



Abstract
Although protein-protein interactions (PPIs) have emerged as an attractive therapeutic target space, the identification of chemicals that effectively inhibit PPIs remains challenging. Here, we identified through library screening a chemical probe (compound 1) that can inhibit the tumor-promoting interaction between the oncogenic factor exon 2–depleted splice variant of aminoacyl–transfer RNA synthetase-interacting multifunctional protein 2 (AIMP2-DX2) and heat shock protein 70 (HSP70). We found that compound 1 binds to the N-terminal subdomain of glutathione S-transferase (GST-N) of AIMP2-DX2, causing a direct steric clash with HSP70 and an intramolecular interaction between the N-terminal flexible region and the GST-N of AIMP2-DX2, which induces masking of the HSP70 binding region during molecular dynamics and mutation studies. Compound 1 thus interferes with the AIMP2-DX2 and HSP70 interaction and suppresses the growth of cancer cells that express high levels of AIMP2-DX2 in vitro and in preliminary in vivo experiment. This work provides an example showing that allosteric conformational changes induced by chemicals can be a way to control pathologic PPIs.
SIGNIFICANCE STATEMENT Compound 1 is a promising protein-protein interaction inhibitor between AIMP2-DX2 and HSP70 for cancer therapy by the mechanism with allosteric modulation as well as competitive binding. It seems to induce allosteric conformational change of AIMP2-DX2 proteins and direct binding clash between AIMP2-DX2 and HSP70. The compound reduced the level of AIMP2-DX2 in ubiquitin-dependent manner via suppression of binding between AIMP2-DX2 and HSP70 and suppressed the growth of cancer cells highly expressing AIMP2-DX2 in vitro and in preliminary in vivo experiment.
Introduction
Since many physiologic and pathologic cellular events are controlled by protein-protein interactions (PPIs), modulating these PPIs promises attractive ways to develop drugs for many diseases (Nero et al., 2014; Ivanov et al., 2013). Although extensive efforts have been made to develop PPI inhibitors that target oncogenic interactions, most have failed due to a lack of structural understanding of the binding interface and limited efficacy using small molecules (Nero et al., 2014; Ran and Gestwicki, 2018; Li et al., 2017). Recently, a few meaningful advances in PPI inhibitor development have been reported using peptide mimetic molecules with molecular weights over 500 Da, but these molecules have shown limitations due to their molecular features including solubility, stability, and cell penetration (Nero et al., 2014; Wójcik and Berlicki, 2016; Touti et al., 2019).
Aminoacyl–transfer RNA synthetase-interacting multifunctional protein 2 (AIMP2) functions as a scaffold for the assembly of the multisynthetase complex (MSC) (Kim, S. et al., 2011). AIMP2 dissociates from the MSC upon UV damage and transforming growth factor-β, tumor necrosis factor-α, and Wnt signaling and functions as a tumor suppressor via its interaction with p53, FUSE-binding protein, Smurf2, tumor necrosis factor receptor–associated factor 2, and dishevelled-1 (Han. et al., 2008; Kim et al., 2003; Kim, et al., 2016; Choi, et al., 2009; Yum et al., 2016). The exon 2–depleted splice variant of AIMP2 (AIMP2-DX2; hereafter referred to as DX2) is an alternative splicing variant of AIMP2 that lacks exon 2 and compromises the tumor suppressive activities of AIMP2 (Choi et al., 2011). DX2 is induced by carcinogens, and the level of DX2 is positively correlated with tumor aggressiveness and poor prognosis in lung, chemoresistant ovarian, and colon cancers (Choi et al., 2011; Choi et al., 2012). Using the same protein structure to bind various signaling molecules, DX2 competitively interacts with the AIMP2-binding proteins p53, FUSE-binding protein, and tumor necrosis factor receptor–associated factor 2, resulting in tumor-promoting functions (Han et al., 2008; Kim et al., 2003; Kim et al., 2016; Choi et al., 2009). DX2 also independently shows oncogenic properties via a decrease in the level of p14 (Oh et al., 2016). Specific short hairpin RNAs against DX2 and DX2-binding small molecules have been used in an attempt to reverse DX2-mediated tumor progression and have shown therapeutic effects in vitro and in vivo (Choi et al., 2011; Lee et al., 2013), suggesting DX2 as a novel therapeutic target against cancer. Recently, DX2 was shown to be stabilized by binding with heat shock protein 70 (HSP70), which blocks Siah1-mediated ubiquitination of DX2, and inhibition of the DX2-HSP70 interaction was found to be effective against DX2-induced tumor progression (Lim et al., 2019). Validation of DX2 and HSP70 as an effective cancer target encouraged us to establish a high-throughput screening method to identify chemicals that can specifically interfere with the interaction of these two oncogenic factors and elucidate effective hits with potentially different modes of action. Here, we investigated an allosteric inhibitor against the DX2 and HSP70 interaction to elucidate its mode of action on the suppression of tumors. We describe the studies on the selection and characterization, mode of action, and inhibition on cancer development of a chemical probe using binding assay, surface plasmon resonance, in vitro pull-down assay, immunoprecipitation, cell viability assay, refolding assay, reverse transcription polymerase chain reaction (PCR), xenograft, mutagenesis, and molecular dynamics (MD) simulation.
Materials and Methods
Chemistry
Commercially available reagents or solvents were used from freshly opened containers without further purification unless otherwise specified. Analytical thin layer chromatography (TLC) analyses were conducted on precoated silica gel 60 GF254 plates. Purification of crude compounds was performed by flash chromatography on Merck silica gel 60 (230–400 mesh). 1H NMR and 13C NMR spectra were obtained on a Bruker 300 MHz NMR spectrometer at ambient temperature. Chemical shifts are given in ppm (δ) referenced to the internal standard tetramethylsilane. Coupling constants (J) are given in Hz. Mass spectra were obtained on a Bruker instrument by using electron impact techniques. Liquid chromatography–mass spectrometry (LC-MS) was employed to determine the purity of the compounds. LC-MS was carried out on a Waters ACQUITY SQD2 Micromass equipped with a BEH C18 column (2.1 × 50 mm, 1.7 μm) high-pressure liquid chromatograph with a linear gradient of 10% to 90% acetonitrile into 0.2% CF3CO2H/H2O over 5 minutes at a flow rate of 0.4 ml/min. UV detection [photo diode array detector (ACQUITY)] was carried out at a wavelength of 254 nm. High-resolution mass spectrometry (HRMS) analysis was performed using an ultrahigh resolution–time-of-flight maXis 4G instrument (Bruker Daltonics, Bremen, Germany).
Compound 1
A mixture of benzaldehyde (6.24 mmol), ethyl 3-oxo-3-phenylpropanoate (5.20 mmol), benzoic acid (0.52 mmol), and piperidine (2.6 mmol) in toluene (30.0 ml) was heated at reflux under Dean-Stark conditions until the starting material disappeared by TLC analysis. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography to afford ethyl (Z)-2-benzoyl-3-phenylacrylate (compound 1) in 43% yield. 1H NMR [300 MHz, deuterated chloroform (CDCl3)] δ 8.0–7.95 (m, 3H), 7.62–7.55 (m, 1H), 7.49–7.42 (m, 2H), 7.41–7.36 (m, 2H), 7.31–7.26 (m, 3H), 4.25 (q, J = 7.1 Hz, 2H), 1.20 (t, J = 7.1 Hz, 3H). LC-MS (M+H)+ 281.2. 13C NMR: (75 MHz, CDCl3) δ 195.67, 165.03, 142.62, 136.16, 133.92, 132.83, 131.32, 130.40, 130.21, 129.16, 128.85, 128.81, 61.58, 14.05. HRMS (EI+) 280.1102.
Compound 12
BBr3 (1 M) in CH2Cl2 (8.9 ml, 8.9 mmol) was added with stirring to ethyl 3-(4-methoxyphenyl)-3-oxopropanoate (1 g, 4.49 mmol) in CH2Cl2 (30 ml) at −78°C. After stirring for 1.5 hours at −15°C, additional 1 M BBr3 in CH2Cl2 (8.9 ml, 8.9 mmol) was added. After additional stirring for 2 hours, methanol (5 ml) and saturated aqueous NaHCO3 were added to quench and neutralize the reaction to pH 7.0. The separated aqueous layer was extracted with CH2Cl2 (2 × 50 ml), and the combined organic layers were dried (MgSO4), rotary evaporated, and chromatographed (ethyl acetate:hexanes 3:7) to afford ethyl 3-(4-hydroxyphenyl)-3-oxopropanoate, compound 12 (Miyatake-Ondozabal and Barrett, 2013). 1H NMR (300 MHz, CDCl3) δ 7.90–7.82 (m, 2H), 6.96–6.88 (m, 2H), 4.23 (q, J = 7.1 Hz, 2H), 3.99 (s, 2H), 1.27 (t, J = 7.1 Hz, 3H). LC-MS (M+H)+ 209.1.
