacological value, there remain important biological questions as to the physiological role of GPR109A, for which the cognate ligand is 3-hydroxy-butyrate (3-HB). 3-HB is a ketone body that is produced from acetyl-CoA in hepatocytes and is an alternative energy source to the brain and, to a lesser extent, the heart when glucose availability is low during fasting or starvation. Activation of GPR109A in adipocytes inhibits fatty acid release, possibly acting as a negative feedback against excessive lipolysis in starvation [71]. Indeed plasma concentration of ketones can change over several orders of magnitude under normal physiology [72]. We have previously shown anti-inflammatory effects of GPR109A activation in both adipocytes [20] and stimulated monocytes [21], those being two principal cell types bearing GPR109A. Each of these cell types is implicated in inflammatory pathologies relating to obesity and/or high caloric intake. It is therefore plausible that a metabolic mediator that is regulated and released by the liver over a large dynamic range in starvation might be involved in the suppression of inflammation in these cell types [73]. This “cross-talk” between the metabolic and inflammatory pathways was further highlighted by a recent study, which showed that high-fat diet and diet-induced obesity in mice significantly reduced the expression of GPR109A and its downstream effector PPARγ in adipose tissue [74].
However, the physiological function of GPR109A in immune cells is still largely unknown. Activation of GPR109A by the bacterial fermentation product, butyrate, was shown to exert anti-inflammatory effect in colonic inflammation [75] and tumour-suppressing effects in inflammation related colon cancer [76]. In colonic mucosa from patients with ulcerative colitis, butyrate was shown to inhibit IFNγ-induced STAT1 activation, and caused Fas-mediated apoptosis of T cells by inhibiting histone deacetylase 1 activity, which was bound to the Fas promotor in T cells. Knocking down GPR109A resulted in altered expression of genes related to multiple inflammatory signalling pathways in mouse colonic epithelial cells [75]. Furthermore, in cerebral hypoxia, 3-HB is also shown to be neuroprotective [77–79]. Although this may reflect adaptations to metabolic substrate utilization or energetics, immune cell modulation via GPR109A is another plausible explanation. In the common immune-mediated skin disorder psoriasis, which was thought to be caused by dysregulated T-cell activation in the skin, one of the therapeutic agents in clinical use, monomethyl fumarate (MMF), and its related compound dimethyl fumarate (DMF), which is a promising novel oral therapeutic option shown to reduce disease activity and progression in patients with relapsing–remitting multiple sclerosis [80], were shown to cause cutaneous flushing by the same mechanism as niacin, mediated by GPR109A [26]. Whether the therapeutic effects of MMF and DMF were also mediated via GPR109A is currently debated, the known antagonism of classic inflammatory pathways by GPR109A activation and both drugs’ immune-modulatory effects in the treatment of psoriasis and multiple sclerosis respectively, however, provides strong hint for its involvement.
GPR109A Agonists in Drug Development
Niacin has been in clinical use for more than half a century. Many previous clinical trials have shown that niacin reduces atherosclerosis, estimated from coronary angiography [81, 82], carotid ultrasound [83–86] and MRI [60]. For a detailed review on historical clinical trials concerning niacin, readers are referred to our recent article [87]. However, despite two recent meta-analyses confirming benefits in cardiovascular (CV) outcomes associated with the use of niacin [88, 89], two large CV outcome trials have recently reported disappointing results.
The Atherosclerosis Intervention in Metabolic syndrome with Low HDL/HIGH Triglyderides: impact on Global Health Outcomes (AIM-HIGH) [90] study was terminated early in 2011 due to a lack of efficacy. Although this study was criticized for its design [91, 92] and the use of small “spike” doses of niacin in the placebo group to conserve blinding, a much larger phase III trial HPS2-THRIVE [93] also reported negative results. This study enrolled 25,673 patients considered to be at high risk for cardiovascular events from the United Kingdom, Scandinavia, and China. Participants received extended release niacin and laropiprant plus statin therapy versus statin therapy alone, with a median follow-up period of 3.9 years. Shortly before this review, it was announced by Merck that the HPS2-THRIVE trial did not reach its primary endpoint [94], and the European Medicines Agency has recommended suspension of niacin/laropiprant products in the EU for adults with dyslipidaemia [95]. With two recent negative outcome trials, one would argue that it appears that niacin does not confer any additional cardiovascular benefit in patients optimally treated with statins. However, these findings have to be interpreted cautiously, as we still do not fully understand how niacin exerts its lipoprotein effects and the pathophysiological significance of GPR109A activation in human atherosclerotic plaque.
