
Abstract
Fenretinide induces apoptosis in neuroblastoma by induction of reactive oxygen species (ROS). In this study, we investigated the role of mitochondria in fenretinide-induced cytotoxicity and ROS production in six neuroblastoma cell lines. ROS induction by fenretinide was of mitochondrial origin, demonstrated by detection of superoxide with MitoSOX, the scavenging effect of the mitochondrial antioxidant MitoQ and reduced ROS production in cells without a functional mitochondrial respiratory chain (Rho zero cells). In digitonin-permeabilized cells, a fenretinide concentration-dependent decrease in ATP synthesis and substrate oxidation was observed, reflecting inhibition of the mitochondrial respiratory chain. However, inhibition of the mitochondrial respiratory chain was not required for ROS production. Co-incubation of fenretinide with inhibitors of different complexes of the respiratory chain suggested that fenretinide-induced ROS production occurred via complex II. The cytotoxicity of fenretinide was exerted through the generation of mitochondrial ROS and, at higher concentrations, also through inhibition of the mitochondrial respiratory chain.
Similar content being viewed by others

Introduction
N-(4-hydroxyphenyl) retinamide (4HPR), also known as fenretinide, is a synthetic retinoic acid derivate that induces apoptosis in a variety of tumors [1–5]. 4HPR induces the production of reactive oxygen species (ROS), depolarization of the inner mitochondrial membrane (Δψm) and apoptosis in neuroblastoma monolayers and spheroids [6–8]. This ROS production was scavenged by Trolox, a cell-permeable and water-soluble vitamin E derivate, and this attenuated the loss of viability by 4HPR, suggesting that ROS plays a role in apoptosis induction by 4HPR in neuroblastoma [6]. The underlying mechanism of 4HPR-induced ROS production and mitochondrial depolarization, however, has still to be elucidated.
The major sources of ROS are the mitochondrial respiratory chain [9, 10], the radical-generating enzymes xanthine/xanthine oxidase [11] and NADPH oxidase [12], and the phospholipase A2-activated arachidonic acid metabolism [13]. The inability of inhibitors of xanthine/xanthine oxidase, NADPH oxidase and phospholipase A2 to suppress 4HPR-induced ROS generation excluded these radical-producing systems from being major sources of 4HPR-induced ROS in cervical cancer cells [14]. In contrast, several lines of evidence indicate that mitochondria play an important role in the cytotoxicity of 4HPR.
Inhibition of the mitochondrial respiratory chain reduces the Δψm, which facilitates the formation of the mitochondrial permeability transition pore. This permeabilization of the inner mitochondrial membrane induced by apoptotic agents is considered to be one of the mechanisms by which proapoptotic proteins are released from mitochondria [15, 16]. Oxidative stress-inducing agents have been shown to increase the permeability of the inner mitochondrial membrane and antioxidants effectively prevented mitochondrial membrane permeabilization, suggesting a relation between ROS production, mitochondrial membrane permeabilization and apoptosis [17–22]. Increase in the mitochondrial membrane permeability, release of cytochrome c and induction of the intrinsic apoptosis pathway have been shown to be key events in 4HPR-induced cell death in some cells [16, 23–25]. Nevertheless, it has been suggested that in neuroblastoma cells, lipoxygenase (12-LOX) was involved in 4HPR-induced ROS formation and that the ROS-inducing process did not take place in mitochondria [26].
In this study, we investigated the mechanism of 4HPR-induced ROS production in general and the effect of 4HPR on the electron flow through the mitochondrial respiratory chain in particular. The electron flow as a result of 4HPR-incubation was studied in six neuroblastoma cell lines by measuring the ATP production in situ in a flux model of digitonin-permeabilized neuroblastoma cells respiring on specific substrates for either complex I (NADH-Q reductase) or complex II (succinate-Q reductase). The data presented in this paper demonstrate the pivotal role of mitochondria in 4HPR-induced ROS production and the inhibition of mitochondrial respiration by 4HPR.
Materials and methods
Chemicals
MitoQ ([10-(4,5-dimethoxy-2-methyl-3,6-dioxo-cyclohexa-1,4-dienyl)-decyl]-triphenyl-phosphonium methnesulfonate) and TPP (decyltriphenylphosphonium bromide) were a kind gift from Antipodean Pharmaceuticals, Dunedin, New Zealand. CM-H2DCFDA (chloromethyldichlorofluorescein diacetate), JC-1 and TMRM were purchased from Invitrogen, Molecular Probes (Carlsbad, CA). All other chemicals were purchased from Sigma (St Louis, MO).
