


Abstract
Chromosomal instability (CIN) is characterized by an increased frequency of changes in chromosome structure or number and is regarded as a hallmark of cancer. CIN plays a prevalent role in tumorigenesis and cancer progression by assisting the cancer cells’ phenotypic adaptation to stress, which have been tightly linked to therapy resistance and metastasis. Both CIN-inducing and CIN-repressing agents are being clinically tested for the treatment of cancer to increase CIN levels to unsustainable levels leading to cell death or to decrease CIN levels to limit the development of drug resistance, respectively. Non-coding RNAs (ncRNAs) including microRNAs and long ncRNAs (lncRNAs) have been fundamentally implicated in CIN. The miR-22, miR-26a, miR-28, and miR-186 target important checkpoint proteins involved in mediating chromosomal stability and their expression modulation has been directly related to CIN occurrence. lncRNAs derived from telomeric, centrosomal, and enhancer regions play an important role in mediating genome stability, while specific lncRNA transcripts including genomic instability inducing RNA called Ginir, P53-responsive lncRNA termed as GUARDIN, colon cancer-associated transcript 2, PCAT2, and ncRNA activated by DNA damage called NORAD have been shown to act within CIN-associated pathways. In this review, we discuss how these ncRNAs either maintain or disrupt the stability of chromosomes and how these mechanisms could be exploited for novel therapeutic approaches targeting CIN in cancer patients.
SIGNIFICANCE STATEMENT Chromosomal instability increases tumor heterogeneity and thereby assists the phenotypic adaptation of cancer cells, causing therapy resistance and metastasis. Several microRNAs and long non-coding RNAs that have been causally linked to chromosomal instability could represent novel therapeutic targets. Understanding the role of non-coding RNAs in regulating different genes involved in driving chromosomal instability will give insights into how non-coding RNAs can be utilized toward modifying chemotherapeutic regimens in different cancers.
Introduction
Non-coding RNAs (ncRNAs) play diverse transcriptional and post-transcriptional regulatory roles across both homeostatic cellular functions and disease states (Cech and Steitz, 2014; Slack and Chinnaiyan, 2019). It is now known that approximately 70% of the human genome is transcribed across different cell types (Djebali et al., 2012) and current annotations describe almost 18,000 lncRNAs loci that produce nearly 50,000 transcripts (Frankish et al., 2021). In addition, the latest miRbase v22 release describes 1,917 hairpin precursors producing over 2,500 mature human microRNAs (miRNAs) (Kozomara et al., 2019). While lncRNAs have been shown to perform diverse transcriptional as well as post-transcriptional gene regulatory functions (reviewed in Kopp and Mendell, 2018; Mohapatra et al., 2021; Statello et al., 2021) the function of miRNAs is more defined, specifically interacting with complementary mRNAs to downregulate their expression via degradation or inhibition of translation (Lim et al., 2005). The prevalent involvement of both, lncRNAs and miRNAs in cancer cell biology has been established (Calin et al., 2002), and miRNA-based therapeutics are actively tested in phase I and II clinical trials (Winkle et al., 2021). In this review, we summarize current knowledge on the causative roles of lncRNAs and miRNAs in chromosomal instability (CIN) in cancer. Here, we specifically make a distinction between genomic instability (i.e., the acquirement of genomic mutations during the replicative cell cycle) and CIN.
CIN is a subtype of genomic instability characterized by changes in chromosome structure and/or count that arise due to chromosome segregation defects, replication stress, defects in the DNA damage response, or telomere dysfunction (Gollin 2004, Burrell et al., 2013; Wilhelm, Said et al., 2020). A further subdivision is made according to the presence of structural (sCIN) or numerical (nCIN) changes (Wilhelm et al., 2020). sCIN are premitotic defects arising during interphase (e.g., due to replication stress, defective DNA damage response, or telomere dysfunction) and are represented by partial deletions, translocations, rearrangements, amplifications, and other aberrations (e.g., dicentric or ring chromosomes). nCIN arise due to chromosomal segregation defects during mitosis because of impairments in regulatory structural components (e.g., centromeres, kinetochores, microtubule, spindle assembly checkpoint) and result in variations of chromosome number (ploidy). There is further distinction to be made between nCIN and aneuploidy: while nCIN is an ongoing process stemming from defects in chromosome segregation that generally leads to aneuploidy, stable aneuploidy can also exist in the absence of nCIN (Schukken and Foijer, 2018).
