


Abstract
Acetyl-CoA carboxylase (ACC) 1 and ACC2 are essential rate-limiting enzymes that synthesize malonyl-CoA (M-CoA) from acetyl-CoA. ACC1 is predominantly expressed in lipogenic tissues and regulates the de novo lipogenesis flux. It is upregulated in the liver of patients with nonalcoholic fatty liver disease (NAFLD), which ultimately leads to the formation of fatty liver. Therefore, selective ACC1 inhibitors may prevent the pathophysiology of NAFLD and nonalcoholic steatohepatitis (NASH) by reducing hepatic fat, inflammation, and fibrosis. Many studies have suggested ACC1/2 dual inhibitors for treating NAFLD/NASH; however, reports on selective ACC1 inhibitors are lacking. In this study, we investigated the effects of compound-1, a selective ACC1 inhibitor for treating NAFLD/NASH, using preclinical in vitro and in vivo models. Compound-1 reduced M-CoA content and inhibited the incorporation of [14C] acetate into fatty acids in HepG2 cells. Additionally, it reduced hepatic M-CoA content and inhibited de novo lipogenesis in C57BL/6J mice after a single dose. Furthermore, compound-1 treatment of 8 weeks in Western diet–fed melanocortin 4 receptor knockout mice—NAFLD/NASH mouse model—improved liver hypertrophy and reduced hepatic triglyceride content. The reduction of hepatic M-CoA by the selective ACC1 inhibitor was highly correlated with the reduction in hepatic steatosis and fibrosis. These findings support further investigations of the use of this ACC1 inhibitor as a new treatment of NFLD/NASH.
SIGNIFICANCE STATEMENT This is the first study to demonstrate that a novel selective inhibitor of acetyl-CoA carboxylase (ACC) 1 has anti–nonalcoholic fatty liver disease (NAFLD) and anti–nonalcoholic steatohepatitis (NASH) effects in preclinical models. Treatment with this compound significantly improved hepatic steatosis and fibrosis in a mouse model. These findings support the use of this ACC1 inhibitor as a new treatment for NAFLD/NASH.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a liver disease characterized by excessive fat accumulation in hepatocytes that is not caused by alcohol consumption (Vernon et al., 2011; Friedman et al., 2018). Nonalcoholic steatohepatitis (NASH) is a subcategory of NAFLD and is defined on the basis of the following liver biopsy histologic features: lobular inflammation, hepatocellular ballooning, fibrosis, and steatosis (Williams et al., 2011; Siddiqui et al., 2018). Since NAFLD and NASH are associated with cirrhosis and hepatocellular carcinoma (Anstee et al., 2013), they also represent important factors that contribute to the recent increase in liver-related morbidity and mortality.
The global prevalence of NAFLD has continued to increase annually with 25% of the world population affected in 2018 (Younossi et al., 2019). Although up to 30% of patients with NAFLD develop NASH (Calzadilla Bertot and Adams, 2016), no anti-NAFLD/anti-NASH drugs have been approved (Sanyal et al., 2010; Cusi et al., 2016; Esler and Bence, 2019), thus resulting in an unmet medical need. Hepatic steatosis is caused by an imbalance in hepatic lipid metabolism favoring the storage of lipids within the liver (Cohen et al., 2011), which triggers hepatic inflammation and, subsequently, fibrosis, which drives NASH progression. Furthermore, metabolic syndromes, such as obesity, insulin resistance, and dyslipidemia, represent the major risk factors for the development of NAFLD/NASH. Therefore, multiple clinical trials are focusing on correcting the dysfunctional lipid metabolism via the application of lipid metabolism pathway modulators, nuclear hormone receptor agonists, and glycemic modulators (Esler and Bence, 2019).