Cell culture
Three MYCN single copy neuroblastoma cell lines (FISK, NASS, SY5Y) and three MYCN amplified neuroblastoma cell lines (IMR32, SJ8, SJNB10) were cultured in RPMI-1640 culture medium supplemented with 10% heat inactivated fetal bovine serum, 50 U/ml penicillin/streptomycin, 4 mM glutamine (Gibco, Invitrogen, Carlsbad, CA) and plasmocin (5.0 mg/l) (InvivoGen, San Diego, CA). Cells were grown at 37° C in humidified air with 5% CO2; all culture flasks and plates were from Corning (Corning, NY). Cells were plated and allowed to adhere overnight before incubations were started. All neuroblastoma cell lines were a generous gift of Prof. R. Versteeg (Department of Human Genetics, Academical Medical Centre, Amsterdam, The Netherlands). Rho zero cells, cultured from 143b osteosarcoma cells, were a kind gift from Dr. L. Nijtmans (Nijmegen Centre for Mitochondrial disorders, Nijmegen, The Netherlands) and cultured on DMEM culture medium supplemented with 10% heat inactivated fetal bovine serum, 50 U/ml penicillin/streptomycin, 1 mM pyruvate, 4 mM glutamine and 50 μg/ml uridine under the conditions described above [27]. All experiments were performed in culture medium unless otherwise specified.
Mitochondrial ROS produced by 4HPR
ROS-production was measured in neuroblastoma and Rho zero cells as described before, using the fluorescent dye CM-H2DCFDA, which reacts with a wide range of radicals, after 2 h incubation with 4HPR (0–20 μM) (neuroblastoma cells and Rho zero cells) with or without MitoQ (1–8 μM) (neuroblastoma cells) [6, 28]. TPP was used as a negative control for MitoQ [29].
IMR32 and SJNB10 cells were incubated with 4HPR (10 μM) and/or the uncoupling agent Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (0–20 μM) for 4 h. Subsequently, ROS production was measured using the CM-H2DCFDA probe as described before [6].
MitoSOX selectively reacts with superoxide in the mitochondria and was used to measure specific mitochondrially generated ROS. All cell lines were incubated for 4 h with 4HPR (0–20 μM), after which medium was replaced by Hanks Buffered Salt Solution (HBSS) (Lonza, Walkersville, MD) containing 1 μM MitoSOX for 10 min. Subsequently, fluorescence was measured in a plate reader (BMG labtech, FLUOstar optima): excitation 510 nm, emission 580 nm. Cells (NASS, SJ8, SJNB10) were cultured and incubated 4 h with 20 μM 4HPR and/or 500 μM Trolox, after which the ROS production was measured with MitoSOX, as described above.
Mitochondrial respiratory chain
The ATP assay was performed essentially as described by Wanders et al. [30] (Fig. 1). Neuroblastoma cells were grown to confluency and incubated 4 h with 4HPR (0–30 μM). This incubation was repeated in the presence of either Trolox (500 μM) or CCCP (5–20 μM). After incubation, the cells were harvested and washed twice with PBS, followed by addition of the reaction medium containing 150 mM KCl, 25 mM Tris–HCl (pH 7.4), 2 mM EDTA, 10 mM potassium phosphate, 1 mM ADP, 0.1% (w/v) bovine serum albumin, 75 μg/ml digitonin and either 10 mM malate (plus 10 mM glutamate) or 10 mM succinate (plus 50 μM rotenone). Reactions were allowed to proceed for 30 min at room temperature, terminated with perchloric acid, protein was removed by centrifugation, and the acid protein-free supernatants were neutralized [30]. Subsequently, ATP was measured fluorimetrically as described previously using a Cobas Fara centrifugal analyzer (Roche, Mannheim, Germany) [31]. Afterwards, the samples were used for aspartate or malate measurements.

Schematic representation of the mitochondrial respiratory chain. After permeabilization of the cells with digitonin, the electron flow through the different complexes was measured. Complex I NADH reductase. Complex II succinate dehydrogenase. Complex III cytochrome c reductase. Complex IV cytochrome c oxidase. Complex V ATP synthase. MDH malate dehydrogenase. GOT glutamate-oxaloacetate transaminase
Aspartate was measured using the end-point method described in Bergmeyer et al. [32]. This method uses the subsequent conversion of aspartate to malate via oxaloacetate in a reaction mixture containing glutamate-oxaloacetate-transaminase, malate dehydrogenase, NADH and α-ketoglutarate. The decrease in NADH, as a consequence of the reduction of oxaloacetate to malate by malate dehydrogenase, was measured fluorimetrically with a Cobas Fara centrifugal analyzer. Quantification was performed by means of a standard curve.