The Role of CIN in Cancer
The vast majority of human tumors show both aneuploidy and chromosomal abnormalities indicative of CIN (Schukken and Foijer, 2018). Aneuploidy in cancer cells is often associated with worse patient survival as the abnormal karyotypes are generally not random but specifically result in gains of oncogenes and losses of tumor suppressor genes thus increasing cancer cell fitness (Nicholson and Cimini, 2013). The role of CIN in cancer cells is more paradoxical, with detrimental effects on tumor fitness in some models (Funk et al., 2016; Zasadil et al., 2016), while leading to tumorigenesis and correlating with drug resistance and metastasis in other models (Schukken and Foijer, 2018). The rate of CIN may play a role conferring these differences as it has been described that a high degree of CIN can be detrimental to tumor cells and can potentially support chemotherapeutic treatment regimens (Funk et al., 2016). Conversely, persistent low-grade CIN contributes to tumorigenesis by increasing intratumoral heterogeneity, and the following clonal selection increases metastatic potential and drug resistance (Schukken and Foijer, 2018). In addition, sCIN can produce translocations creating oncogenic fusion genes that may play a pivotal role in early tumorigenesis (Mitelman et al., 2007). CIN has furthermore been directly linked to innate immune reactions through the release of double-stranded DNA in the cytosol. While this mechanism was linked to increased tumor invasion and metastasis (Bakhoum et al., 2018), defects in type I interferon and other immune pathways within tumor cells and/or their microenvironment are thought to lead to immune evasion of such CIN-introduced immunostimulatory effects (Bakhoum and Cantley, 2018, Tijhuis et al., 2019).
MicroRNAs Implicated in CIN
Several miRNAs have been functionally implicated in CIN through their regulation of specific target genes (i.e., checkpoint proteins regulating DNA repair and mitosis) that have a direct link to CIN (Fig. 1). Other miRNAs targeting important structural components such as the cohesin complex (involved in sister chromatid cohesion and the DNA damage response; reviewed in Kuru-Schors et al., 2021; Yamada et al., 2021) could also promote CIN although the direct causal relationship requires further study.
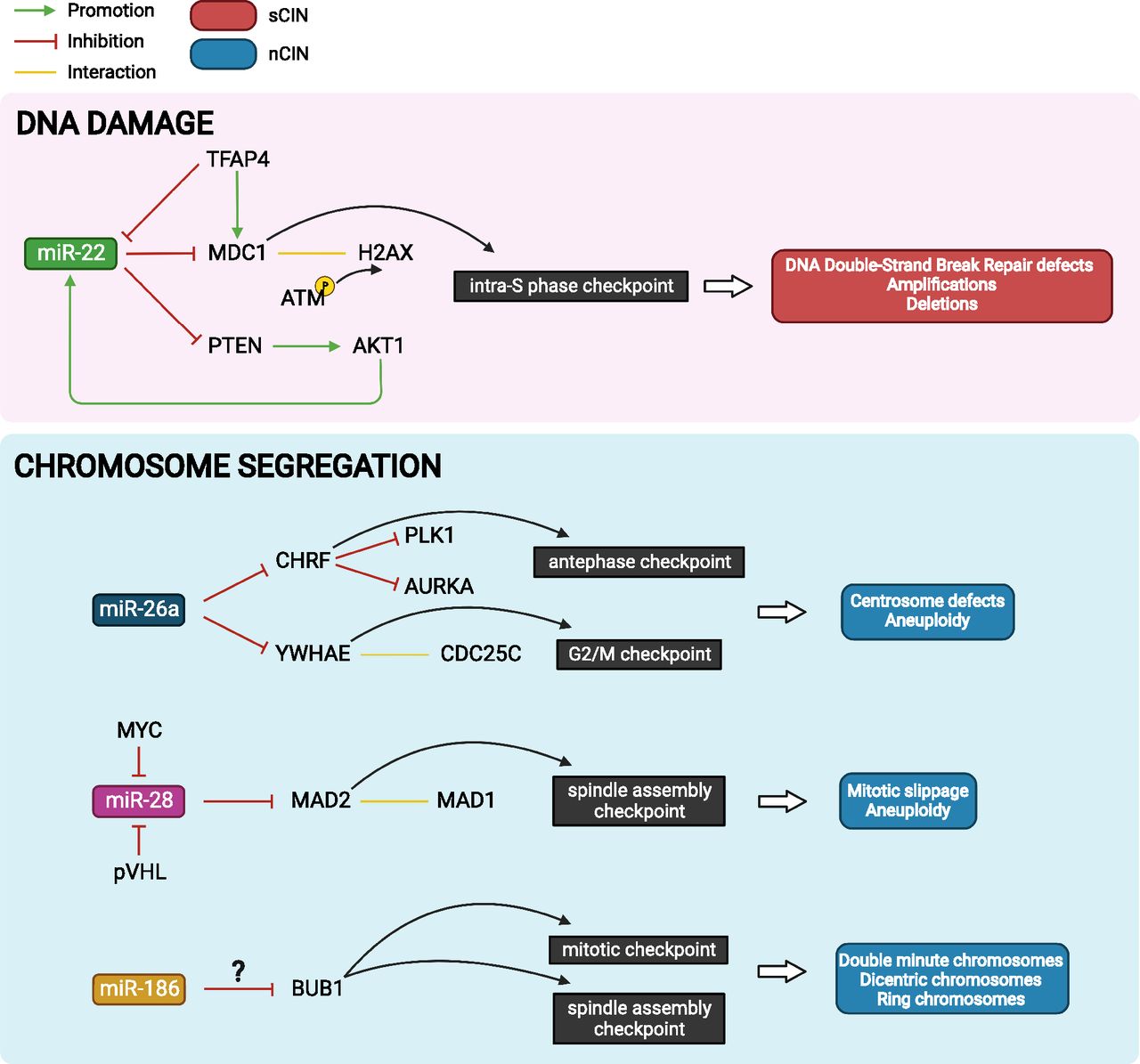
MicroRNAs implicated in the generation of CIN. miR-22 directly regulates MDC1 through HR-mediated DNA repair and contributes to CIN. miR-26a, miR-28 and miR-186 regulates genes involved in cell cycle checkpoints and contributes to centrosome defect, aneuploidy, double minute, dicentric, and ring chromosomes rendering CIN.