Compound 13
The same reaction to prepare compound 1 was performed to provide ethyl (Z)-2-(4-hydroxybenzoyl)-3-phenylacrylate (compound 13) (Sun et al., 2009) in 25% yield. 1H NMR (300 MHz, CDCl3) δ 7.94 (s, 1H), 7.91–7.83 (m, 2H), 7.39–7.34 (m, 2H), 7.27–7.22 (m, 2H), 6.87–6.83 (m, 2H), 4.24 (q, J = 7.1 Hz, 2H), 1.20 (t, J = 7.1 Hz, 3H). LC-MS (M+H)+ 297.2.
Compound 15
To a flame-dried round-bottom flask, 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-day]imidazol-4-yl)pentanoic acid (compound 14; 0.1 g, 0.409 mmol) and two drops of dimethylformamide were added to dry dichloromethane (2 ml). The mixture was cooled to 0°C, and oxalyl chloride (0.075 ml, 0.409 mmol) was added dropwise to the flask. The reaction was stirred for 3 hours, after which a solution of compound 13 (0.181 g, 0.613 mmol) and pyridine (0.050 ml, 0.613 mmol) in CH2Cl2 (2 ml) was added dropwise, and the reaction was allowed to stir overnight. When TLC showed completion of the reaction, the reaction mixture was diluted with water (10 ml) and washed with brine (2 × 15 ml). The organic layer was concentrated in vacuo and purified by automated column chromatography (eluting with ethyl acetate:hexanes 4:6) to afford 4-((Z)-2-(ethoxycarbonyl)-3-phenylacryloyl)phenyl 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoate (compound 15) (Ackerman et al., 2018) (20 mg, 10% yield). 1H NMR (300 MHz, CDCl3) δ 8.93 (d, J = 0.9 Hz, 1H), 8.02–7.96 (m, 3H), 7.36 (dd, J = 7.9, 1.9 Hz, 2H), 7.30 (dd, J = 5.6, 1.3 Hz, 1H), 7.28–7.23 (m, 1H), 7.21–7.15 (m, 2H), 6.90 (s, 1H), 4.86 (dd, J = 7.5, 5.2 Hz, 1H), 4.30 (dd, J = 5.4, 2.8 Hz, 1H), 4.24 (q, J = 7.0 Hz, 2H), 3.22 (dd, J = 10.7, 5.1 Hz, 2H), 2.98 (dd, J = 13.6, 5.3 Hz, 1H), 2.61 (t, J = 7.3 Hz, 2H), 1.87–1.71 (m, 5H), 1.58 (q, J = 7.5 Hz, 2H), 1.21 (t, J = 7.1 Hz, 3H). LC-MS (M+H)+ 523.2. 13C NMR: (101 MHz, CD3OD) δ 194.67 (d, J = 13.3 Hz), 171.57, 164.95 (d, J = 32.5 Hz), 163.51, 155.45, 142.30, 141.50, 133.54, 133.00, 132.81, 131.74, 131.60, 131.13, 130.95, 130.51, 130.31, 130.09, 129.80, 128.55, 128.44, 127.86, 122.11, 115.30, 61.96, 61.51–60.73 (m), 60.23, 55.61, 39.66, 34.37–31.94 (m), 29.79–26.84 (m), 25.47–23.48 (m), 13.02 (d, J = 2.4 Hz). HRMS (FAB+) 523.1884.
Cell Culture and Materials
The CHO-K1 and A549 cell lines were purchased from the American Type Culture Collection and Korea Cell Line Bank, respectively. The H460, H358, H441, H1299, Calu-6, HCC-1588, H226, H1650, WI-26, and 293T cell lines were kindly gifted from Biocon (Medicinal Bioconvergence Research Center, Yonsei University). CHO, A549, H460, H358, H441, H1299, Calu-6, HCC-1588, H226, and H1650 cells were cultivated in RPMI medium supplemented with 10% FBS and 1% penicillin/streptomycin in 5% CO2 at 37°C. 293T and WI-26 cells were cultured in Dulbecco’s modified Eagle’s medium under the same conditions as above. DX2 was cloned into the EcoRI/XhoI sites of the pcDNA3.0 and pEGFP-C2 vectors to express FLAG- and GFP-tagged DX2, respectively. Mutagenesis was performed with Quik-ChangeII (Promega) following the manufacturer’s instructions. The purified human HSP70 (#ADI-NSP-555) protein was purchased from Enzo. The purified human tag-free DX2 proteins were kindly provided by the laboratory of Prof. Young Ho Jeon (Korea University). MG-132 (#474790) and doxycycline (dox; #D3447) were purchased from Millipore and Sigma, respectively. Specific antibodies against DX2, AIMP2, Lysyl-T RNA Synthetase 1, Methionyl-T RNA Synthetase 1, Glutamyl-Prolyl-T RNA Synthetase 1, and Aminoacyl t-RNA synthetase complex-interacting multifunctional protein 1 were kindly provided by Biocon. The anti-Siah1 (#ab2237) antibody was purchased from Abcam, and specific antibodies against actin (#A1978) and FLAG (#F3165) were purchased from Sigma. Antibodies against HSP70 (#sc-24), ubiquitin (#sc-8017), CDK4 (#sc-23896), and GFP (#sc-9996) were purchased from Santa Cruz Biotechnology.
Molecular Modeling
For the relaxation of the protein (apo system) in the solvent system, we performed a 200 ns MD simulation using the DX2 structure consisting of residues L50 through K251 without the disordered N-terminal flexible region (NFR). To examine the involvement of the NFR and binding mode of compound 1, the NFR consisting of A38 to A49 was added into the protein structure in an inhibitor-bound state, followed by another 300 ns of simulation. The 300 ns simulation was repeated three times. The crystal structure of the AIMP2 glutathione S-transferase (GST) domain (Protein Data Bank identifier: 5A34) (Cho et al., 2015) with a resolution of 2.6 Å was used for the molecular modeling study. The DX2 structure was constructed using the built and edited protein tools implemented in Discovery Studio 2018 software (https://www.3ds.com/products-services/biovia/products/molecular-modelingsimulation/ biovia-discovery-studio). The binding site located in the N-terminal subdomain of GST (GST-N) was defined by chemical shift perturbation data obtained from NMR binding experiments (Lim et al., 2019). To generate initial docking poses subjected to the following MD simulation study, the docking simulation of compound 1 was performed using CDOCKER (Wu et al., 2003) implemented in Discovery Studio 2018 software. The MD simulations were performed using GROMACS 2016 with the Plumed plugin version 2.4 (Tribello et al., 2014). The Chamm36 all-atom force field was used for the protein and ligand, and transferableintermolecular potential with 3 points was used for water. CHARMM-GUI (Jo et al., 2008) was used for the generation of input for the simulations. The topologies and parameters of the ligands were generated by the CHARMM General Force Field program (Vanommeslaeghe et al., 2010). A fully solvated cubic water box 10 Å thick was constructed for each system. The systems were energy minimized by the steepest descent method to remove possible bad contacts until a tolerance of 1000 kJ/mol. The constant particle number, volume, and temperature equilibration process was conducted for the minimized structures for 25 ps with a 1 fs time step at 303.15 K. The LINCS algorithm (Hess, B. et al., 1997) was used to constrain the bonds involving hydrogen atoms by their equilibrium bond lengths. Finally, the production runs were performed at a temperature of 303.15 K and a pressure of 1 bar by constant particle number, pressure, and temperature dynamics achieved with the Nosé-Hoover thermostat (Hoover, 1985) and Parrinello-Rahman (Parrinello and Rahman, 1981) barostat. The length of the time step was set to 2 fs for the production runs, and the trajectory was saved every picosecond. The cutoff values of short-range electrostatic interactions and van der Waals interactions were set to 12 Å. The particle mesh Ewald method (Essmann et al., 1995) was used for long-range electrostatic interactions. To analyze protein-ligand interactions, the snapshot with the lowest nonbonded energy between the protein and ligand and a highly populated conformation of the ligand during the last 200 ns was selected as a representative structure. To avoid the ligand escaping from the binding site into the bulk solvent region, an upper-wall restraint force was applied to the system when the distance between the center of mass of the GST-N and the center of mass of compound 1 was greater than the cutoff limit (dup) of 12 Å. For the wall, the harmonic potential was set with a force constant κ = 200 kJ/mol⋅nm−2.