There are at least 5 novel candidates of selective GPR109A agonists from Merck, Arena, Schering-Plough, Glaxo Smith-Kline (GSK), and Incyte [96]. GSK was the first pharmaceutical company to develop small molecule agonist for GPR109A. Their first clinical candidate of GPR109A agonist was GSK-256073; however, neither the chemical structure nor its clinical data were published. Incyte’s clinical candidate INCB-19602 reduced FFA in a phase I clinical trial but subsequent phase II trial in diabetic patients was terminated (ClinicalTrials.gov identifier: NCT00698789). Merck, Arena, and Schering-Plough have also developed a couple of interesting GPR109A agonists. MK-0354 was developed as a partial GPR109A agonist [70, 97]. Phase I pharmacodynamic studies revealed potent FFA effect with MK-0354 compared to extended release niacin. But in a phase II lipid efficacy study, no clinically meaningful changes of plasma LDL-c, HDL-c, and triglyceride in dyslipidaemic patients after MK-0354 treatment for 4 weeks [69]. This was initially thought to be a partial agonist effect of MK-0354 but two other recent clinical candidates MK-1903 and SCH900271, which are both full GPR109A agonists, were found to show similar effects.[29••] Given the dissociation revealed in GPR109A knockout mice between niacin’s GPR109A-mediated FFA effect and GPR109A-independent plasma lipoprotein effect, this lack of lipid efficacy from selective GPR109A agonists is perhaps unsurprising. Nevertheless, with modern molecular imaging techniques [98], some of these advanced candidates may be reinvestigated in selective patient cohorts, especially in those with accelerated vascular inflammation [99].
Conclusion
The pathophysiological and pharmacological roles of GPR109A still remain unclear. There is firm in vitro and in vivo evidence in animals that GPR109A activation is anti-inflammatory; but with the latest data from clinical trials, clinicians are certainly doubtful in the “HDL hypothesis” with regards to the benefits of raising HDLs, either by niacin or by other agents such as CETP inhibitors, in the treatment of atherosclerosis. The presence of GPR109A in adipocytes and immune cells, both cell types contribute to the inflammatory states in obesity, which is regulated by a systemic metabolites 3-HB synthesised by the liver over a large dynamic range, suggests a potential role in the “cross-talk” between metabolic and inflammatory pathways. By discovering the downstream signalling pathways of GPR109A, one may unlock the true potential of GPR109A activation not just in atherosclerosis and vascular inflammation, but also in other systemic inflammatory processes and metabolic diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Offermanns S, Colletti SL, Lovenberg TW, Semple G, Wise A, Ijzerman AP. International union of basic and clinical pharmacology. Lxxxii: nomenclature and classification of hydroxy-carboxylic acid receptors (gpr81, gpr109a, and gpr109b). Pharmacol Rev. 2011;63:269–90. This review comprehensively discussed the discovery of HCA family of receptors, their putative biological roles, as well as development of pharmaceutical agents to target GPR109A, GPR109B, and GPR81.
Altschul R, Hoffer A, Stephen JD. Influence of nicotinic acid on serum cholesterol in man. Arch Biochem Biophys. 1955;54:558–9.
Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, et al. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303:364–9.
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. Puma-g and hm74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–5.
Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, et al. Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem. 2003;278:9869–74.
Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (d)-beta-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor puma-g. J Biol Chem. 2005;280:26649–52.
Cai TQ, Ren N, Jin L, Cheng K, Kash S, Chen R, et al. Role of gpr81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun. 2008;377:987–91.
Ahmed K, Tunaru S, Langhans CD, Hanson J, Michalski CW, Kolker S, et al. Deorphanization of gpr109b as a receptor for the beta-oxidation intermediate 3-oh-octanoic acid and its role in the regulation of lipolysis. J Biol Chem. 2009;284:21928–33.
LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35.
Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504.
Pedersen TR, Faergeman O, Kastelein JJ, Olsson AG, Tikkanen MJ, Holme I, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the ideal study: a randomized controlled trial. JAMA. 2005;294:2437–45.
Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective american studies. Circulation. 1989;79:8–15.
Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–9.
Yvan-Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, et al. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–8.
Rosenson RS, Brewer Jr HB, Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–19.
Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2:309–15.
Shalhoub J, Falck-Hansen MA, Davies AH, Monaco C. Innate immunity and monocyte-macrophage activation in atherosclerosis. J Inflamm. 2011;8:9.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74.
Hewing B, Fisher EA. Preclinical mouse models and methods for the discovery of the causes and treatments of atherosclerosis. Expert Opin Drug Discov. 2012;7:207–16.
Digby JE, McNeill E, Dyar OJ, Lam V, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in adipocytes demonstrated by suppression of fractalkine, rantes, and mcp-1 and upregulation of adiponectin. Atherosclerosis. 2010;209:89–95.
Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai JT, Wamil M, et al. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by gpr109a dependent mechanisms. Arterioscler Thromb Vasc Biol. 2012;32:669–76.
Kamanna VS, Kashyap ML. Mechanism of action of niacin on lipoprotein metabolism. Curr Atheroscler Rep. 2000;2:36–46.
Peterson MJ, Hillman CC, Ashmore J. Nicotinic acid: studies on the mechamism of its antilipolytic action. Mol Pharmacol. 1968;4:1–9.
Yousefi S, Cooper PR, Mueck B, Potter SL, Jarai G. Cdna representational difference analysis of human neutrophils stimulated by gm-csf. Biochem Biophys Res Commun. 2000;277:401–9.
Schaub A, Futterer A, Pfeffer K. Puma-g, an ifn-gamma-inducible gene in macrophages is a novel member of the seven transmembrane spanning receptor superfamily. Eur J Immunol. 2001;31:3714–25.
Hanson J, Gille A, Zwykiel S, Lukasova M, Clausen B, Ahmed K, et al. Nicotinic acid and monomethyl fumarate induced flushing involves gpr109a expressed by keratinocytes and cox-2 dependent prostanoid formation in mice. J Clin Investig. 2010;120:2910–9.
Zhang Y, Schmidt RJ, Foxworthy P, Emkey R, Oler JK, Large TH, et al. Niacin mediates lipolysis in adipose tissue through its g-protein coupled receptor hm74a. Biochem Biophys Res Commun. 2005;334:729–32.
Digby JE, Lee JM, Choudhury RP. Nicotinic acid and the prevention of coronary artery disease. Curr Opin Lipidol. 2009;20:321–6.
•• Lauring B, Taggart AKP, Tata JR, Dunbar R, Caro L, Cheng K, et al. Niacin lipid efficacy is independent of both the niacin receptor gpr109a and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115–148ra115. This fascinating study first used a genetic, humanised, mouse model to show that the effect of niacin on FFA lipolysis is dependent on GPR109A but its effect of plasma lipoprotein is not. The authors then went on to show that selective GPR109A agonists can inhibit FAA release but has no effect on plasma lipoprotein. To confirm applicability in human subjects, the authors conducted separate human trials to show that selective GPR109A agonists, while reducing plasma FAA, did not exhibit anticipated effect on plasma lipoprotein; hence refuting the previous “FFA hypothesis” regarding niacin’s mechanism of action.
Offermanns S. It ain’t over 'til the fat lady sings. Sci Transl Med. 2012;4:148fs130.
Jin FY, Kamanna VS, Kashyap ML. Niacin accelerates intracellular apob degradation by inhibiting triacylglycerol synthesis in human hepatoblastoma (hepg2) cells. Arterioscler Thromb Vasc Biol. 1999;19:1051–9.
Zhang LH, Kamanna VS, Zhang MC, Kashyap ML. Niacin inhibits surface expression of atp synthase beta chain in hepg2 cells: implications for raising hdl. J Lipid Res. 2008;49:1195–201.
Ganji SH, Tavintharan S, Zhu D, Xing Y, Kamanna VS, Kashyap ML. Niacin noncompetitively inhibits dgat2 but not dgat1 activity in hepg2 cells. J Lipid Res. 2004;45:1835–45.
Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, et al. Identification of a gene encoding an acyl coa:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A. 1998;95:13018–23.
Chung S, Sawyer JK, Gebre AK, Maeda N, Parks JS. Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo. Circulation. 2011;124(15):1663–72.
Wu Z-H, Zhao S-P. Niacin promotes cholesterol efflux through stimulation of the pparγ-lxrα-abca1 pathway in 3t3-l1 adipocytes. Pharmacology. 2009;84:282–7.
Knowles H, Poole R, Workman P, Harris A. Niacin induces pparγ expression and transcriptional activation in macrophages via hm74 and hm74a-mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol. 2006;71:646–56.
Navab M, Imes SS, Hama SY, Hough GP, Ross LA, Bork RW, et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88:2039–46.
Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:1987–94.
Calabresi L, Franceschini G, Sirtori CR, De Palma A, Saresella M, Ferrante P, et al. Inhibition of vcam-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun. 1997;238:61–5.
Park SH, Park JH, Kang JS, Kang YH. Involvement of transcription factors in plasma hdl protection against tnf-alpha-induced vascular cell adhesion molecule-1 expression. Int J Biochem Cell Biol. 2003;35:168–82.
Xia P, Vadas MA, Rye KA, Barter PJ, Gamble JR. High density lipoproteins (hdl) interrupt the sphingosine kinase signaling pathway. A possible mechanism for protection against atherosclerosis by hdl. J Biol Chem. 1999;274:33143–7.
Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of hdl. Circ Res. 2004;95:764–72.
Cockerill GW, Huehns TY, Weerasinghe A, Stocker C, Lerch PG, Miller NE, et al. Elevation of plasma high-density lipoprotein concentration reduces interleukin-1-induced expression of e-selectin in an in vivo model of acute inflammation. Circulation. 2001;103:108–12.
Wu BJ, Yan L, Charlton F, Witting P, Barter PJ, Rye KA. Evidence that niacin inhibits acute vascular inflammation and improves endothelial dysfunction independent of changes in plasma lipids. Arterioscler Thromb Vasc Biol. 2010;30:968–75.
Bisoendial RJ, Hovingh GK, Levels JH, Lerch PG, Andresen I, Hayden MR, et al. Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation. 2003;107:2944–8.
Warnholtz A, Wild P, Ostad MA, Elsner V, Stieber F, Schinzel R, et al. Effects of oral niacin on endothelial dysfunction in patients with coronary artery disease: results of the randomized, double-blind, placebo-controlled inef study. Atherosclerosis. 2009;204:216–21.
•• Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor gpr109a expressed by immune cells. J Clin Investig. 2011;121:1163–73. This elegant study showed that niacin reduces progression of atherosclerosis in a mouse model independent of its lipoprotein effect. Importantly, this anti-atherosclerotic effect of niacin is dependent on the expression of GPR109A in myeloid cells and that bone-marrow transplantation of GPR109A-deficient myeloid cells abrogated the beneficial effect of niacin. This convincingly illustrated that GPR109A mediates the anti-inflammatory effect of niacin independent of its effect on plasma lipoprotein.
Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis. 2009;202:68–75.
Lukasova M, Hanson J, Tunaru S, Offermanns S. Nicotinic acid (niacin): New lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol Sci. 2011;32(12):700–7.
Digby JE, Martinez FO, Jefferson A, Ruparelia N, Wamil M, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotnic acid: Mechanisms of action in human monocytes. Circ Suppl. 2011;124:A14830.
Tavintharan S, Lim SC, Sum CF. Effects of niacin on cell adhesion and early atherogenesis: biochemical and functional findings in endothelial cells. Basic Clin Pharmacol Toxicol. 2009;104:206–10.
Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74:213–20.
Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ ccr2, ccr5, and cx3cr1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94.
Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–66.
Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter abca1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–43.
Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, et al. Macrophage abca1 reduces myd88-dependent toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51:3196–206.
Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, et al. Atp-binding cassette transporters and hdl suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–93.
Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–16.
Lee JMS, Robson MD, Yu L-M, Shirodaria CC, Cunnington C, Kylintireas I, et al. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular functiona randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol. 2009;54:1787–94.