Malate was determined fluorimetrically (excitation at 365 nm, emission at 450 nm) on a Cobas Fara centrifugal analyzer by the end-point measurement of the reduction of NAD + analogue 3-acetylpyridine adenine dinucleotide (APAD) to APADH by malate dehydrogenase. The reaction was performed in a 100 mM Tris–HCl buffer at pH 9 containing 0.1% Triton-X-100 and 1 mM APAD. Malate was quantified by means of a standard curve. Protein concentration was determined as described before, using the BCA assay [6].
Inhibiting mitochondrial respiratory chain complexes
Cells were incubated with 10 μM 4HPR in combination with rotenone (0–80 μM) (complex I inhibitor), Carboxin (0–2 mM), TTFA (0–2 mM) (thenoyltrifluoroacetone) (both complex II inhibitors) or antimycine A (0–40 μM) (complex III inhibitor) for 4 h. After incubation, both ROS generation and mitochondrial membrane potential were measured with CM-H2DCFDA and JC-1, respectively, as described before [6].
Measuring mitochondrial membrane potential
Cells were cultured and incubated with 4HPR (20 μM) for 4 h in combination with either Trolox (500 μM) or MitoQ (4 and 8 μM), after which the medium was replaced by medium containing 10 μg/ml JC-1 and mitochondrial membrane depolarization was measured [6]. In addition, the mitochondrial membrane potential was measured using 4 μM TMRM to confirm the JC-1 data. Fluorescence was measured with excitation 550 nm and emission 600 nm.
Measurement of cell viability
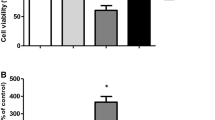
SJNB10 cells were incubated with different concentrations 4HPR (0–40 μM) and/or Trolox (500 μM). After 24 h, the viability of the cells was measured using MTS assay (Promega, Madison, WI) according to the manufacturer’s protocol. MTS incubation lasted for 4 h at 37°C. The experiments were performed in quadruplicate.
Results
ROS production using mitoSOX and the mitochondrial antioxidant MitoQ
Mitochondrial ROS production was measured using the mitoSOX probe, a ROS probe that specifically detects superoxide in mitochondria. A concentration dependent increase of mitochondrial ROS after 4 h 4HPR incubation was observed in all cell lines except FISK and SY5Y (Fig. 2a). MitoQ is a specific mitochondrial antioxidant that attaches to the mitochondrial membrane. To demonstrate that the ROS production induced by 4HPR incubation is indeed generated mitochondrially, cells were pre-incubated with MitoQ followed by co-incubation with 4HPR. 4HPR-induced ROS production, measured using the CM-H2DCFDA probe, was scavenged when cells were pre-incubated for 2 h with 1 μM MitoQ (Fig. 2b). The degree of protection against ROS varied considerably between the various cell lines. A moderate protective effect of MitoQ on ROS scavenging was observed in SJNB10 and SY5Y. However, increasing the concentration of MitoQ up to 8 μM resulted in a complete protection against 4HPR-induced ROS in SJNB10 cells and SY5Y cells (Fig. 3a). The protective effect of mitoQ in SY5Y cells was not observed when cells were incubated with mitoQ and 4HPR separately (Fig. 3b), indicating a low retention of mitoQ in SY5Y cells. The apparent inability of mitoSOX to detect superoxide in mitochondria in SY5Y cells might be caused by low accumulation or retention of mitoSOX in mitochondria in SY5Y cells, as observed for mitoQ. Thus, our data suggest that ROS production induced by 4HPR is formed mitochondrially in our panel of neuroblastoma cells.

Mitochondrial ROS generation after 4HPR incubation. a Mitochondrial superoxide production measured by mitoSOX after 4 h incubation with 0 μM (black bars), 10 μM (grey bars) or 20 μM (white bars) 4HPR. b The effect of the specific mitochondrial probe mitoQ against ROS in cell lines incubated for 2 h with (open square) or without (black diamond) 1 μM mitoQ followed by co-incubation for 2 h with 0–20 μM 4HPR. ROS was measured using the DFCDA probe. Each figure represents the mean ± SD of three experiments

Scavenging of mitochondrial ROS by mitoQ. a SJNB10 and SY5Y cells were incubated for 2 h with MitoQ followed by co-incubation for 2 h with 20 μM 4HPR. Control black bars; 4HPR dark grey bars; 4HPR and MitoQ (4 μM) light grey bars; 4HPR and MitoQ (8 μM) white bars. b SJNB10 and SY5Y cells were pre-incubated for 2 h with MitoQ followed by culturing in mitoQ-free medium for 1 h and subsequently incubation with 20 μM 4HPR for 2 h. Control black bars; 4HPR dark grey bars; 4HPR and MitoQ (4 μM) light grey bars; 4HPR and MitoQ (8 μM) white bars. Each bar represents the mean ± SD of three experiments
ROS-production studied in Rho zero cells
In Rho zero cells, cultured from osteosarcoma cells, the 4HPR-induced ROS production was measured to investigate whether ROS production is due to a specific effect of 4HPR on the respiratory chain. After 4 h incubation with 5 μM 4HPR, hardly any ROS production was observed in Rho zero cells when compared to control osteosarcoma cells (Fig. 4). In addition, 4HPR-induced ROS production, measured using the CM-H2DCFDA probe, was scavenged when the control osteosarcoma cells were pre-incubated for 2 h with 1 μM MitoQ followed by co-incubation with 10 μM 4HPR. Thus, a functional mitochondrial respiratory chain is required for 4HPR-induced ROS production.