The g_mmpbsa tool (Kumari et al., 2014) was used to calculate the binding energies of the system with the molecular mechanics Poisson-Boltzmann surface area method. The calculation was performed with 3000 snapshots extracted from the 300 ns trajectory every 100 ps. We calculated the final binding free energy and the energetic contribution of each residue by python scripts “MmPbSaStat.py” and “MmPbSaDecomp.py,” which are the part of g_mmpbsa.
High-Throughput Screening
DX2 and HSP70 were cloned into pBiT1.1-N[TK/LgBiT] and pBiT2.1-N[TK/SmBiT], respectively. LgBiT fused with protein kinase A type 2A regulatory subunit (PRKAR2A) and SmBiT fused with protein kinase A catalytic subunit (PRKACA) were obtained from Promega. CHO-K1 cells transfected with LgBiT-DX2 and SmBiT-HSP70 were seeded into 96-well white-bottom plates. After incubation for 24 hours, the cells were treated with 6186 chemicals [5 μM each, Korea Chemical Bank (KCB)] in serum-free media for 4 hours. Luciferase activity was detected using a NanoBiT assay system following the manufacturer’s protocol (Promega). Ninety-nine chemicals showing over 60% inhibition at 5 μM were subjected to secondary screening using LgBiT-PRKAR2A and SmBiT-PRKACA for negative screening via the same experimental procedure as above. Ten compounds that showed no effect on the negative screening were subjected to a third screening involving in vitro pull-down assays and cell viability assays using DX2-inducible cell lines.
Surface Plasmon Resonance
To measure the binding affinity of compound 1 to the DX2 protein, we used a Reichert SR7500DC instrument (Reichert Technologies, Depew, NY). Thioredoxin-tagged DX2 and thioredoxin proteins were immobilized at levels of 11,500 and 4000 response units, respectively, on a carboxymethyl dextran chip (Reichert, Depew, NY) with buffer containing 10 mM sodium acetate (pH 5.5). Compound 1 (1.5–50 μM) in 2% DMSO-containing PBS binding buffer (pH 7.4) flowed at a rate of 30 μl/min. Sensorgrams were fitted to a simple 1:1 Langmuir interaction model (A + B ⇌ AB) using the Scrubber 2.0 analysis program (BioLogic Software, Australia, and Kaleida Graph Software, Australia) to calculate the values of Ka and KD.
Fluorescence-Based Equilibrium Binding Assay
To determine the binding affinity of compound 1 to the DX2 protein, we performed fluorescence-based equilibrium binding experiments. All titration experiments were conducted at 20°C using a Jasco FP 6500 spectrofluorometer (Easton, MD). Purified human tag-free DX2 proteins were equilibrated with various concentrations of ligand 1 before fluorescence emission was measured. Ligand stock solutions were titrated into a protein sample dissolved in phosphate buffer (pH 7.4) containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4. Protein samples were excited at 280 nm, and the decrease in fluorescence emission upon ligand binding was measured at 335 nm as a function of the ligand concentration. All titration data were fitted to a hyperbolic binding equation to obtain the KD values.
Biomolecular Fluorescence Complement Assay
For the biomolecular fluorescence complement (BiFC) assay, DX2 and HSP70 were cloned into the EcoRI/XhoI sites of the pCE-BiFC-VN173 and pCE-BiFC-VC155 vectors to tag Venus-N173 and Venus-C155, respectively. CHO cells expressing VN173-DX2 and VC155-HSP70 were treated with compound 1 in a dose-dependent manner for 4 hours. After incubation, the cells were washed with cold PBS three times and fixed with 100% cold methanol for 10 minutes, and the nuclei were stained with 4′,6-diamidino-2-phenylindole for cell counting. The BiFC signal was determined by confocal microscopy, and signal-positive cells were counted in the same number of cells. The experiment was independently repeated three times.
In Vitro Pull-Down Assay
DX2 was cloned into the EcoRI/XhoI sites of the pGEX4T-1 vector to express GST-tagged DX2. The purified GST or GST-DX2 proteins were mixed with the HSP70 protein (#ADI-NSP-555, Enzo) in 50 mM Tris-HCl (pH 7.4) buffer containing 100 mM NaCl, 0.5% Triton X-100, 10% glycerol, 1 mM EDTA, and protease inhibitor (Calbiochem), followed by treatment with or without compound 1 for 4 hours. After incubation, proteins coprecipitated with beads were washed three times with incubation buffer excluding 0.5% Triton X-100, subjected to SDS-PAGE, and detected by Coomassie staining. To examine the binding of compound 1 to the DX2 or HSP70 proteins, we incubated compound 15 with 100 μg of tag-free DX2 or HSP70 proteins as starting amounts for 12 hours in incubation buffer and performed the same procedure described above. Biotin alone was used as a negative control compound for product 15.
Immunoprecipitation
The cells were lysed with 50 mM Tris-HCl (pH 7.4) lysis buffer containing 100 mM NaCl, 0.5% Triton X-100, 0.1% SDS, 10% glycerol, 1 mM EDTA, and protease inhibitor (Calbiochem). Two hundred micrograms of total cell lysates as starting amounts were mixed with specific antibodies and preincubated with agarose beads for 12 hours against the proteins of interest. After mixing, proteins coprecipitated with agarose beads were gently washed with cold lysis buffer excluding 0.5% Triton X-100 and 0.1% SDS three times and separated by SDS-PAGE. Proteins were detected by immunoblotting using specific antibodies against the proteins of interest. To determine the compound 1–mediated ubiquitination of DX2, the cells were treated with compound 1 and MG-132 (50 μM) for 12 hours. The cells were then lysed with 50 mM Tris-HCl (pH 7.4) buffer containing 100 mM NaCl, 0.5% Triton X-100, 1% SDS, 10% glycerol, 1 mM EDTA, protease inhibitor (Calbiochem), and the pan-deubiquitinase inhibitor PR-619 (Sigma-Aldrich), boiled at 100°C for 10 minutes, and then diluted in lysis buffer including 0.1% SDS (Crespo-Yàñez et al., 2018). The following experimental procedures were performed as described above.
Xenograft
H460 cells (1 × 107) were subcutaneously injected into the left and right sites of the backs of 7-week-old female BALB/cSLCnu/nu mice (Central Laboratory Animal Inc., Korea). Five or 10 mg/kg compound 1 was intraperitoneally administered to the mice (n = 3 mice per group) every other day for 10 days. The volumes of the embedded tumors and body weights were measured five times over the experimental period. After 10 days, all mice were sacrificed, and the embedded tumors were excised. The weights of the harvested tumors were measured, and photos of the whole-body embedded tumors and excised tumors were taken. To determine the level of the DX2 protein in the embedded tumors, the excised tumors were homogenized and lysed in PBS containing 1% Triton X-100, 0.1% SDS, and protease inhibitor (Calbiochem). Supernatants after centrifugation at 15,000 rpm at 4°C for 30 minutes were subjected to SDS-PAGE and immunoblotting using a specific antibody against DX2. Animal experiments were approved and performed in compliance with the University Animal Care and Use Committee guidelines at Yonsei University.
Three-Dimensional Culture
H460 cells in two-dimensional culture conditions were detached from the culture plate using Accumax (EMD Millipore). After cell counting using the disposable hemocytometer C-Chip (INCYTO), we diluted the cells to a concentration of 5000 cells per 100 µl in medium supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were then seeded onto ultralow attachment 96-well three-dimensional (3D) culture plates (Corning). After incubation for 48 hours, the spheroids and cancer cells cultivated under 3D culture conditions were treated with compound 1 for 72 hours, and cell viability was determined using a CellTiter-Glo 3D cell viability assay (Promega) following the manufacturer’s instructions. The experiment was independently repeated three times.
Anchorage-Independent Colony Formation Assay
To generate stable cells, we introduced FLAG-tagged DX2 wild type (WT) and mutants into A549 or H460 cells for cultivation in medium containing G418 (800 μg/ml, #G0175, Duchefa) for the selection of cells stably expressing the ectopically introduced plasmids. After culture for two weeks, the settled colonies were selected, and the level of overexpressed DX2 was determined by immunoblotting using an anti-FLAG antibody. Each of the stable cells was subjected to an anchorage-independent colony formation assay using a cell transformation assay kit (Cell Biolabs, Inc.) following the manufacturer’s instructions. The number of colonies stained by hematoxylin (Sigma) was counted. The experiment was independently repeated three times.