miR-22 directly binds and represses the levels of mediator of DNA damage checkpoint 1 (MDC1) in three different cell lines (HEK293T, HeLa, U2OS) (Lee et al., 2015). MDC1 is an intra-S phase checkpoint protein that functions early in the DNA damage response, interacting with H2A histone family member X at DNA double-strand breaks and recruiting ataxia-telangiectasia mutated kinase to mediate H2A histone family member X phosphorylation. Loss of MDC1 in mice confers CIN (i.e., chromosome and chromatid breaks, fragmentation, dicentric chromosomes) (Lou et al., 2006), and the tumor suppressor protein is frequently mutated, lost, or repressed in human cancers (Ruff et al., 2020). Multiple chromosomal abnormalities including recurring clonal amplifications and deletions were detected in miR-22 overexpressing GM00637 (fibroblast) cells. Furthermore, miR-22 overexpression increased the frequency of chromosome breaks in U2OS (osteosarcoma) cells, and this effect was fully rescued by reconstitution of MDC1 containing a mutated miR-22 binding site. In addition, it was shown that overexpression of AKT serine/threonine kinase 1 (AKT1), a positive upstream regulator of miR-22, reduced homologous recombination (HR) via miR-22–mediated suppression of MDC1 (Lee et al., 2015). Suppression of HR is known to be causative for aneuploidy and CIN (Griffin, 2002). Of note, miR-22 was shown to form a feed-forward loop with AKT1 via repression of phosphatase and tensin homolog, a negative regulator of AKT1 (Bar and Dikstein, 2010). The miR-22-MDC1 regulatory axis and its effect on CIN was further confirmed in colorectal cancer (CRC) cell lines. In this context, transcription factor AP-4 (TFAP4) was identified as a dual regulator of MDC1 expression via direct transcriptional induction of MDC1 and via negative regulation of miR-22, which targets MDC1. TFAP4-deficient cells accordingly showed a marked decrease in HR activity and increased micronuclei formation and gains in chromosome numbers, indicative of CIN. Treatment of TFAP4-deficient cells with miR-22 antagomirs (or ectopic MDC1) could restore HR-mediated DNA damage repair, while TFAP-4 proficient cells treated with miR-22 mimics (or MDC1 small interfering RNA) showed defective HR-mediated DNA damage repair (Chou et al., 2022). These studies show that miR-22 directly regulates MDC1 in multiple cellular contexts, and this strongly affects HR-mediated DNA repair and, consequently, causes CIN.