ROS production in Rho zero cells (open square) and control cells (black diamond) measured after 4 h incubation with 0–20 μM 4HPR. ROS was measured using the DFCDA probe. The amount of ROS was expressed relative to that in untreated cells. Each figure represents the mean ± SD of three experiments
Inhibition of the mitochondrial respiratory chain complexes
To investigate which complexes of the mitochondrial respiratory chain are involved in 4HPR-induced ROS production, we studied the effect of 4HPR on the electron flux through the respiratory chain in digitonin-permeabilized cells (Fig. 1).
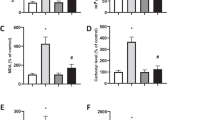
The effect on aspartate formation and ATP synthesis was measured using malate as a substrate for the mitochondrial respiratory chain in combination with glutamate. The effect of 4HPR on the formation of aspartate, which is the end-product of malate oxidation when malate oxidation is studied in the presence of glutamate (Fig. 1), and ATP synthesis was investigated. 4HPR inhibited aspartate production in a similar concentration-dependent manner as observed for the ATP production in all six cell lines (Fig. 5a).

The effect of 4HPR on ATP synthesis and aspartate formation (complex I) and ATP synthesis and malate formation (complex II) in digitonin-permeabilized neuroblastoma cells. All cell lines were treated with 0–30 μM 4HPR for 4 h. To measure the flux through complex I–V and II–V, malate (plus glutamate) and succinate (plus rotenone) were used as substrates, respectively, and the products a Aspartate (black diamond)-ATP (open square) and b malate (black square)-ATP (open square) were measured. Each figure represents the mean ± SD of three experiments
In order to demonstrate that the inhibition of 4HPR was not restricted to complex I-mediated ATP synthesis, we subsequently determined the effect of 4HPR on ATP synthesis when the complex II substrate succinate was used as electron donor for the mitochondrial respiratory chain (Fig. 1). Rotenone was included in the reaction medium to inhibit the electron flow through complex I. In all cell lines a concentration-dependent decrease of ATP and malate synthesis was observed as a consequence of 4HPR incubation (Fig. 5b), although in FISK cells a less potent decrease of malate compared to that of ATP was observed.
To investigate whether the decreased flux through the mitochondrial respiratory chain combined with the decreased ATP production was due to complex V (ATP synthase) inhibition by 4HPR, the cells were incubated with the uncoupling agent CCCP. ATP production followed by CCCP incubation was decreased to <20% (Fig. 6). As expected, aspartate and malate production was not decreased when incubated with only CCCP. In the presence of CCCP, a decrease was still observed in malate and aspartate production after 4HPR incubation (Fig. 6). Therefore, the decreased flux through the mitochondrial respiratory chain combined with the decreased ATP production was not due to complex V (ATP synthase) inhibition by 4HPR.

The effect of 4HPR on ATP and aspartate, and ATP and malate synthesis in digitonin-permeabilized SY5Y and SJNB10 cells incubated for 4 h with the uncoupling agent CCCP (5 μM for malate–ATP, 15 μM for aspartate–ATP in SJNB10, 20 μM for aspartate–ATP in SY5Y) and 0–30 μM 4HPR. a Aspartate (black triangle) –ATP(open square) and b malate (black triangle)–ATP (open square). Each figure represents the mean ± SD of three experiments
Surprisingly, the 4HPR-induced inhibition of the mitochondrial respiratory chain also occurred in FISK cells, which hardly produced any mitochondrial ROS following 4HPR treatment. However, the 4HPR concentrations required for inhibition of the electron flux were much higher than the optimal 4HPR concentrations inducing mitochondrial ROS production, which were 10–30 and 5–10 μM, respectively. These results demonstrate that 4HPR inhibited the electron flux through the mitochondrial respiratory chain between Co-enzyme Q and complex IV and that this inhibition was not required for the mitochondrial ROS production.