Cell Viability Assay
DX2-inducible or empty vector–inducible A549 isogenic cells (2 × 103 cells per well) were cultured in 96-well flat-bottom plates and pretreated with dox (Sigma) to induce DX2 expression. After pretreatment with dox for 24 hours, the cells were cultured in serum-free medium diluted with chemicals for 48 hours. To check the resistance vulnerability, H460 cells pretreated with compound 1 (0.5 μM) for three weeks were treated with chemical in a dose-dependent manner for 72 hours. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) solution (5 mg/ml, Sigma) was added to each well followed by incubation for 1.5 hours. After the medium was discarded, the precipitated formazan crystals were solubilized with DMSO (Duchefa), and the absorbance was measured at 560 nm using a microplate reader (Sunrise, Tecan). All of the experiments were repeated three times independently.
Refolding Assay
Using a Protein Refolding Kit (Boston Biochem) following the manufacturer's instructions, the effects of the compound on the protein refolding process were examined. Purified proteins HSP70 and heat shock protein 40 and Glow-Fold Substrate were mixed with compound 1 in reaction buffer containing Mg2+ATP, and all of the reagents were incubated for 5 minutes at room temperature. To unfold the substrate, the mixture was heated at 50°C for 8 minutes and left on ice for 10 minutes. The refolding reaction was conducted at 30°C for 1.5 hours, and the luminescence signal from the refolded substrate was measured with a plate reader (Glomax, Promega). Experiments were independently repeated three times.
Reverse Transcription PCR
Total RNA extracted from H460 cells using the RNeasy Mini Kit (Qiagen) was used for reverse transcription PCR with deoxynucleotide triphosphates, random hexamers, and Moloney murine leukemia virus. To determine the mRNA expression of DX2, AIMP2, and actin, we used 2 μl of cDNA in the PCR with the following specific primers: DX2, CTGGCCACGTGCAGGATTACGGGG and AAGTGAATCCCAGCTGATAG; AIMP2, ATGCCGATGTACCAGGTAAAG and CTTAAGGAGCTTGAGGGCCGT; actin, CCTTCCTGGGCATGGAGTCCT and GGAGCAATGATCTTGATCTT. Actin was used as a loading control.
Statistics
Statistical tests were performed with Prism (GraphPad). A value of P < 0.05 was considered statistically significant. All error bars represent S.D. For quantitative data, statistical parameters are reported in the figure legends.
Results
Selection and Characterization of a Chemical Probe
Our previous report demonstrated that DX2 is stabilized by its interaction with HSP70 and that inhibition of the interaction between these two proteins reduces DX2-mediated cancer proliferation (Lim et al., 2019). Therefore, we searched for an inhibitor of this interaction to suppress DX2-dependent cancer progression using a luciferase-based complementation assay system (Dixon et al., 2016) established for high-throughput screening (Fig. 1A). In a primary screening using 6186 compounds from KCB, 99 compounds showed over 60% inhibition at 5 μM (Fig. 1B, upper; Supplemental Fig. 1A). The specificity of these 99 compounds for DX2 was determined using the binding pair PRKACA and PRKAR2A (Dixon et al., 2016), and 10 compounds were found to specifically suppress the binding between DX2 and HSP70 but not PRKACA-PRKAR2A (Fig. 1, B and C). Further validation of the 10 compounds was carried out using in vitro pull-down and cell viability assays. An in vitro pull-down assay using the purified HSP70 and GST-DX2 proteins demonstrated that compound 1 significantly inhibited the interaction of these two proteins (Fig. 1D). Furthermore, compound 1 suppressed DX2-dependent cell proliferation in a DX2-inducible system (Fig. 1, B and E, bottom). Using this three-step screening procedure, we identified compound 1 as a potential PPI inhibitor of DX2 and HSP70 (Fig. 1F).

Identification of compound 1, a PPI inhibitor of DX2 and HSP70. (A) NanoBiT-based screening system. (B) Flowchart of the binding inhibitor screening. Ninety-nine chemicals showing over 60% inhibition at 5 μM were selected from the primary screening. To examine the specificity, we used the binding pair PRKACA and PRKAR2A as a secondary screening, and 10 chemicals were chosen. (C) Comparison of the inhibitory effects of the 10 chemicals (10 μM) on the binding of DX2-HSP70 and PRKACA-PRKAR2A via the NanoBiT assay. (D) Inhibitory efficacy of the chemicals on the direct binding of DX2 and HSP70 determined by an in vitro pull-down assay. Quantified values of binding are presented at the bottom of the gel image. (E) Cell viability was determined upon treatment with the 10 chemicals (10 μM) in DX2-inducible A549 cells. DX2 was induced by treatment with dox. (F) Structure of compound 1. The experiments in (C) and (E) were independently repeated three times with error bars denoting S.D. Student’s two-tailed t test was performed for statistical analysis. *P < 0.05; **P < 0.01; ***P < 0.005. EV, empty vector.
Additionally, the usefulness of compound 1 as a starting point for the optimization of the discovery of an anticancer agent was validated through the examination of preliminary structure-activity relationships on a series of 8 compounds in KCB libraries (Table 1). There were eight compounds (2–9) with enedione moieties in the KCB whose biologic activities on DX2 were evaluated at 5 μM. Compounds 2–4 substituted at the 3 or 4 positions of the R1 benzene ring showed 66%, 76%, and 53% inhibition, respectively. Replacement of the ester at R2 with amide 5 (56% inhibition), arylketo 8 (58% inhibition), and alkylketo 9 (58%) retained the compound’s ability to inhibit the interaction between DX2 and HSP70. Additionally, the introduction of biaryl groups (compounds 8 and 9) at R3 was tolerable for activity, and a variety of variations at R3 might be possible. Additionally, compound 1 can form a covalent bond with DX2 by a nucleophilic addition. Then, we examined compounds 10 and 11 without the site required for nucleophilic addition among the KCB libraries, resulting in retention of the inhibitory activities (Table 2). Based on this preliminary structure-activity relationship study, we synthesized compound 1 and its biotinylated derivative compound 15 by conventional procedures (Miyatake-Ondozabal and Barrett, 2013; Sun et al., 2009; Ackerman et al., 2018) (Fig. 2) for biologic evaluation, including dose dependency, functional assays in cell lines, and studies on the mode of action. Because compound 4 with a (4-phenylpropoxy)benzyl group at R1 was active (53% inhibition) in the screening, we primarily attempted to prepare compound 15, where the 4-position of the benzene ring at R1 is biotinylated. Fortunately, compound 15 showed similar activity to compound 1 and was used to study the mode of action (Supplemental Fig. 3, A and B). In summary, compounds with three or four substituents on the benzoyl moiety retained activity, and the introduction of a biaryl group at C3 instead of a benzene group was tolerable for efficacy. The replacement of the ester group with a more metabolically stable keto or amide group also maintained activity, suggesting that the structure of compound 1 can be broadly modified to improve efficacy and safety and should be useful as a chemical probe to elucidate the mode of action on DX2.

Preparation of the biotinylated compound 15 reagents and conditions: (a) BBr3 2 equiv, CH2Cl2, −15°C, 1.5 hours, 82%; (b) piperidine 0.5 equiv, benzaldehyde 1.2 equiv, toluene, reflux, 43 and 25%: (c) (COCl)2 1 equiv, cat. dimethylformamide, CH2Cl2, 0°C; (d) pyridine 1 equiv, CH2Cl2, room temperature, overnight, 10%.
We examined the inhibitory activity of compound 1 for further validation as a chemical probe (Fig. 3). The IC50 value of compound 1 was determined to be 2.9 μM for inhibiting the binding between DX2 and HSP70, but the IC50 was over 100 μM for the inhibition of PRKACA and PRKAR2A binding (Fig. 3A), indicating that compound 1 specifically interrupted the binding between DX2 and HSP70. Next, time-dependent binding inhibition of compound 1 was also confirmed (Supplemental Fig. 2A). Compound 1 did not show time-dependent inhibitory activity on the binding between DX2 and HSP70, which indirectly indicates that inhibition of DX2 by compound 1 is not based on covalent modification. To exclude the possibility of artifacts of our screening system, we determined the effects of compound 1 in a complementary fluorescence-based assay, and the same result was observed (Supplemental Fig. 2B). The endogenous interaction of the two proteins was found to be abrogated by treatment with compound 1 by reciprocal immunoprecipitation, implying its physiologic function (Fig. 3B). We also examined whether compound 1 suppresses the direct binding of these two proteins in a dose-dependent manner via a pull-down assay; similar to the other assays, we observed dose-dependent inhibition of their direct binding (Fig. 3C). Additionally, we tested whether depletion of compound 1 induces the binding of DX2 and HSP70. The compound 1–mediated decrease in the binding of the two proteins was recovered by adding fresh medium (Fig. 3D), indicating that compound 1 inhibits the binding of the two proteins. Together, these observations indicate that compound 1 specifically interferes with the binding of DX2 with HSP70.