Another example is miR-26a that is ubiquitously expressed from three distinct genomic loci as miR-26a-1, miR-26a-2, and miR-26b from chromosome 3, 12, and 2, respectively. miR-26a has been described as both oncomiR and tumor suppressor miRNA depending on the cancer type and target gene repertoire involved (e.g., in proliferation, cell cycle regulation, apoptosis, and metabolism) (Rizzo et al., 2017; Li et al., 2021). A study applying sustained miR-26a overexpression in breast cancer cells and mouse embryonic fibroblasts reported the occurrence of aneuploidy as well as centrosome defects such as fap1multipolar, monopolar, and defective bipolar cells (Castellano et al., 2017). Multipolar spindles are strongly associated with chromosome mis-segregation (Silkworth et al., 2009). Mechanistically, this effect is likely attributed to miR-26a directly targeting multiple genes involved in mitosis and cytokinesis [i.e., checkpoint with forkhead and ring finger domains (CHFR), LARP1, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein epsilon (14-3-3 epsilon; YWHAE)]. The restoration of CHFR expression rescued the occurrence of multipolar spindles in two miR-26a overexpressing breast cancer cell lines, while restoration of YWHAE showed this rescue effect in one of two breast cancer cell lines. CHFR is an antephase checkpoint protein that delays entry into mitosis (via inhibition of polo-like kinase 1 and aurora kinase A if mitotic stress inhibits centrosome segregation) (Scolnick and Halazonetis, 2000, Sanbhnani and Yeong, 2012). YWHAE is associated with the G2/M checkpoint (via interaction with CDC25C) (Telles et al., 2009) during DNA replication and is thought to associate with centrosomes and/or microtubules during mitosis (Pietromonaco et al., 1996; Abdrabou et al., 2020). Of note, miR-26a-1 is located within the 3p21.1 region that is frequently deleted in epithelial malignancies (Diederichs and Haber, 2006); however, whether it affects the frequency of CIN has not been investigated.
miR-28 has been shown to potently regulate the levels of mitotic arrest deficient 2 (MAD2) protein via translational inhibition in various human and mouse cell types (i.e., HeLa embryonic kidney, HCT116 colon cancer, RPE-1 retinal pigment epithelial, and IMCD-3 murine kidney cells) (Hell et al., 2014). MAD2 is a mitotic spindle assembly checkpoint protein, deregulation of which is majorly involved in causing CIN through its regulatory roles in spindle assembly and kinetochore-microtubule attachments (Kabeche and Compton, 2012; Schuyler et al., 2012). CIN can result from both up- and downregulation of MAD2 due to the requirement of a delicate balance between mitotic arrest deficient 1 and 2 for spindle checkpoint function (Schuyler et al., 2012). Overexpression of miR-28 in HeLa, RPE-1, and IMCD-3 cells abolished pro-metaphase arrest in the presence of the mitotic checkpoint activator nocodazole, caused chromosome mis-segregation events (e.g., lagging chromosomes) and mitotic slippage, and led to change in chromosome number. This effect was completely rescued upon expression of MAD2 lacking the 3′UTR required for regulation by miR-28. Furthermore, depletion or loss of the tumor suppressor gene Von Hippel–Lindau tumor suppressor (VHL) known to reduce MAD2 levels and introduce CIN was shown to induce the expression of miR-28 at the transcriptional level. Consequently, miR-28 inhibition could rescue the occurrence of CIN in pVHL (VHL protein)-deficient cells, implying the suitability of anti–miR-28 therapy to target CIN in VHL-deficient cancers such as renal cell carcinomas (Hell et al., 2014). The miR-28–MAD2 regulatory axis was independently confirmed in Burkitt lymphoma (P3HR and RAJI) cells, where overexpression of miR-28 caused G1 arrest and growth reductions (Schneider et al., 2014), but whether this effect was directly related to CIN remains to be elucidated. In this context, the proto-oncogene MYC (hereafter: MYC) was identified as a negative regulator of miR-28 causing its reduced expression in Burkitt lymphomas. Restoration of miR-28 expression could prevent MYC-induced transformation of MCF10A breast epithelial cells, indicating that miR-28 reconstitution therapy could be suitable for the treatment of MYC-positive cancers (Schneider et al., 2014). However, the therapeutic targeting of the miR-28–MAD2 axis should be explored with caution due to the exact dosing of MAD2 required for correct function of the spindle assembly checkpoint (Schuyler et al., 2012).
The role of miR-186 overexpression in the presence and absence of arsenite has been investigated (Wu et al., 2019). Arsenic exposure is known to introduce CIN via interference with DNA damage repair and disruption of mitotic progression (States, 2015; Sage et al., 2017). In two of three miR-186 overexpressing HaCaT (keratinocyte) clones, the occurrence of double minute, dicentric, and ring chromosomes was increased. Exposure to arsenite further increased the number of double minute and dicentric chromosomes in miR-186 overexpressing clones. The authors suggest a functional link between miR-186 overexpression and BUB1 mitotic checkpoint serine/threonine kinase (BUB1) downregulation, however, direct regulation of BUB1 by miR-186 is not experimentally supported (Wu et al., 2019). Opposing this hypothesis, a recent report notes a link between BUB1 overexpression and mitotic segregation errors and CIN in multiple myeloma (Fujibayashi et al., 2020).