ROS scavenging and the effects on the mitochondrial respiratory chain
Co-incubation with 4HPR and CCCP did not result in a decreased ROS production (data not shown). Thus, uncoupling the mitochondrial membrane did not prevent ROS production, which indicates that 4HPR-induced ROS production is not the result of inhibition of complex V of the mitochondrial respiratory chain. Trolox is an antioxidant that scavenged the 4HPR-induced ROS in neuroblastoma [6]. The decrease of the mitochondrial membrane potential induced by 4HPR could not be prevented by Trolox or MitoQ (data not shown) [6]. The activity of the mitochondrial respiratory chain in cells incubated with the combination of 4HPR and Trolox was investigated, and Trolox did not prevent the decrease of aspartate-ATP and malate-ATP (data not shown). In contrast, in cells incubated with low concentrations 4HPR (0–10 μM), the viability loss is attenuated by Trolox. However, when cells were incubated with higher 4HPR concentrations (20–40 μM), the loss of viability could not be fully prevented by Trolox (Fig. 7). These data suggest that the cytotoxicity of 4HPR is exerted through the generation of ROS at low concentrations of 4HPR, whereas inhibition of the mitochondrial respiratory chain might play a role at high concentrations of 4HPR.

Effect of Trolox on loss of viability in 4HPR-treated cells. SJNB10 cells were incubated with 0–40 μM 4HPR with (grey bars) or without (black bars) 500 μM Trolox for 24 h. Viability was measured using the MTS method and was depicted as percentage of control. Each figure represents the mean ± SD of four experiments
Specific inhibition of the mitochondrial respiratory chain complexes
To investigate the role of each complex in the 4HPR-induced ROS production, the neuroblastoma cells were incubated with specific inhibitors of complex I, II and III. Carboxin and TTFA, two inhibitors of complex II, were the only mitochondrial respiratory chain complex inhibitors that reduced the 4HPR-induced ROS production (Fig. 8). Rotenone and antimycin A, inhibitors of complex I and complex III (cytochrome reductase), respectively, did not reduce the ROS production (data not shown). A similar protective effect of the complex II inhibitor TTFA on 4HPR-induced ROS formation was observed in other neuroblastoma cell lines such as IMR32, SJNB10, NASS, SY5Y and FISK (as well as in the osteosarcoma control cells). This result is in line with the observation that mitochondrial ROS production and inhibition of the electron flux through the mitochondrial respiratory chain are two independent mechanisms of 4HPR.

The effect of complex II inhibitors on ROS production in SJNB10 cells. Cells were incubated with (grey bars) or without (black bars) 10 μM 4HPR for 4 h. a In combination with 0–2 μM TTFA. b In combination with 0–2 mM Carboxin. After incubation, ROS production was measured using CM-H2DCFDA probe. Percentage of ROS induction compared to control. Each figure represents the mean ± SD of three experiments
Discussion
In this paper, we have performed an in depth analysis of the role of mitochondria in 4HPR-induced ROS formation. Using the specific mitochondrial ROS-probe, MitoSOX, we showed that 4HPR-induced ROS is of mitochondrial origin in our panel of neuroblastoma cell lines. As MitoSOX detects primarily superoxide, our results are in apparent contrast with those of others who suggested that hydrogen peroxide is produced as a result of 4HPR incubation [14, 24]. The pivotal role of mitochondria in 4HPR-induced ROS formation was further strengthened by the scavenging effect of MitoQ, a specific mitochondrial antioxidant. Furthermore, the fact that Rho zero osteosarcoma cells were unable to produce as much ROS as their control cells indicated that for ROS production to occur a functional mitochondrial respiratory chain is required.
Our finding that ROS production is reduced by the complex II inhibitors Carboxin and TTFA suggests that complex II is the major source of 4HPR-induced ROS formation in neuroblastoma. It should be noted that the difference between the efficacy of the complex I and II inhibitors in ROS formation might be due to the fact that electrons channelled through complex II produce about four times more superoxide than those channelled through complex I [33, 34]. Suzuki et al. [14] suggested that 4HPR activated ROS generation in mitochondria at the Co-enzyme Q site, complex III or both in cervical cancer. In leukemia cells 4HPR-induced ROS production has been suggested to occur between complex I and II [35]. Our results demonstrate a major role for complex II in the generation of ROS.
Our results are in apparent contrast with those of Lovat et al. [36, 37], who stated that lipoxygenase (12-LOX) was the mediator of 4HPR-induced ROS in SY5Y neuroblastoma cells and that the ROS-inducing process did not take place in the mitochondria. Inhibitors of 12-LOX and phospholipase A2 blocked 4HPR-induced apoptosis. However, in cervical cells inhibition of phospholipase A2 was unable to suppress 4HPR-induced ROS [14].