Characterization of compound 1. (A) NanoBiT assay for the binding between DX2 and HSP70 and the binding between PRKACA and PRKAR2A upon treatment with compound 1. The experiments were independently repeated three times with error bars denoting the S.D. Student’s two-tailed t test was performed for statistical analysis (***P < 0.005). (B) Inhibitory effects of compound 1 on the endogenous binding of DX2 with HSP70. HSP70 (left) or DX2 (right) from the same cells were precipitated with a specific antibody. Actin was used as a loading control. (C) In vitro pull-down assay showing the inhibitory effects of compound 1 on the interaction between DX2 and HSP70. Quantified values of binding are presented at the bottom of the gel image. (D) H460 cells were treated with compound 1, and then the chemicals were removed by addition of fresh medium at the indicated time. The cells were precipitated by using an anti-HSP70 antibody. (E) Protein and mRNA levels from H460 cells treated with compound 1. (F) Ubiquitination assay of H460 cells treated with compound 1. (G) Significance of Siah1 on HSP70- or compound 1–dependent DX2 levels. H460 cells knocked down by introducing specific Siah1-specific siRNA (si-Siah1) were ectopically expressed with HSP70 (upper) or treated with compound 1 (bottom). The quantification of the protein level for each experiment is presented at the bottom of each blot. EV, empty vector; IP, immunoprecipitation; WCL, whole cell lysates; Ub, ubiquitin.
Because inhibition of the interaction between DX2 and HSP70 has been reported to lead to the turnover of DX2 via ubiquitination (Lim et al., 2019), we examined whether compound 1 destabilizes the DX2 protein. First, we monitored the protein and mRNA levels of DX2 upon treatment with different doses of compound 1 and found that compound 1 specifically reduced the DX2 protein level but not the mRNA level (Fig. 3E). We also observed increased ubiquitination of DX2 upon treatment with compound 1, which was found to be mediated by its increased binding to Siah1, an E3 ligase against DX2 (Lim et al., 2019), and decreased binding to HSP70 (Fig. 3F), implying that compound 1 induces the binding of Siah1 to DX2 by interfering with HSP70 access, resulting in DX2 turnover. The significance of Siah1 on compound 1–mediated degradation of DX2 was also investigated by knockdown of Siah1 via its specific small interfering RNA (siRNA). When Siah1 was knocked down, the level of DX2 basically increased without HSP70 expression, and there was no decrease in DX2 despite treatment with compound 1 (Fig. 3G), suggesting that Siah1 is critical for the compound 1–dependent decrease in DX2. We further validated that compound 1 did not affect the protein level of AIMP2 or other aminoacyl–transfer RNA synthetases, which are AIMP2-interacting proteins in the MSC (Kim et al., 2011) (Fig. 3E; Supplemental Fig. 2C, upper). We also determined the level of CDK4, a known downstream molecule of HSP70 (Lim et al., 2019), and the protein-folding activity of HSP70 upon treatment of compound 1 to elucidate the possibility that compound 1 could inhibit the interaction of DX2 and HSP70 based on the interaction with HSP70. We confirmed that there was no effect on the level of CDK4 or on the folding activity of HSP70 (Supplemental Fig. 2, C, bottom, and D), suggesting the PPI inhibition based on the interaction with DX2, not on the interaction with HSP70 of compound 1. Altogether, these data led to the conclusion that compound 1 inhibited the interaction between DX2 and HSP70, resulting in DX2 degradation via recruitment of Siah1.
Mode of Action Study of Compound 1
To unveil the mode of action of compound 1, we used biotin-conjugated compound 15. First, we compared the function of compound 15 with that of the original compound 1. Treatment of H460 cells with compound 15, similar to compound 1, reduced the binding of HSP70 to DX2 and destabilized the DX2 protein (Supplemental Fig. 3, A and B), suggesting that the conjugated biotin compound retained its function. We then examined whether compound 15 bound directly to DX2 or HSP70 using an in vitro pull-down assay and observed direct binding between the compound and DX2, not HSP70 (Fig. 4A). We further performed a competitive binding assay using compounds 15 and 1 to determine the specific binding of the chemical to DX2 or HSP70. The DX2 protein, not HSP70, was pulled down by compound 15, and the amount of coprecipitated DX2 was reduced by the addition of compound 1, implying the specific binding of compound 1 to DX2 and not HSP70 (Fig. 4B). We also confirmed the competitive binding of compounds 1 and 15 to endogenous DX2 proteins as above via the treatment of H460 cells with the two compounds (Fig. 4C). Next, we measured the binding affinity of compound 1 to DX2 via a surface plasmon resonance assay and determined a KD value of 12 μM (Fig. 4D), which was similar to the IC50 value obtained for the inhibition of the interaction between DX2 and HSP70 (Fig. 3A), implying the significance of the binding of the compound to DX2 for inhibition of the DX2-HSP70 interaction. We further confirmed the binding affinity via a fluorescence-based equilibrium binding experiment (Breen et al., 2016) and obtained a binding affinity similar to that stated above (Supplemental Fig. 3C). The results from two other binding assays suggested that direct binding of compound 1 to DX2 could affect protein function. We also determined which domain of DX2 bound to compound 1. DX2 was divided into the NFR, GST domain (GST), and C terminus of the GST domain (Lim, S. et al., 2019) (Fig. 4E, right), and each fragment was mixed with compound 1. The in vitro pull-down assay using streptavidin-Sepharose beads revealed that the NFR and GST-N, a region known for binding with HSP70 (Lim et al., 2019), were bound by compound 1 (Fig. 4E, left), further validating compound 1 as a PPI inhibitor. All of the binding data imply that compound 1 inhibits the interaction between DX2 and HSP70 via direct binding with DX2.
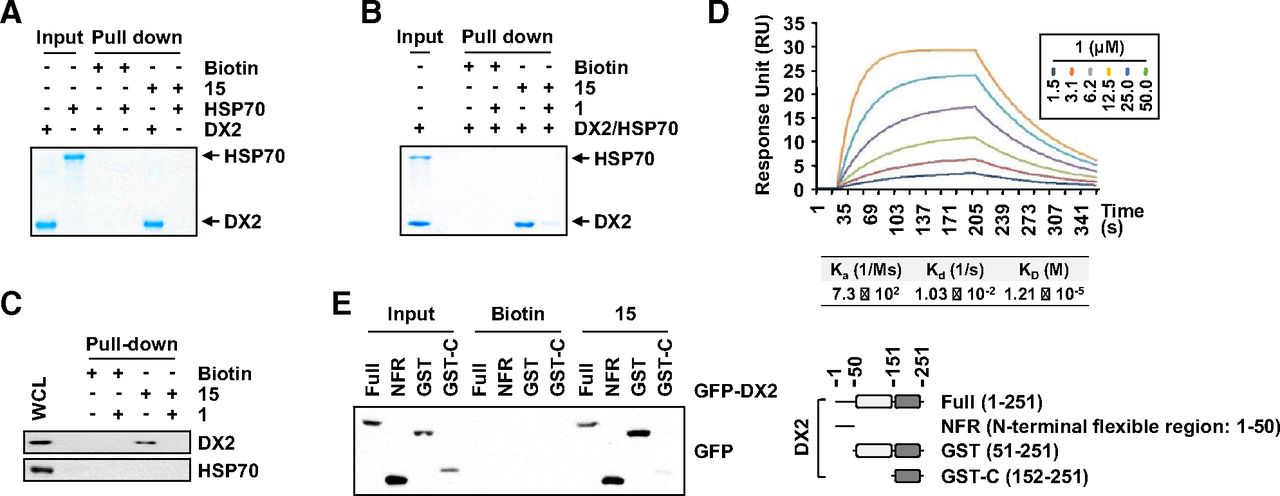
Direct binding of compound 1 to DX2. (A) Direct binding of compound 1 to DX2. The purified DX2 or HSP70 proteins were mixed with compound 15 or biotin. Biotin was used as a negative control. (B) Specific binding of compound 1 to DX2, not HSP70. The proteins DX2 and HSP70 were incubated with compound 15, and compound 1 was additionally added. (C) Specific binding of compound 1 to endogenous DX2. H460 cells were treated with biotin, compound 15, or compound 1 as indicated and analyzed by immunoblotting. (D) Surface plasmon resonance analysis for calculating the binding affinity between DX2 and compound 1. (E) Determination of the domain responsible for the binding between compound 1 and DX2. The DX2 protein was divided as shown on the right. GST-C, C terminus of the GST domain; RU, response unit; WCL, whole cell lysates.