Long ncRNAs Implicated in CIN
Multiple long ncRNAs have causal relationships for CIN through regulation of all CIN-related pathways (i.e., telomere integrity, chromosomal segregation, and DNA damage response) (Table 1). One of the mechanisms maintaining chromosomal integrity and stability during cell division is the telomere, a region of repetitive DNA complexed with specialized ribonucleoproteins (O’Sullivan and Karlseder, 2010). Abnormal shortening of telomere length can cause CIN including abnormal structural alterations such as chromosome end-to-end fusions (Baird, 2018, Turner et al., 2019). Telomere length is regulated by telomerase, a telomeric DNA-synthesizing ribonucleotide enzyme, the shelterin complex, and various other proteins. In addition, telomeres are transcribed into lncRNAs of varying length termed telomeric repeat-containing RNAs (TERRA) (Azzalin et al., 2007), which also regulate telomere length through various mechanisms. TERRA contain G-rich repetitive sequences complementary to the RNA component of telomerase [i.e., telomerase RNA component (Terc)], the interaction with which blocks the Terc template region and prevents telomere–telomerase interaction (Schoeftner and Blasco, 2008). TERRA have furthermore been shown to bind to the telomerase reverse transcriptase polypeptide (Redon et al., 2010). Both of these TERRA functions, being a competitive inhibitor of Terc and an allosteric inhibitor of telomerase reverse transcriptase, negatively regulate telomerase activity and block telomere extension (Schoeftner and Blasco, 2008, Redon et al., 2010). In yeast, it was furthermore found that TERRA regulate the length of telomeres independently of telomerase through interaction with the Ku70/80 dimer. Ku70/80 binds to the telomere to protect the chromosome ends that have 3′-overhangs from degradation by exonuclease 1. TERRA binding of Ku70/80 interfered with its ability to inhibit exonuclease 1 (Pfeiffer and Lingner, 2012). TERRA has been shown to interact with various other telomere-associated proteins including the shelterin complex component telomere repeat factor 2 (TRF2) through its N-terminal Gly/Arg-rich domain, which assisted the recruitment of origin recognition complex subunit 1. Depletion of TERRA caused decreased binding of the origin recognition complex at telomeres and loss of histone H3 lysine 9 trimethylation and thereby affected heterochromatin formation and caused telomere dysfunction (Deng et al., 2009). A follow-up study showed that the interaction between TRF2 and TERRA is mediated through G-quadruplex structures within the lncRNA. Interestingly, a G-quadruplex targeting compound, N-methyl mesoporphyrin IX, was found to specifically inhibit this interaction while also downregulating TERRA expression (Mei et al., 2021). In addition, the formation of DNA:RNA hybrid structures involving TERRA has been identified at telomeres. Such hybrids were enriched in immunodeficiency (centromeric instability and facial anomalies) syndrome cells (that have short telomeres and increased TERRA) and were associated with increased levels of DNA damage at chromosome ends (Sagie et al., 2017). Involvement of TERRA in the regulation of telomere dynamics makes it an attractive therapeutic target in cancer that may experience both shortened and elongated telomeres depending on cancer type and stage (Fernandes et al., 2020).
lncRNAs causative for CIN
Similar to lncRNAs derived from telomeric sequences, the repetitive DNA regions of the centromere also produce RNA transcripts. The centromere is made up of alpha satellite DNA, (i.e., repeated ∼171-bp monomer units), which associates with a complex of proteins during mitosis to form the kinetochore responsible for attaching spindle microtubules. The transcriptional activity of the centromere was first discovered in rice (Nagaki et al., 2004) and later also identified in humans, producing centromeric RNAs (cenRNAs) (Wong, Brettingham-Moore et al., 2007; Chan et al., 2012; McNulty et al., 2017; Ishikura et al., 2020). CenRNAs were shown to physically interact with centromere proteins, specifically centromere protein A (CENP-A) and its chaperone Holliday junction recognition protein (Quénet and Dalal, 2014). CENP-A is a histone H3 variant that epigenetically specifies centromere identity and function (Fachinetti et al., 2013) and depletion of cenRNAs lead to a loss of CENP-A and Holliday junction recognition protein at centrosomes (Quénet and Dalal, 2014). An independent study similarly identified loss of CENP-A loading upon cenRNAs depletion and additionally detected a loss of colocalization with centromere protein C (CENP-C), which was shown to form a stable complex together with cenRNA and CENP-A (McNulty et al., 2017). Other proteins associated with cenRNA are inner centromere protein and aurora kinase B (AURKB), both subunits of chromosome passenger complex that regulates the attachment of microtubules to the kinetochore (Wong et al., 2007; Ideue, Cho et al., 2014). CenRNAs depletion disrupted the localization of inner centromere protein, its interactor survivin, and AURKB at kinetochores, resulting in abnormal chromosome segregation (Wong et al., 2007; Ideue et al., 2014). In addition, reduction in cenRNA transcription induced AURKB activation (Ideue et al., 2014), which is known to dysregulate microtubule attachment to the kinetochore (Murata-Hori and Wang, 2002; Portella et al., 2011). The overexpression of cenRNAs has been detected in several human cancers, which lead to increased CIN, indicating cenRNA expression