Acetyl-CoA carboxylase (ACC) is an essential rate-limiting enzyme in fatty acid metabolism that catalyzes the carboxylation of acetyl-CoA to form malonyl-CoA (M-CoA) (Brownsey et al., 1997). Mammals have two ACC isoforms. ACC1 is localized in the cytosol and predominantly expressed in lipogenic tissues, such as the liver and adipose tissue, whereas ACC2 is localized in the mitochondrial surface and is predominantly expressed in oxidative tissues, including skeletal muscle and heart, and to some extent in the liver and adipose tissues (Abu-Elheiga et al., 2001). M-CoA produced by ACC1 is an intermediate in de novo lipogenesis (DNL) and acts as a substrate for fatty acid synthase in acyl-chain elongation (Mao et al., 2006). Furthermore, M-CoA produced by ACC2 inhibits carnitine palmitoyltransferase 1, which participates in regulating fatty acid β-oxidation (Abu-Elheiga et al., 2001, 2003). Indeed, several studies have shown that liver-specific ACC1 knockout (KO) mice have reduced hepatic DNL, M-CoA, and triglyceride (TG) accumulation (Mao et al., 2006; Harada et al., 2007), whereas ACC2 KO mice exhibit increased fatty acid oxidation coupled with elevated whole-body energy expenditure and improved whole-body adiposity compared with wild-type mice (Abu-Elheiga et al., 2001; Choi et al., 2007).
Recently, several ACC1/2 dual inhibitors have been investigated for treating NAFLD/NASH in preclinical and clinical studies (Chen et al., 2019; Esler and Bence, 2019), many of which have reported that these dual inhibitors induce undesirable effects, including plasma TG elevation, which may be caused by excessive suppression of ACC activity. However, the anti-NAFLD/anti-NASH effect of selective ACC1 inhibitors has not been evaluated in preclinical or clinical studies to understand whether the therapeutic window for ACC1 inhibitors would be greater than that for ACC1/2 inhibitors.
To support the efficacy of an ACC1 inhibitor, selective ACC1 suppression using antisense oligonucleotides demonstrated both reduced triglyceride synthesis and increased fat oxidation in primary rat hepatocytes (Savage et al., 2006). Furthermore, hepatic DNL is markedly higher in patients with NAFLD than in healthy subjects (Donnelly et al., 2005; Fabbrini et al., 2008; Lambert et al., 2014). Lastly, ACC1 but not ACC2 expression has been reported as upregulated in the liver of patients with NAFLD (Kohjima et al., 2007). These data suggest that ACC1 inhibition alone could have the potential to improve hepatic steatosis and fibrosis.
Recently, Mizojiri et al. (2018) generated a systemic selective human ACC1 inhibitor, compound-1 (selectivity >17,000-fold over that for human ACC2). To investigate the anti-NAFLD/anti-NASH effect of ACC1 inhibition, we evaluated the effect of compound-1 on M-CoA production and hepatic DNL in vitro and in vivo. Additionally, we investigated the effects of compound-1 on hepatic steatosis and fibrosis by using Western diet (WD)-fed melanocortin 4 receptor (MC4R) KO mice, a genetic and obesogenic dietary model that mimics the human pathophysiology of NAFLD/NASH with obesity, insulin resistance, excessive lipid accumulation, and enhanced liver fibrosis (Itoh et al., 2011; Konuma et al., 2015; Shiba et al., 2018). Taken together, these data are supportive of the critical role of ACC1 in the pathophysiology of liver diseases.
Materials and Methods
Compounds
Compounds used in the study were synthesized by Takeda Pharmaceutical Company Limited (Kanagawa, Japan). In the animal studies, compounds were suspended in 0.5% methylcellulose (MC).