To investigate the binding mode of compound 1 with DX2, we performed a molecular modeling study because the disordered NFR was not visible in other structural assays (Lim et al., 2019). Due to the disordered nature of the NFR, the replicated run of 300 ns MD simulations was conducted three times. The initial pose of compound 1 for MD simulation with the highest docking score of 34.94 was obtained from molecular docking simulation using CDOCKER. To increase the chance of determining the binding event and to properly adjust the position of compound 1 to fit into the GST-N, we performed MD simulations with the upper-wall restraint force. Because ligand binding is a rare event in the simulation considering the protein flexibility in the solvent system, this binding event was observed in only one trajectory among three replicated systems with the upper wall. From this trajectory, we found that compound 1 sought the proper hydrophobic fit into the GST-N for the beginning 100 ns (Fig. 5A). The DX2 was well stabilized at around 0.45 nm of Cα-root mean square deviation (Supplemental Fig. 4). Although the Cα-root mean square deviation values are relatively fluctuating during beginning 200 ns, the binding conformation of compound 1 was well maintained in the GST-N for the last 200 ns, showing stable interactions with three significant residues, Y47, N56, and K129 (Fig. 5B). Cα-RMSF on DX2 represented the significant conformational change of the N-terminal region during the beginning 200 ns simulation time. Not only the most residues in binding region but also the N-terminal region, which is generally fluctuating, seemed to be very stable during the last 100 ns (Supplemental Fig. 4B). The snapshot at 211.3 ns, having the lowest nonbonded energy (Supplemental Fig. 4C) between DX2 and a highly populated conformer of compound 1 during the last 200 ns (Supplemental Fig. 4A, inset), was selected as a representative structure for further interaction analysis. For the energetics of the binding mode between compound 1 and DX2, we calculated the total binding energy using the molecular mechanics Poisson-Boltzmann surface area method. The binding structure was stabilized from initial −21.33 kJ/mol to an average −37.21 ± 13.08 kJ/mol of total binding energy over the last 100 ns (Supplemental Fig. 4C). The energy of representative structure at 211.3 ns was −46.31 kJ/mol. To validate the important amino acids in the protein-ligand interaction, per-residue energy decomposition analysis was performed with the 300 ns simulation trajectory. From this analysis, we found that the three key residues (Y47, N56, and K129) are located at the lowest nonbonded interaction energies (Supplemental Fig. 4D), which is well correlated with the mutation analysis. The carbonyl groups of the benzoyl and ester moieties on compound 1 interact with the side chain amines of N56 and K129, respectively, by H-bonding (Supplemental Fig. 4B). There was a π-π interaction between the styrene of compound 1 with Y47 of DX2. To validate the MD simulation results, we generated alanine mutants of the DX2 amino acids that were suggested to be critical for the binding of compound 1. First, we examined the binding of the alanine mutants to the chemicals using compound 15 via an in vitro pull-down assay. DX2 WT and most of the mutants tested showed strong binding, but the binding of the DX2 mutants Y47A, N56A, and K129A to compound 15 was significantly reduced (Fig. 5C). Next, we determined whether compound 1 diminished the binding between HSP70 and DX2. Based on the immunoprecipitation results using the expressed mutants in 293T cells, we determined that compound 1 abrogated the interaction between HSP70 and DX2 WT but not Y47A, N56A, or K129A, as described above (Fig. 5D). Because Y47, N56, and K129 are critical residues of DX2 for binding with compound 1, we tested the compound 1–mediated ubiquitination of the selected DX2 mutants. From the ubiquitination assay, we observed that DX2 WT was ubiquitinated upon treatment with compound 1, resulting in a decreased level of DX2, but DX2 ubiquitination and DX2 protein expression of the tested mutants were not affected by treatment with compound 1 (Fig. 5E). Together, these observations indicate that the pocket surrounded by Y47, N56, and K129 in the NFR and GST-N of DX2 is critical for binding with compound 1.

Prediction and validation of the binding site of compound 1. (A) Distance distribution of compound 1 with three significant residues displaying stable interactions during the last 200 ns. The blue, magenta and green lines represent the distances of ligand:O3, ligand:C13, and ligand:O1 toward K129:NZ, Y47:CG, and N56:ND2, respectively. Compound 1 is represented as green sticks. (B) Binding mode of compound 1 with DX2 obtained from the molecular modeling study. The electrostatic surface model of DX2 taken from the representative structure at 211.3 ns (right) and the zoomed-in view showing the detailed interactions with compound 1 (green) with the three significant binding residues (red) shown by the stick model (left). (C) Binding of compound 1 to DX2 via mutagenesis studies. Cell extracts expressing each of the FLAG-tagged DX2 mutants were mixed with compound 15 and subjected to an in vitro pull-down assay. PD denotes pull-down. (D) DX2-HSP70 binding inhibition of compound 1 via mutagenesis studies. 293T cells expressing FLAG-tagged DX2 were incubated with or without compound 1 for 6 hours and then subjected to immunoprecipitation. Actin was used as a loading control. The quantification of the protein level for each experiment is presented as a bar graph on the right of (C) and (D). (E) Ubiquitination assay using DX2 mutants. The amounts of ubiquitinated DX2 were assessed by SDS-PAGE and immunoblotting using an anti-ubiquitin (Ub) antibody. The quantification of the protein level for each experiment is presented as a bar graph on the right of (C) and (D). IP, immunoprecipitation; WCL, whole cell lysates.
The NFR was shown to be involved in the binding of compound 1 to the GST-N of DX2 (Fig. 6; Supplemental Movies 1 and 2). Among the three significant interacting residues, Y47 is located in the NFR and has a stable hydrophobic interaction with compound 1. Moreover, a stable hydrophobic interaction between L50 and T117 was shown in the chemically bound system but not in the apo system (Fig. 6A), indicating that the disordered NFR containing L50 has no chance to interact with the GST-N containing T117 without compound 1 binding. Interestingly, along with being involved in the binding of compound 1, the NFR binds with the surface of DX2 through intramolecular hydrophobic interactions, meaning that the binding of the NFR to GST-N could mask the L97 and T117 residues, which are significant residues for the binding to HSP70 (Lim et al., 2019). In particular, hydrophobic interactions of G40, Y47, and L50 in the NFR with L97 and T117 on the surface of GST-N were observed in the representative structure of the system (Fig. 6B). The induced binding of the NFR and GST-N in the presence of compound 1 seemed to significantly increase the area of the bumping region to interfere with the interaction between DX2 and HSP70 (Fig. 6C). These analyses revealed that the interaction of compound 1 in the pocket of DX2 surrounding Y47, N56, and K129 induces a steric clash against HSP70 by direct binding interference and a conformational change in DX2, resulting in turnover of the DX2 protein.