may be an early event in cancer development and could be a useful marker for neoplastic cells (Ting et al., 2011; Chan et al., 2017; Ichida et al., 2018). In tumor cells, CENP-A is often overexpressed and can mislocalize to regions outside centromeres altering the recruitment of centromere- and kinetochore-associated proteins and leading to CIN (Shrestha et al., 2017). Ectopic accumulation of CENP-A is, for example, observed at chromosomal region 8q24, which harbors the frequently translocated oncogene c-Myc. Five lncRNAs are expressed from region 8q24: colon cancer-associated transcripts 1 and 2, prostate cancer–associated transcripts 1 and 2, and plasmacytoma variant translocation 1. Interestingly, knockdown of several of these lncRNAs decreased the ectopic localization of CENP-A and colocalization of CENP-C, an effect that was most prominent upon knockdown of PCAT2. A direct interaction between PCAT2 and CENP-A that was dependent on transcriptionally coupled H3.3 chaperones histone cell cycle regulator and death domain–associated protein caused this ectopic placement of CENP-A/C in colon cancer cells. Insertion of transgenic PCAT2 at a naïve chromosome locus 4q31 was sufficient to cause ectopic CENP-A/C localization (Arunkumar et al., 2022). This study thus shows that oncogenic lncRNAs may mimic cenRNAs to ectopically recruit centromere-associated proteins, thereby causing genomic fragility.
In B cells, tumorigenic genomic translocations are frequently associated with the off-targeting of activation-induced cytidine deaminase (AID). AID mediates somatic hypermutation and class switch recombination, mechanisms designed to diversify B cell antigen receptors, by mediating deamination of cytosines, leading to mutations or double-strand breaks within the immunoglobulin genes. AID is known to have recurrent off-targets such as proto-oncogenes BCL6 (B-cell lymphoma 6) and MYC, causing their translocation and juxtaposition to potent Ig enhancers, which, in turn, results in their overexpression. AID off-targeting hotspots were shown to be characterized by convergent transcription stemming from antisense transcription of enhancer RNAs within sense transcribed genes (Meng et al., 2014; Qian et al., 2014). Such regions of convergent transcription were more prone to genomic instability upon depletion of the RNA exosome (region that regulates the ncRNA expression initiating from enhancers), which was shown to resolve RNA/DNA hybrid structures (R loops) stemming from regions of convergent transcription (Pefanis et al., 2015). Thus, the active transcription of enhancer RNAs and their RNA exosome-mediated degradation play a major role in B cell–specific chromosomal translocations arising from AID off-targeting.
The lncRNA genomic instability inducing RNA (Ginir) and its antisense transcript (Ginir-as) display a tight and balanced spaciotemporal expression pattern during mouse embryonic development. Ginir expression was higher in proliferating cells during development, particularly in neuronal tissues, while Ginir-as expression was higher in nonproliferating cells of major organs in the adult mouse. The overexpression of Ginir (but not Ginir-as or the Ginir–Ginir-as combination) induced oncogenic transformation of NIH/3T3 (mouse fibroblast) cells in vitro and in murine xenograft models, inducing increased proliferation and invasion potential. DNA double-strand breaks, activation of the DNA damage response, as well as mitotic defects were noted in Ginir-overexpressing cells, including multipolar spindles resulting in multinucleated cells. Mechanistically, this effect was mediated through interaction of Ginir with centrosomal protein 112 (Cep112) and breast cancer type 1 susceptibility protein (Brca1) (Panda et al., 2018). Brca1 is, in addition to its prominent role in the DNA damage response, involved in centrosome regulation and its dysfunction causes increases in centrosome number (Yoshino et al., 2021). High levels of Ginir disrupted the interaction between Cep112 and Brca1 and downregulated their expression, consequently leading to centrosome amplification and this effect that was phenocopied by the individual knockdown of both Cep112 and Brca1 (Panda et al., 2018). Ginir thus acted as an oncogene in adult murine cells by introduction of DNA damage and CIN.
LncRNA P53-responsive lncRNA (GUARDIN) was discovered as a p53-responsive lncRNA, facilitating DNA damage response and modulating the p53 cytotoxic effect. Knockdown of GUARDIN reduced the proliferation and carcinogenic potential of HCT116 (colon cancer) cells. A dual role was defined for this lncRNA: on one hand, it sequestered miR-23a (i.e., acted as an endogenous competing RNA), thereby affecting expression of the miR-23a target gene TRF2. TRF2 is a part of the shelterin complex, essential for telomere integrity, and its levels are maintained through the GUARDIN–miR-23a–TRF2 axis. Consequently, GUARDIN depletion resulted in DNA damage at telomeres and end-to-end fusion of chromosomes. On the other hand, GUARDIN directly interacted with BRCA1 as well as BRCA1-associated RING domain 1 and depletion of GUARDIN resulted in downregulation of BRCA1 through proteosomal degradation. This affected the DNA damage response and markedly reduced the activity of HR and nonhomologous end-joining pathways (Hu et al., 2018). The BRCA1- BRCA1-associated RING domain 1 interaction is also essential for centrosome regulation during mitosis (Yoshino et al., 2021), but whether GUARDIN affects centrosome number and function remains to be elucidated.