Analysis of the electron flux through the mitochondrial respiratory chain in situ, using digitonin-permeabilized neuroblastoma cells, showed a strong concentration-dependent decrease in ATP synthesis in mitochondria selectively respiring on either the complex I substrate malate (plus glutamate) or the complex II substrate succinate (plus rotenone). Parallel measurement of either the complex I-mediated production of aspartate or the complex II-mediated production of malate showed a decrease in synthesis of both products. Combining 4HPR with the uncoupling agent CCCP demonstrated that the inhibiting effect did not take place in complex V, as the inhibition in aspartate and malate oxidation by 4HPR is not alleviated by CCCP. These data suggest that 4HPR inhibits the mitochondrial respiratory chain between Co-enzyme Q and complex IV (cytochrome oxidase). Previously, the effect of 4HPR-induced ROS generation on the mitochondrial respiratory chain has been examined to some extent in cervical cancer cells and leukemia cells, and the main target site of 4HPR was located ‘downstream’ of mitochondrial respiratory chain complexes I and II and ‘upstream’ of complexes III and IV, possibly at the coenzyme Q site of the mitochondrial respiratory chain [14, 35].
No scavenging effect of CCCP on 4HPR-induced ROS production was observed in our study. This is in contrast with the fact that CCCP inhibits ROS formation induced by 4HPR in cervical cancer cells, which strongly supports the conclusion that 4HPR acts as a pro-oxidant via the mitochondrial respiratory chain [14]. The uncoupling proton ionophore CCCP can ‘short-circuit’ the proton current so that both the proton gradient and membrane potential across the inner mitochondrial membrane collapse. Consequently, no phosphorylation of ADP can take place [38].
Cytochrome c oxidase (Complex IV) subunit VI overexpression in glioblastoma cells led to decreased ROS production followed by decreased apoptosis by 4HPR. In addition, cytochrome c oxidase subunit III was down-regulated by 4HPR in hepatoma cells, which was accompanied by ROS production and apoptotic cell death [39, 40]. In this respect, it is worthwhile to note that the oxidation of cardiolipin by 4HPR has been suggested to occur, which would result in inhibition of complex IV of the mitochondrial respiratory chain [35]. An association between mitochondrial cardiolipin peroxidation and cytochrome c release has been established [10, 41], which would be in line with the 4HPR-induced cytochrome c release in cervical carcinoma cells, head and neck squamous carcinoma cells, leukemia cells and neuroblastoma cells [14, 16, 23, 35, 37].
Previously, it has been shown that in leukemia cells and cervical cancer cells, 4HPR decreased the mitochondrial membrane potential in an antioxidant-sensitive pathway, which indicated that 4HPR-induced ROS generation is required for mitochondrial membrane depolarization [14, 42]. However, in neuroblastoma cells, depolarization of the mitochondrial membrane potential was not attenuated by Trolox or by MitoQ, nor was the inhibition of 4HPR on the mitochondrial respiratory chain scavenged by Trolox. This indicates that in neuroblastoma cells ROS is not the cause of the inhibition of the mitochondrial respiratory chain and the mitochondrial depolarization. This is in contrast with Asumendi et al. [35] who suggested ROS production to be the cause of mitochondrial alterations in leukemia. Our results indicated that low concentrations of 4HPR (<10 μM) induced ROS-production at complex II of the mitochondrial respiratory chain in most cell lines; this effect and the loss of viability is scavenged by Trolox. High concentrations 4HPR (>20 μM) inhibited the mitochondrial respiratory chain, which is not scavenged by Trolox, nor is the loss of viability in cells treated with high concentrations 4HPR. In addition, the inhibition of the mitochondrial respiratory chain might result in the disappearance of the mitochondrial membrane potential.
In HeLa cells, 4HPR does not induce mitochondrial membrane permeabilization through a direct action of 4HPR on mitochondria or on the mitochondrial permeability transition pore complex. Thus, in HeLa cells the ability of 4HPR to generate ROS, mitochondrial depolarization, and permeabilization must involve mediators generated outside of mitochondria, such as ceramide, lipoxygenase and ROS [23].
In this paper, we showed that 4HPR-induced ROS production in neuroblastoma cells mainly takes place in the mitochondria, more precisely, at complex II of the mitochondrial respiratory chain. At low concentrations of 4HPR, the cytotoxicity of 4HPR was exerted through the generation of mitochondrial ROS, whereas high concentrations of 4HPR were associated with inhibition of the mitochondrial respiratory chain.