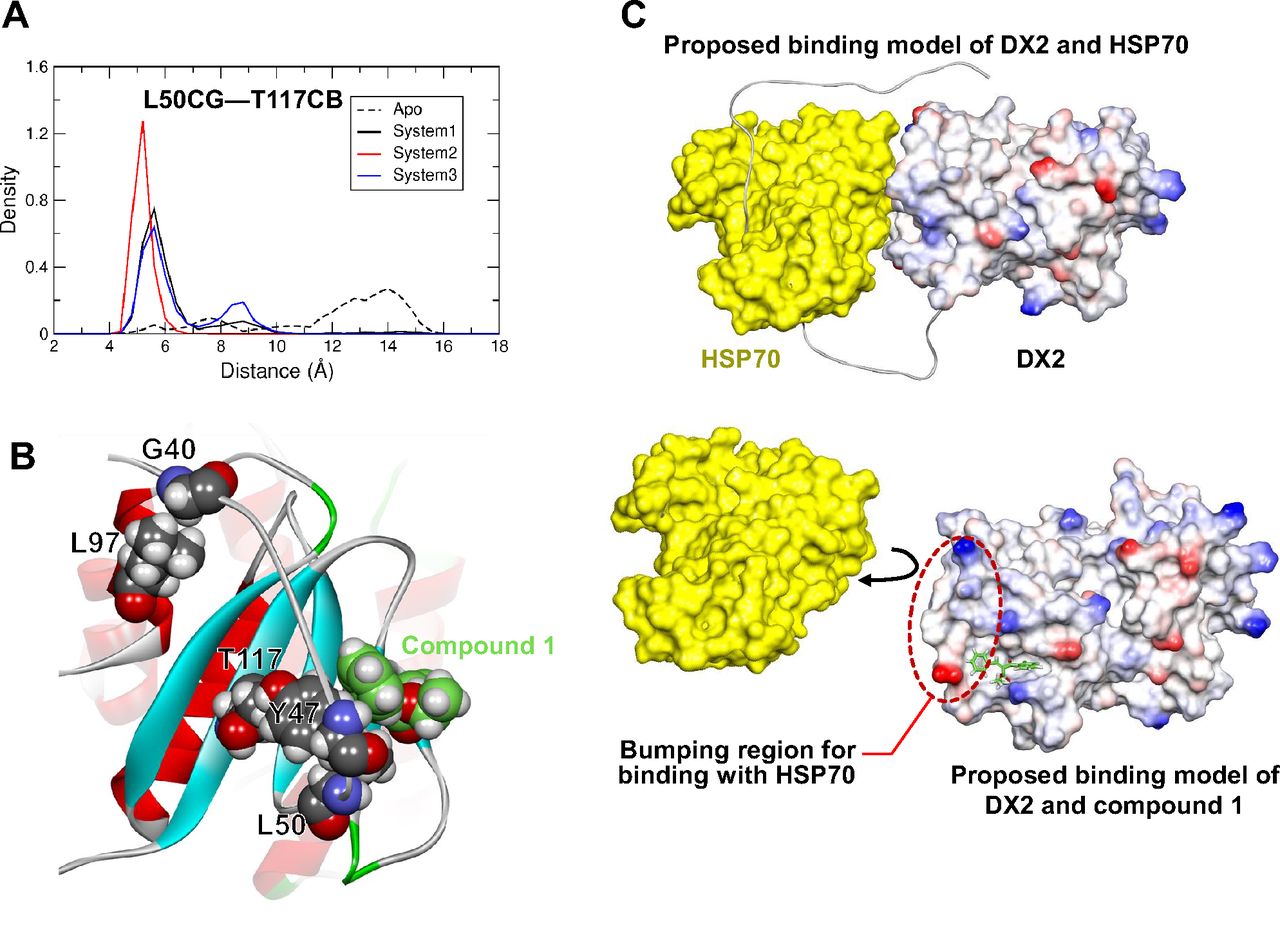
Determination of the mode of action of compound 1. (A) Distance distribution of L50 in the NFR and T117 in the GST-N of DX2. The red and blue lines indicate the results obtained from the apo and chemical-bound systems, respectively. (B) The representative structure of chemically bound DX2 showing that the binding region with HSP70 can be masked by significant hydrophobic interactions. The key residues of DX2 (gray) and compound 1 (green) are represented as the van der Waals model. (C) The proposed binding models of DX2 and HSP70 (upper) and DX2 and compound 1 (bottom). The DX2-NFR (residues 1–50) is depicted as a line in the absence of compound 1 (upper), but the NFR (residues 38–50) is represented as a surface to highlight the bumping region in the presence of compound 1 (bottom). The structures of DX2 and HSP70 are shown as electrostatic and yellow surfaces, respectively.
Inhibition of Cancer Development via Compound 1
Because compound 1 leads to the degradation of DX2 via inhibition of binding with HSP70, we examined whether compound 1 suppresses cancer cell proliferation. H460 cells showing a high level of DX2 were treated with compound 1, and cell viability was shown to decline in a dose-dependent manner (Fig. 7A, red circle). We further determined whether compound 1–mediated suppression of cell viability was dependent on the level of DX2. After examining the levels of DX2 and HSP70 in various lung cells (Supplemental Fig. 5A), the cells were subjected to a cell viability assay upon treatment with compound 1, and the 50% inhibition concentration (EC50) of cell proliferation was calculated. The cells with the highest levels of DX2, H460 and H358, showed the lowest EC50 values among the tested cell lines (Fig. 7A, red), and the EC50 values were very similar to the IC50 and KD previously determined for the inhibition of binding between DX2-HSP70, implying that suppression of the interaction between DX2 and HSP70 using compound 1 leads to cell death. Normal (WI-26) and cancerous (H1650) lung cells, those with the lowest level of DX2, were not affected by compound 1 (Fig. 7A, yellow), further suggesting that DX2 is required for compound 1 to affect cell death. The significance of the DX2 level on compound 1–mediated suppression of cell proliferation was also determined in DX2-knockdown H460 and H358 cells by introducing specific siRNA (Supplemental Fig. 5B). A time-dependent inhibitory effect of cancer cell proliferation via compound 1 was also confirmed (Supplemental Fig. 5C). Before determining the in vivo efficacy of this compound, we treated H460 cells with compound 1 in a 3D cell culture system. Compound 1 dose-dependently decreased the proliferation of H460 spheroids in the 3D environment (Fig. 7B), implying that compound 1 could function in an in vivo model. The IC50 of compound 1 in 3D culture was much higher than that in the two-dimensional system, which might be due to the poor penetration of compound into the spheroids as mentioned in publications (Park et al., 2016; Langhans, 2018). We further validated the effects of compound 1 on cancer cell progression in vivo via a xenograft model using H460 cells. Compound 1 was intraperitoneally injected at doses of 5 and 10 mg/kg, and tumor growth and body weight were monitored over the experimental periods. Even though these data are preliminary due to small number of mice per each group, administration of compound 1 significantly reduced tumor size in a dose-dependent manner with little effect on body weight (Fig. 7C, left; Supplemental Fig. 5D). A reduction in tumor weight was also observed (Fig. 7C, middle). We further validated the endogenous levels of the DX2 and HSP70 proteins in the excised tumors from the mice. The level of DX2, but not HSP70, was found to be decreased after injection of compound 1, and the quantified level is shown as a graph excluding the confusion from object variation (Supplemental Fig. 5E), implying that treatment with compound 1 led to inhibition of cancer proliferation via degradation of the DX2 protein.

Inhibitory effects of compound 1 on cancer cell proliferation. (A) Cell viability assay on various normal and cancerous lung cells. Each color indicates the different protein levels of DX2 (red: high, blue and green: median, yellow: low) (Supplemental Fig. 5A). (B) Inhibition of H460 cell spheroids in 3D culture conditions upon treatment with compound 1 (96 hours). Cell viability was determined by a CellTiter-Glo 3D assay. (C) The effect of compound 1 on the in vivo cancer development model. A total of 5 or 10 mg/kg (mpk) compound 1 was intraperitoneally administered for 10 days every other day to mice (n = 3) subcutaneously embedded with H460 cells. Tumor size was monitored for the injection period (left). The weights of the excised tumors are shown as a bar graph (middle). Representative images of mice bearing tumors and the excised tumors are shown (right). (D) Cell viability assays using A549 cells stably expressing the indicated DX2 mutants treated with compound 1. Red and blue colors indicate WT and mutants of DX2, respectively. (E) Anchorage-independent colony-forming assay using H460 cells stably expressing DX2 WT or mutants treated with compound 1. Representative images are shown on the right. The experiments were independently repeated three times with error bars denoting the S.D. in (A), (B), (D), and (E). Student’s two-tailed t test was performed for statistical analysis. *P < 0.05; **P < 0.01; ***P < 0.005. EV, empty vector.
To unveil the dependency of the effects of compound 1 on its binding with DX2, we generated A549 cell lines stably expressing DX2 WT or binding-defective mutants Y47A, N56A, and K129A and treated these cells with various doses of compound 1. The EC50 value of these A549 stable cells expressing DX2 WT (0.92 μM) was very similar to the EC50 found in H460 cells (0.97 μM) (Fig. 7D, red; Fig. 7A), emphasizing the significance of the DX2 level on the function of compound 1. However, there was no effect from compound 1 in cell lines expressing DX2 binding-defective mutants (Fig. 7D, blue), suggesting that the decreased cell viability induced by treatment with compound 1 is mediated by its binding with DX2. We further examined the significance of the binding of compound 1 to DX2 via an anchorage-independent colony-forming assay. As expected, the number of colonies and the efficacy of compound 1 both increased in cells stably expressing DX2 WT but not in the mutant cell lines (Fig. 7E). The above two assays showed that abrogation of the binding between DX2 and compound 1 prevented the chemical-mediated inhibition of cancer cell development, leading us to the conclusion that the binding of compound 1 to DX2 is critical for the function of compound 1. To examine the resistance vulnerability of compound 1, we determined the EC50 of compound 1 in H460 cells pretreated with chemicals for three weeks and compared it with that of native H460 cells. A small decrease in EC50 was observed, but compound 1 efficiently decreased cell viability even though it was used as a pretreatment (Supplemental Fig. 5F), meaning that compound 1 could also function for a long time.