Colon cancer-associated transcript 2 (CCAT2) was first identified as a lncRNA arising from the cancer-associated 8q24 gene desert and was selectively overexpressed in microsatellite-stable CRC patient samples (Ling et al., 2013), which generally have a worse prognosis compared with microsatellite-instable CRCs (Boland and Goel, 2010). In vivo xenograft models showed that CCAT2 overexpression promoted tumor growth and metastasis and upregulated expression of the oncogene MYC. A strong link to the development of CIN was identified upon in vitro overexpression of CCAT2 in HCT116 cells, which caused the occurrence of aberrant metaphases resulting in both sCIN and nCIN and. consequently, a dramatic increase in the amount of polyploid cells. CCAT2-overexpressing clones were marked by the presence of three or more centrosomes, causing faulty chromosome segregation (Ling et al., 2013). In a follow-up study, the precise functional role of CCAT2 in CIN was further unraveled, identifying the lncRNA as a positive regulator of BOP1 ribosomal biogenesis factor (BOP1), either via induction of MYC or directly. BOP1 overexpression in CRC cell lines phenocopied the effects of CCAT2, leading to abnormal spindles during metaphase as well as anaphase bridges, consequently causing chromosome fusions, breaks, and fragmentation. In the functional model further established, BOP1 increased the active (i.e., phosphorylated) form of AURKB, while CCAT2 was found to form a complex with AURKB, possibly mediating the interaction between BOP1 and AURKB (Chen et al., 2020). AURKB is a part of the chromosomal passenger complex associated with centrosomes and controls spindle assembly and cytokinesis via different substrates (Hindriksen et al., 2015). Increased active AURKB disrupts chromosome-microtubule attachments and causes premature collapse of the mitotic spindle (Muñoz-Barrera and Monje-Casas, 2014).
ncRNA activated by DNA damage (NORAD) is a ubiquitously expressed, highly conserved lncRNA that is strongly induced by DNA damage. Knockout of NORAD was shown to result in a high frequency of mitotic errors (e.g., anaphase bridges, mitotic slippage) leading to nCIN (chromosome gains/losses, aneuploidy) as well as sCIN (rearrangements) in HCT116 (CRC) cells, and this phenotype could be rescued by NORAD restoration. Mechanistically, this effect was mediated by the sequestration of PUMILIO proteins (PUMILIO homolog 2 and, to a lesser extent, PUMILIO homolog 1) and 15 binding motifs (i.e., PUMILIO response elements) were identified within NORAD lncRNA (Lee et al., 2016). An independent study suggested that the interaction between PUM2 and NORAD is mediated via KH RNA binding domain containing, signal transduction associated 1 (SAM68), which also has recurring binding sites within the lncRNA (Tichon et al., 2018). PUMILIO proteins post-transcriptionally regulate gene expression by binding to these specific response elements in the 3′UTRs of mRNAs (Wickens et al., 2002). Among PUMILIO target genes are key mitotic DNA repair and DNA replication factors mediating genomic stability, which were repressed following the lack of PUMILIO homolog 2/PUMILIO homolog 1 sequestration in NORAD knockout cells (Lee et al., 2016). A follow-up study showed that the expression of a circular RNA containing 4 to 8 PUMILIO response elements could rescue the chromosome segregation defects caused by CRISPR-mediated NORAD depletion, causing phase-separation of PUM proteins into punctate cytoplasmic foci (i.e., NP bodies) (Elguindy and Mendell, 2021).