References
Delia D, Aiello A, Lombardi L, Pelicci PG, Grignani F, Grignani F, Formelli F, Menard S, Costa A, Veronesi U (1993) N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res 53:6036–6041
Hsieh TC, Ng C, Wu JM (1995) The synthetic retinoid N-(4-hydroxyphenyl) retinamide (4-HPR) exerts antiproliferative and apoptosis-inducing effects in the androgen-independent human prostatic JCA-1 cells. Biochem Mol Biol Int 37:499–506
Mariotti A, Marcora E, Bunone G, Costa A, Veronesi U, Pierotti MA, Della VG (1994) N-(4-hydroxyphenyl)retinamide: a potent inducer of apoptosis in human neuroblastoma cells. J Natl Cancer Inst 86:1245–1247
Oridate N, Lotan D, Mitchell MF, Hong WK, Lotan R (1995) Inhibition of proliferation and induction of apoptosis in cervical carcinoma cells by retinoids: implications for chemoprevention. J Cell Biochem Suppl 23:80–86
Pellegrini R, Mariotti A, Tagliabue E, Bressan R, Bunone G, Coradini D, Della VG, Formelli F, Cleris L, Radice P (1995) Modulation of markers associated with tumor aggressiveness in human breast cancer cell lines by N-(4-hydroxyphenyl) retinamide. Cell Growth Differ 6:863–869
Cuperus R, Tytgat GA, Leen R, Brites P, Bras J, Caron HN, van Kuilenburg AB (2008) Pleiotropic effects of fenretinide in neuroblastoma cell lines and multicellular tumor spheroids. Int J Oncol 32:1011–1019
Lovat PE, Ranalli M, Bernassola F, Tilby M, Malcolm AJ, Pearson AD, Piacentini M, Melino G, Redfern CP (2000) Synergistic induction of apoptosis of neuroblastoma by fenretinide or CD437 in combination with chemotherapeutic drugs. Int J Cancer 88:977–985
Lovat PE, Ranalli M, Annichiarrico-Petruzzelli M, Bernassola F, Piacentini M, Malcolm AJ, Pearson AD, Melino G, Redfern CP (2000) Effector mechanisms of fenretinide-induced apoptosis in neuroblastoma. Exp Cell Res 260:50–60
Richter C, Schweizer M (1997) Oxidative stress in mitochondria. In: Scandalios JG (ed) Oxidative stress and the molecular biology of antioxidant defenses. Cold Spring Harbor Laboratory Press, Plainview, pp 169–200
Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y (2000) Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 351:183–193
McCord JM (1985) Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312:159–163
Trudel S, Paquet MR, Grinstein S (1991) Mechanism of vanadate-induced activation of tyrosine phosphorylation and of the respiratory burst in HL60 cells. Role of reduced oxygen metabolites. Biochem J 276(Pt 3):611–619
Henderson LM, Chappell JB, Jones OT (1989) Superoxide generation is inhibited by phospholipase A2 inhibitors. Role for phospholipase A2 in the activation of the NADPH oxidase. Biochem J 264:249–255
Suzuki S, Higuchi M, Proske RJ, Oridate N, Hong WK, Lotan R (1999) Implication of mitochondria-derived reactive oxygen species, cytochrome C and caspase-3 in N-(4-hydroxyphenyl)retinamide-induced apoptosis in cervical carcinoma cells. Oncogene 18:6380–6387
Roberson KM, Penland SN, Padilla GM, Selvan RS, Kim CS, Fine RL, Robertson CN (1997) Fenretinide: induction of apoptosis and endogenous transforming growth factor beta in PC-3 prostate cancer cells. Cell Growth Differ 8:101–111
Kim HJ, Chakravarti N, Oridate N, Choe C, Claret FX, Lotan R (2006) N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of MAPKs in head and neck squamous carcinoma cells. Oncogene 25:2785–2794
Tiwari BS, Belenghi B, Levine A (2002) Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol 128:1271–1281
Higuchi M, Proske RJ, Yeh ET (1998) Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene 17:2515–2524
Hail N Jr, Youssef EM, Lotan R (2001) Evidence supporting a role for mitochondrial respiration in apoptosis induction by the synthetic retinoid CD437. Cancer Res 61:6698–6702
Halestrap AP, McStay GP, Clarke SJ (2002) The permeability transition pore complex: another view. Biochimie 84:153–166
Tafani M, Schneider TG, Pastorino JG, Farber JL (2000) Cytochrome c-dependent activation of caspase-3 by tumor necrosis factor requires induction of the mitochondrial permeability transition. Am J Pathol 156:2111–2121
Tiwari M, Kumar A, Sinha RA, Shrivastava A, Balapure AK, Sharma R, Bajpai VK, Mitra K, Babu S, Godbole MM (2006) Mechanism of 4-HPR-induced apoptosis in glioma cells: evidences suggesting role of mitochondrial-mediated pathway and endoplasmic reticulum stress. Carcinogenesis 27:2047–2058