Krupnick JG, Benovic JL. The role of receptor kinases and arrestins in g protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol. 1998;38:289–319.
Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Beta-arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–50.
Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–90.
• Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, et al. Β-arrestin1 mediates nicotinic acid–induced flushing, but not its antilipolytic effect, in mice. J Clin Investig. 2009;119:1312–21. This study beautifully dissected the role of G-protein versus β-arrestin signalling downstream of GPR109A activation by niacin. It demonstrated that while its therapeutic effect on FFA release may be mediated through the classic G-protein cascade, the side-effect of cutaneous flushing can be dissociated as they are mediated by an alternative signalling pathway via β-arrestins. The implication of this ‘biased agonism’ may be exploited in guiding future drug development.
Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a g protein-coupled receptor stimulated regulator of nf-kb pathways. Mol Cell. 2004;14:303–17.
Witherow DS. β-arrestin inhibits nf- b activity by means of its interaction with the nf- b inhibitor i b. Proc Natl Acad Sci. 2004;101:8603–7.
Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, et al. A nuclear function of β-arrestin1 in gpcr signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–47.
Richman JG, Kanemitsu-Parks M, Gaidarov I, Cameron JS, Griffin P, Zheng H, et al. Nicotinic acid receptor agonists differentially activate downstream effectors. J Biol Chem. 2007;282:18028–36.
Lai E, Waters MG, Tata JR, Radziszewski W, Perevozskaya I, Zheng W, et al. Effects of a niacin receptor partial agonist, mk-0354, on plasma free fatty acids, lipids, and cutaneous flushing in humans. J Clin Lipidol. 2008;2:375–83.
Semple G, Skinner PJ, Gharbaoui T, Shin YJ, Jung JK, Cherrier MC, et al. 3-(1h-Tetrazol-5-yl)-1,4,5,6-tetrahydro-cyclopentapyrazole (mk-0354): a partial agonist of the nicotinic acid receptor, g-protein coupled receptor 109a, with antilipolytic but no vasodilatory activity in mice. J Med Chem. 2008;51:5101–8.
Senior B, Loridan L. Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature. 1968;219:83–4.
Offermanns S. The nicotinic acid receptor gpr109a (hm74a or puma-g) as a new therapeutic target. Trends Pharmacol Sci. 2006;27:384–90.
Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov. 2012;11:603–19.
Wanders D, Graff EC, Judd RL. Effects of high fat diet on gpr109a and gpr81 gene expression. Biochem Biophys Res Commun. 2012;425:278–83.
Zimmerman MA, Singh N, Martin PM, Thangaraju M, Ganapathy V, Waller JL, et al. Butyrate suppresses colonic inflammation through hdac1-dependent fas upregulation and fas-mediated apoptosis of t cells. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1405–15.
Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. Gpr109a is a g-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69:2826–32.
Masuda R, Monahan JW, Kashiwaya Y. D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res. 2005;80:501–9.
Prins ML, Lee SM, Fujima LS, Hovda DA. Increased cerebral uptake and oxidation of exogenous betahb improves atp following traumatic brain injury in adult rats. J Neurochem. 2004;90:666–72.
Samoilova M, Weisspapir M, Abdelmalik P, Velumian AA, Carlen PL. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem. 2010;113:826–35.
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–107.
Cashin-Hemphill L, Mack WJ, Pogoda JM, Sanmarco ME, Azen SP, Blankenhorn DH. Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up. JAMA. 1990;264:3013–7.
Brown BG, Zhao XQ, Chait A, Fisher LD, Cheung MC, Morse JS, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–92.
Villines TC, Stanek EJ, Devine PJ, Turco M, Miller M, Weissman NJ, et al. The arbiter 6-halts trial (arterial biology for the investigation of the treatment effects of reducing cholesterol 6-hdl and ldl treatment strategies in atherosclerosis): Final results and the impact of medication adherence, dose, and treatment duration. J Am Coll Cardiol. 2010;55:2721–6.
Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial biology for the investigation of the treatment effects of reducing cholesterol (arbiter) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–7.
Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: Arbiter 3. Curr Med Res Opin. 2006;22:2243–50.
Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–22.
Digby JE, Ruparelia N, Choudhury RP. Niacin in cardiovascular disease: recent preclinical and clinical developments. Arterioscler Thromb Vasc Biol. 2012;32:582–8.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Lavigne PM, Karas R. The role of niacin in the aftermath of aim-high: a meta-analysis. J Am Coll Cardiol. 2012;59:E1687.
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, et al. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Nicholls SJ. The aim-high (atherothrombosis intervention in metabolic syndrome with low hdl/high triglycerides: impact on global health outcomes) trial: to believe or not to believe? J Am Coll Cardiol. 2012;59:2065–7.
Rosenson RS. Clinical trials of hdl cholesterol-raising therapy: what have we learned about the hdl hypothesis from aim-high? Curr Atheroscler Rep. 2012;14:190–2.
Hps2-thrive: A randomized trial of the long-term clinical effects of raising hdl cholesterol with extended release niacin/laropiprant. Http://www.Thrivestudy.Org.
Merck news release “merck announces hps2-thrive study of tredaptive (extended release niacin/laropiprant) did not achieve primary endpoint” released 20th december 2012.
European medicines agency confirmes recommendation to suspend tredaptive, pelzont and trevaclyn. Press release 18th january 2013. European medicines agency.
Shen HC, Colletti SL. Novel patent publications on high-affinity nicotinic acid receptor agonists. Expert Opin Ther Pat. 2009;19:957–67.
Semple G, Fioravanti B, Pereira G, Calderon I, Uy J, Choi K, et al. Discovery of the first potent and orally efficacious agonist of the orphan g-protein coupled receptor 119. J Med Chem. 2008;51:5172–5.
Lindsay AC, Choudhury RP. Form to function: current and future roles for atherosclerosis imaging in drug development. Nat Rev Drug Discov. 2008;7:517–29.
Marnane M, Merwick A, Sheehan OC, Hannon N, Foran P, Grant T, et al. Carotid plaque inflammation on (18) f-fluorodeoxyglucose positron emission tomography predicts early stroke recurrence. Ann Neurol. 2012;71:709–18.
Li G, Shi Y, Huang H, Zhang Y, Wu K, Luo J, et al. Internalization of the human nicotinic acid receptor gpr109a is regulated by gi, grk2, and arrestin3. J Biol Chem. 2010;285:22605–18.
Prusty D, Park B-H, Davis KE, Farmer SR. Activation of mek/erk signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma and c/ebp alpha gene expression during differentiation of 3t3-l1 preadipocytes. J Biol Chem. 2002;277:46226–32.
Chawla A, Boisvert WA, Lee C-H, Laffitte BA, Barak Y, Joseph SB, et al. A pparg-lxr-abca1 pathway in macrophages in involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–71.
Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. Ppar-α and ppar-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the abca1 pathway. Nat Med. 2001;7:53–8.
Acknowledgments
RPC, JED, JTC acknowledge the support of the BHF Centre of Research Excellence, Oxford. RPC is a Wellcome Trust Senior Research Fellow in Clinical Science. JTC is a Medical Research Council Clinical Research Training Fellow. Our laboratory is supported by the Oxford Comprehensive Biomedical Research Centre NIHR funding scheme.
Conflicts of Interest
Joshua T Chai has had travel/accommodations expenses covered or reimbursed by Roche.
Janet E Digby declares that she has no conflicts of interest.
Robin Choudhury has board membership with Astra Zeneca, GlaxoSmithKline, Merck/MSD, and Roche, is a consultant to AstraZeneca, GlaxoSmithKline and Merck/MSD, has grants/grants pending with Novartis, AstraZeneca, GlaxoSmithKline, Merck/MSD, and Roche, received honoraria from Astra Zeneca, and has had travel/accommodations expenses covered or reimbursed by Astra Zeneca, Merck/MSD and Roche.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Vascular Biology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Chai, J.T., Digby, J.E. & Choudhury, R.P. GPR109A and Vascular Inflammation. Curr Atheroscler Rep 15, 325 (2013). https://doi.org/10.1007/s11883-013-0325-9
Published:
DOI: https://doi.org/10.1007/s11883-013-0325-9