Conclusions and Future Perspectives
As CIN is an event occurring early in tumor development and has strong implications in tumor resistance to cytotoxic anticancer drugs (Pikor et al., 2013; Vargas-Rondón et al., 2017), its exploitation for therapeutic purposes requires fundamental understanding of the abnormal molecular pathways driving CIN. CIN may be exploited therapeutically in multiple ways: (1) by reducing CIN to hinder tumor adaptability and development of drug resistance, (2) by increasing CIN to produce unsustainable karyotypes leading to cell death, or (3) by targeting the CIN-tolerance mechanisms acquired by tumor cells (Thompson et al., 2017; Sansregret et al., 2018). Multiple compounds such as anaphase promoting complex/cyclosome inhibitors (reduce CIN by prolonging metaphase/mitotic exit), spindle assembly checkpoint inhibitors (induce CIN by premature mitotic exit) or Aurora kinase inhibitors (induce CIN by inhibiting chromosome alignment and spindle assembly) show promising results in preclinical and/or early clinical studies (Thompson et al., 2017). Multiple of the ncRNAs described here could similarly be targeted to increase or decrease the basal level of CIN in cancer cells. ncRNAs are furthermore involved in CIN tolerance mechanisms. One such mechanism to compensate for the negative effects of aneuploidy is gene dosage compensation. For example, the OncomiR-1 cluster miRNAs miR-17, miR-19a, and miR-20a have been found to be involved in the compensation for increased copy numbers of proto-oncogene MYC. Accordingly, the inhibition of these miRNAs caused cytotoxicity that was stronger in cells with higher copy numbers of MYC (Acón et al., 2021). The OncomiR-1 cluster has furthermore been shown to form a feedback loop with signal transducer and activator of transcription 3 (Jo et al., 2014; Acón et al., 2021), indicating a broader role for this cluster in gene dosage compensation.
The different modes of targeting CIN in cancer further imply that good biomarkers for CIN are urgently needed for patient stratification. Monitoring of ncRNAs such as cenRNAs, the levels of which are thought to increase prior to CIN occurrence (Ting et al., 2011; Chan et al., 2017; Ichida et al., 2018) could deliver such much needed markers in the near future.
Thus, the ncRNAs described here not only grant a better understanding of mechanisms leading to CIN but also further expand the possibilities for its therapeutic targeting. Increasing use of large-scale sequencing has immensely helped in a better understanding of the chromosomal changes incorporated in different cancers. However, different emerging factors including ncRNAs have been reported in regulating chromosomal stability directly or indirectly, thereby fueling the tumor heterogeneity and propagating the karyotype diversity that is important to understand to find better treatment strategies.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Mohapatra, Winkle, Ton, Nguyen, Calin.
Footnotes
- Received June 15, 2022.
- Accepted August 17, 2022.
G.A.C.’s laboratory is supported by National Institutes of Health National Cancer Institute [Grants 1R01-CA182905-01 and 1R01-CA222007-01A1], National Institute of General Medical Sciences [Grant 1R01-GM122775-01], National Institute of Dental and Craniofacial Research [Grant 5R01-DE032018-02], DoD Idea Award [W81XWH-21-1-0030], a Team DOD grant in Gastric Cancer [W81XWH-21-1-0715], a Chronic Lymphocytic Leukemia Moonshot Flagship project, a CLL Global Research Foundation 2019 grant, a CLL Global Research Foundation 2020 grant, The G. Harold & Leila Y. Mathers Foundation, two grants from Torrey Coast Foundation, an Institutional Research Grant, an Institutional Bridge Fund, and a development grant associated with the Brain SPORE [2P50CA127001]. S.M. is supported by the CPRIT Research Training [Grant RP210028].
G.A.C. is the scientific founder of Ithax Pharmaceuticals. No author has an actual or perceived conflict of interest with the contents of this article.
Abbreviations
- AID
- activation-induced cytidine deaminase
- AKT1
- AKT serine/threonine kinase 1
- AURKB
- aurora kinase B
- BOP1
- BOP1 ribosomal biogenesis factor
- Brca1
- breast cancer type 1 susceptibility protein
- BUB1
- BUB1 mitotic checkpoint serine/threonine kinase
- CCAT2
- colon cancer-associated transcript 2
- CENP-A
- centromere protein A
- CENP-C
- centromere protein C
- cenRNAs
- centromeric RNAs
- Cep112
- centrosomal protein 112
- CHFR
- checkpoint with forkhead and ring finger domains
- CIN
- chromosomal instability
- CRC
- colorectal cancer
- Ginir
- genomic instability inducing RNA
- Ginir-as
- antisense genomic instability inducing RNA
- GUARDIN
- P53-responsive long non-coding RNA
- HR
- homologous recombination
- lncRNAs
- long non-coding RNAs
- MAD2
- mitotic arrest deficient 2
- MDC1
- mediator of DNA damage checkpoint 1
- miRNAs
- microRNAs
- nCIN
- numerical chromosomal instability
- ncRNAs
- non-coding RNAs
- NORAD
- non-coding RNA activated by DNA damage
- sCIN
- structural chromosomal instability
- Terc
- telomerase RNA component
- TERRA
- telomeric repeat-containing RNAs
- TFAP4
- transcription factor AP-4
- TRF2
- telomere repeat factor 2
- VHL
- Von Hippel–Lindau tumor suppressor
- YWHAE
- tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein epsilon
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}