Discussion
DX2 is considered a novel target for cancer therapeutics because of its oncogenicity, and specific siRNA and small molecules targeting DX2 have been studied as therapeutic strategies for DX2-expressing tumors (Choi et al., 2011; Lee et al., 2013). Despite many trials, none of the examined tools have been shown to efficiently target DX2. Recently, HSP70 was shown to significantly affect the protein level of DX2 by blocking the access of Siah1, a specific E3 ligase, leading to the suggestion that a PPI inhibitor between DX2 and HSP70 may be a good tool to target DX2 (Lim et al., 2019). Here, we suggest compound 1 as a novel PPI inhibitor for the binding of DX2 and HSP70 and fully address the functional mode of action of this compound. We identified compound 1 as a hit compound for a PPI inhibitor targeting the interaction of DX2 and HSP70 using a luciferase-based complementation system (NanoBiT) (Lim et al., 2019). Compound 1 has the possibility of forming a covalent bond with DX2 by nucleophilic addition. Then, we examined compound 13 and compound 14 [5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-day]imidazol-4-yl)pentanoic acid] without the site for nucleophilic addition, resulting in retention of the inhibitory activity (Table 2). Additionally, compound 1 did not show time-dependent inhibitory activity on the binding of DX2 and HSP70 (Supplemental Fig. 2A). These results indirectly indicate that inhibition of DX2 by compound 1 is not based on covalent modification. Compound 1 was found to inhibit and induce the binding of HSP70 and Siah1 to DX2, respectively, resulting in ubiquitination-dependent degradation of DX2. Furthermore, compound 1 was shown to reduce cancer cell development in vitro and in preliminary in vivo experiment in a DX2-dependent manner. We further analyzed the mode of action of compound 1. The binding of compound 1 to DX2 is important for DX2 cellular function, and DX2 residues Y47, N56 and K129 appeared to be significant for binding via MD simulation and mutational analysis. Two modes of action of compound 1 were proposed: allosteric modulation of the DX2 structural conformation and direct steric clashing of DX2-HSP70 binding.
Because a structural understanding of target proteins is critical for drug discovery, many target proteins have been analyzed using techniques such as X-ray crystallography and NMR analysis. However, assessing the structure of the full-length protein via the above traditional analyses is difficult because many proteins have an unstructured flexible region. This flexible region has been reported to be significant for protein function (Babu, 2016); therefore, there have been many efforts to overcome this limitation. DX2 has a disordered NFR consisting of 50 amino acids. To determine whether the mode of action of compound 1 was associated with the NFR, we conducted MD simulations. The MD simulations suggested that compound 1 binds the pocket surrounding the NFR (Y47) and GST-N (N56 and K129), which was indirectly confirmed via mutagenesis experiments. Through the MD study, we also unveiled the mode of action of compound 1, which directly interferes with access to HSP70 and masks the HSP70 binding surface of DX2 via a hydrophobic interaction between the NFR and the GST-N binding site in the bound state of compound 1. From these results, we demonstrated that the NFR and GST-N might be critical for the binding of DX2 to HSP70, further supporting the described mode of action of compound 1. Although some DX2 exists in its free form, the possibility that DX2 could be constitutively bound to HSP70 still exists. Therefore, structural analysis with compound 1 via MD simulations needs to be considered with HSP70 bound to DX2 for competitive binding, and this will be our further study.
Many of the 650,000 PPIs estimated from proteomics tools have recently been reported to be oncogenic (Ran and Gestwicki, 2018; Li et al., 2017). Therefore, PPIs have been considered to be a significant therapeutic target, especially for cancer, and many trials have sought to discover PPI inhibitors that interfere with oncogenic PPIs (Nero et al., 2014; Ivanov et al., 2013). The discovery of small molecule PPI inhibitors is now a research focus because of the importance of PPIs in cancer. Here, we identified compound 1, with a molecular weight of less than 500 Da, as a PPI inhibitor that efficiently suppresses the oncogenic binding between DX2 and HSP70. Our results showing PPI inhibition using a small molecule with a molecular weight of less than 500 Da could provide new encouragement to continue the search for the discovery of PPI inhibitors using small molecules, which exhibit fewer problems in the clinical setting than large molecules. Cancer patients suffer from the side effects of therapeutic agents against cancer. Treatment with compound 1 did not affect normal cell proliferation (Supplemental Table 1) or the body weights of the tested mice, suggesting that compound 1 has no or only small side effects. This advantage may be due to the PPI inhibition mode of action; specific inhibition of the interaction between DX2 and HSP70 led cancer cell regression without affecting normal cells. We also evaluated metabolic stability of compound 1. The remaining amount of compound 1 was determined using liquid chromatography–tandem mass spectrometry analysis (mass spectroscope: Agilent6460; high pressure liquid chromatograph: Agilent1261) upon incubation with rat and human liver microsomes for 30 minutes, which provided 8.76% and 7.02%, respectively (Supplemental Table 2). The metabolic stability of compound 1 is very poor, and further optimization is needed to improve its physicochemical properties as well efficacy for the discovery of a sustainable lead on antitumor agent based on the inhibition of DX2 and HSP70 interaction.
Conclusion
We identified compound 1 to inhibit the tumor-promoting protein-protein interaction between DX2 and HSP70 through allosteric modulation as well as competitive binding. We performed MD simulations, mutagenesis analyses, and characterization of biologic function using compound 1 and its biotin-conjugated chemical 15 to elucidate the mode of action of compound 1. Compound 1 seems to induce allosteric conformational changes in the DX2 protein and direct binding clashes between DX2 and HSP70. Compound 1 reduced the DX2 protein level in a ubiquitin-dependent manner via suppression of the binding between DX2 and HSP70 and suppressed the growth of cancer cells highly expressing DX2 in vitro and in preliminary in vivo experiment. This paper presents a novel chemical route to interfere with oncogenic PPIs.
Authorship Contributions
Participated in research design: D.G. Kim, Huddar, Lim, Y. Lee, Park, K. Lee, S. Lee, S. Kim.
Conducted experiments: D.G. Kim, Huddar, Lim, Kong, Park, S. Lee, M. Kim.
Contributed new reagents or analytical tools: D.G. Kim, Lim, Park.
Performed data analysis: D.G. Kim, Huddar, Y. Lee, Park, Suh, K. Lee, S. Lee, S. Kim.
Wrote or contributed to the writing of the manuscript: D.G. Kim, Huddar, Y. Lee, Park, S. Lee, S. Kim.
Footnotes
- Received May 27, 2021.
- Accepted September 1, 2021.
↵1 D.G.K. and S.H. contributed equally to this work.
This work was supported by Global Frontier Project grants [NRF-M3A6A4-2010-0029785 and NRF-2013M3A6A4944802], an IMRCT R grant [NRF-2018R1A5A2023127], NRF-2021R1A3B1076605 of the National Research Foundation of Korea (NRF), SI-1951-30 funded by the Ministry of Science and ICT (MSIT), and the Yonsei University Research Fund [2020-22-0356].
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AIMP2
- aminoacyl–transfer RNA synthetase-interacting multifunctional protein 2
- AIMP2-DX2 (DX2)
- exon 2–depleted splice variant of aminoacyl–transfer RNA synthetase-interacting multifunctional protein 2
- BiFC
- biomolecular fluorescence complement
- CDCl3
- deuterated chloroform
- compound 1
- ethyl (Z)-2-benzoyl-3-phenylacrylate
- compound 13
- ethyl (Z)-2-(4-hydroxybenzoyl)-3-phenylacrylate
- compound 15
- 4-((Z)-2-(ethoxycarbonyl)-3-phenylacryloyl)phenyl 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoate
- 3D
- three-dimensional
- dox
- doxycycline
- GST
- glutathione S-transferase
- GST-N
- N-terminal subdomain of glutathione S-transferase
- HRMS
- high-resolution mass spectrometry
- HSP70
- heat shock protein 70
- KCB
- Korea Chemical Bank
- LC-MS
- liquid chromatography–mass spectrometry
- MD
- molecular dynamics
- MSC
- multisynthetase complex
- NFR
- N-terminal flexible region
- PPI
- protein-protein interaction
- PRKACA
- protein kinase A catalytic subunit
- PRKAR2A
- protein kinase A type 2A regulatory subunit
- siRNA
- small interfering RNA
- TLC
- thin layer chromatography
- WT
- wild type
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}