Boya P, Morales MC, Gonzalez-Polo RA, Andreau K, Gourdier I, Perfettini JL, Larochette N, Deniaud A, Baran-Marszak F, Fagard R, Feuillard J, Asumendi A, Raphael M, Pau B, Brenner C, Kroemer G (2003) The chemopreventive agent N-(4-hydroxyphenyl)retinamide induces apoptosis through a mitochondrial pathway regulated by proteins from the Bcl-2 family. Oncogene 22:6220–6230
Hail N Jr, Lotan R (2001) Mitochondrial respiration is uniquely associated with the prooxidant and apoptotic effects of N-(4-hydroxyphenyl)retinamide. J Biol Chem 276:45614–45621
Poot M, Hosier S, Swisshelm K (2002) Distinct patterns of mitochondrial changes precede induction of apoptosis by all-trans-retinoic acid and N-(4-hydroxyphenyl)retinamide in MCF7 breast cancer cells. Exp Cell Res 279:128–140
Lovat PE, Oliverio S, Ranalli M, Corazzari M, Rodolfo C, Bernassola F, Aughton K, Maccarrone M, Hewson QD, Pearson AD, Melino G, Piacentini M, Redfern CP (2002) GADD153 and 12-lipoxygenase mediate fenretinide-induced apoptosis of neuroblastoma. Cancer Res 62:5158–5167
Appleby RD, Porteous WK, Hughes G, James AM, Shannon D, Wei YH, Murphy MP (1999) Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur J Biochem 262:108–116
James AM, Cocheme HM, Murphy MP (2005) Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech Ageing Dev 126:982–986
Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RA, Murphy MP (2008) Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J 411:633–645
Wanders RJ, Ruiter JP, Wijburg FA (1993) Studies on mitochondrial oxidative phosphorylation in permeabilized human skin fibroblasts: application to mitochondrial encephalomyopathies. Biochim Biophys Acta 1181:219–222
Williamson JR, Corkey BE (1969) Assays of intermediates of the citric acid cycle and related compounds by fluorometric enzyme methods. Methods Enzymol 13:434–513
Bergmeyer HU, Bernt E, Möllering H, Pfeiderer G (1974) l-aspartate and l-asparagine. In: Bergmeyer HU (ed) Methods of enzymatic analysis. Academic Press, New York, pp 1696–1700
Forman HJ, Boveris A (1982) Free radicals in biology. Academic Press, New York
Higuchi M, Honda T, Proske RJ, Yeh ET (1998) Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene 17:2753–2760
Asumendi A, Morales MC, Alvarez A, Arechaga J, Perez-Yarza G (2002) Implication of mitochondria-derived ROS and cardiolipin peroxidation in N-(4-hydroxyphenyl)retinamide-induced apoptosis. Br J Cancer 86:1951–1956
Lovat PE, Corazzari M, Di SF, Piacentini M, Redfern CP (2005) The role of gangliosides in fenretinide-induced apoptosis of neuroblastoma. Cancer Lett 228:105–110
Lovat PE, Corazzari M, Goranov B, Piacentini M, Redfern CP (2004) Molecular mechanisms of fenretinide-induced apoptosis of neuroblastoma cells. Ann N Y Acad Sci 1028:81–89
Skulachev VP (1996) Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys 29:169–202
You KR, Wen J, Lee ST, Kim DG (2002) Cytochrome c oxidase subunit III: a molecular marker for N-(4-hydroxyphenyl)retinamise-induced oxidative stress in hepatoma cells. J Biol Chem 277:3870–3877
Eun SY, Woo IS, Jang HS, Jin H, Kim MY, Kim HJ, Lee JH, Chang KC, Kim JH, Seo HG (2008) Identification of cytochrome c oxidase subunit 6A1 as a suppressor of Bax-induced cell death by yeast-based functional screening. Biochem Biophys Res Commun 373:58–63
Nakagawa Y (2004) Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann N Y Acad Sci 1011:177–184
Morales MC, Perez-Yarza G, Rementeria NN, Boyano MD, Apraiz A, Gomez-Munoz A, Perez-Andres E, Asumendi A (2007) 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free Radic Res 41:591–601
Acknowledgments
This work was supported by a grant from the Stichting Kindergeneeskundig Kankeronderzoek (SKK) of The Netherlands. We would like to thank Jos Ruiter for his help on the respiratory chain studies. The mitoQ was kindly provided by Antipodean Pharmaceuticals, Dunedin, New Zealand.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cuperus, R., Leen, R., Tytgat, G.A.M. et al. Fenretinide induces mitochondrial ROS and inhibits the mitochondrial respiratory chain in neuroblastoma. Cell. Mol. Life Sci. 67, 807–816 (2010). https://doi.org/10.1007/s00018-009-0212-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0212-2