Mouse and Human ACC Enzyme Assays
Compounds were dissolved in DMSO and then diluted with an enzyme reaction buffer [50 mM HEPES (pH 7.5), 10 mM MgCl2, 10 mM tripotassium citrate, 2 mM dithiothreitol, and 0.001% fatty acid–free bovine serum albumin]. Recombinant mouse ACC1 or ACC2 produced by Sf-9 cells was diluted with the enzyme reaction buffer to 0.8 µg/ml and 0.1 µg/ml, respectively. A 5-µl aliquot of the compound solution was added to each well of a 384-well assay plate, and 10 µl of the enzyme mixture was added to each well. The mixture was incubated at room temperature for 60 minutes. Next, a substrate solution (50 mM KHCO3, 200 µM ATP, and 5 µl of 200 µM acetyl-CoA) was added to each well, and the mixture was reacted at room temperature for 15 minutes. The reaction was stopped by adding 40 µl of stop solution (1.3% formic acid, 0.2 µM malonyl-13C3-CoA; 655759; Fujifilm Wako Pure Chemical Industries, Osaka, Japan) to each of the obtained reaction mixtures. The production of M-CoA was detected using RapidFire mass spectrometry (API4000; Sciex, Tokyo, Japan) and corrected with malonyl-13C3-CoA. The IC50 values were calculated using XLfit from the data expressed as inhibition (%) by using fit Model 204 (4 Parameter Logistic Model). The response of vehicle control was considered as 0% inhibition, and the response without enzyme was considered as 100% inhibition. The human ACC enzyme assay was performed after the protocol reported in a previous study (Mizojiri et al., 2018).
Cell Culture
The human hepatoma-derived cell line HepG2 was purchased from the American Type Culture Collection (HB-8065; Manassas, VA). The cells were incubated in Dulbecco’s modified Eagle medium (DMEM) (10567014; Thermo Fisher Scientific, Tokyo, Japan) containing 10% fetal bovine serum (SH30084.03; Hyclone Laboratories Inc., Logan, UT), penicillin, and streptomycin under 5% CO2 at 37°C.
Measurement of M-CoA Content in HepG2 Cells
HepG2 cells were plated in a 12-well plate at 2.4 × 105 cells/well and incubated for 48 hours under 5% CO2 at 37°C. After the medium was removed, cells were incubated with DMSO or compound-1 in the assay medium (DMEM containing 0.09% fatty acid–free bovine serum; 017-22231; Fujifilm Wako Pure Chemical Industries, Osaka, Japan) for 2 hours. Next, the cells were washed with 500 μl of ice-cold PBS, and 1200 μl of ice-cold 6% hydrogen peroxide was added to lyse the cells. After the cell lysates were collected in tubes, samples were immediately stored at −80°C until analysis. Refer to the experimental section on ''Measurement of Cellular or Hepatic M-CoA'' for the measurements.
Measurement of DNL in HepG2 Cells
HepG2 cells were plated in a 24-well plate at 1.5 × 105 cells/well and incubated for 24 hours under 5% CO2 at 37°C. The cells were washed twice with 500 μl of PBS and incubated with DMSO or compound-1 in assay medium (DMEM containing 0.09% fatty acid–free bovine serum) for 1 hour. Subsequently, 5 μl of 14C-acetate (25 mM, 10 μCi/μmol) was added and incubated for 2 hours at 37°C. After the cells were washed twice with 500 μl of PBS, 500 μl of 0.5 N NaOH was added to lyse the cells. Next, 200 μl of 50% KOH and 1000 μl of ethanol were added to the cell lysate and incubated for 1 hour at 70°C for saponification. Next, 1000 μl of H2O was added followed by the addition of 4000 μl of petroleum ether. After the organic layers were collected, HCl was added to the aqueous layer to adjust the pH to below 4. Subsequently, 4000 μl of petroleum ether was added, and the organic layers were collected. The pooled organic layers were dried using N2 gas and resuspended in a scintillation A cocktail. Radioactivity was measured using liquid scintillation counting in 3110 TR (PerkinElmer Inc., Waltham, MA).
Animals
Animal experiments were approved by the Institutional Animal Care and Use Committee of Shonan Research Center, Takeda Pharmaceutical Company Limited (Kanagawa, Japan), and conformed to the guidelines of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Male C57BL/6J mice were purchased from Charles River Laboratories Japan (Yokohama, Japan) and fed normal chow (CE-2; CLEA Japan, Inc., Tokyo, Japan). Male Mc4r null (MC4R KO) mice were generated after a previous study (Matsumoto et al., 2020). MC4R KO mice and wild-type littermate mice were fed CE-2. All animals were housed in a group under controlled conditions including a 12-hour/12-hour light/dark cycle at 20–26°C and humidity of 40%–70%; they were allowed free access to food and tap water.
Evaluation of the Effect of a Single Dose of Compound-1 on Hepatic M-CoA in CE-2–Fed C57BL/6J Mice (Dose-Response Study)
Seven-week-old CE-2–fed C57BL/6J mice were divided into eight groups of five mice each based on body weight (BW) and BW change during habituation. Mice were orally administered (10 ml/kg volume) with vehicle (0.5% MC), compound-1, or compound-2 and sacrificed after 2 hours of treatment under isoflurane anesthesia. Blood samples were collected from the abdominal vena cava and centrifuged at 13,000 × g for 5 minutes at 4°C to collect plasma, which was stored at −80°C for pharmacokinetic analysis. After the liver weight was measured, liver samples were immediately frozen in liquid nitrogen and stored at −80°C for M-CoA measurement and pharmacokinetic (PK) analysis. Refer to the M-CoA and PK experimental section for the measurements.
Evaluation of the Effect of a Single Dose of Compound-1 on Hepatic DNL in WD-Fed C57BL/6J Mice
Seven-week-old mice were divided into four groups of three mice each based on BW after they were fed WD (D12079B; Research Diets, Inc., New Brunswick, NJ) for 3 days. Mice were orally administered (10 ml/kg volume) vehicle (0.5% MC) or compound-1 (10 mg/kg) and intraperitoneally administered 14C-acetate (400 μCi/5 ml/kg) after 1 hour of treatment. Mice were sacrificed after 2 or 5 hours of drug treatment under isoflurane anesthesia. The radioactivity of the fatty acid fraction in the liver was measured according to a previous study (Harwood et al., 2003).
Repeated Dose Study of Compound-1 in WD-Fed MC4R KO Mice
Nine-week-old MC4R KO mice were fed WD, and wild-type (WT) mice were fed CE-2. At the age of 23 weeks, WD-fed MC4R KO mice were divided into four groups of eight mice each based on BW, food intake (FI), plasma glucose , TG, total cholesterol (TC), aspartate aminotransferase (AST), alanine aminotransferase (ALT), and insulin at day −3. Mice were orally administered (5 ml/kg volume) vehicle (0.5% MC) or compound-1 (3, 10, and 30 mg/kg) once daily for 8 weeks.
BW and FI were monitored every 3–4 days. On days 17, 31, and 56, blood samples were collected from the tail vein for the measurement of plasma parameters. Plasma glucose, TG, TC, AST, and ALT were enzymatically measured using an AutoAnalyzer 7180 (Hitachi High-Technologies Corporation, Tokyo, Japan). Plasma insulin levels were measured using sandwich ELISA (Shibayagi, Gunma, Japan, or Morinaga Institute of Biologic Science, Kanagawa, Japan).
For pharmacokinetic analysis, blood samples were collected from the tail vein at day 56. Blood samples were centrifuged at 13,000 × g for 5 minutes at 4°C to collect plasma, which was stored at −80°C for pharmacokinetic analysis. Refer to the PK experimental section for the measurements.
Mice were sacrificed under isoflurane anesthesia on day 0 (pre) and day 60. Blood samples were collected from the abdominal vena cava and centrifuged at 13,000 × g for 5 minutes at 4°C to collect plasma, which was stored at −80°C for the measurement of parameters. After the liver weight was measured, liver samples were immediately frozen in liquid nitrogen and stored at −80°C. For histopathological analysis, liver samples were stored in 10% neutral buffered formalin.
The hepatic TG content was measured as follows. First, 900 μl of saline was added to 100 mg liver tissue and homogenized (27 Hz, 2 minutes) with zirconia beads. The homogenate (200 μl) was mixed thoroughly with 600 μl of CHCl3:MeOH (1:2) for 30 minutes. After 200 μl of CHCl3 and 200 μl of H2O were added to the mixed homogenate, it was stirred for 10–30 minutes. After centrifugation (12,000 × g, 2 minutes, room temperature), the CHCl3 layers were collected, and 50 μl was dried under N2 gas and resuspended in isopropanol (80 μl for WT mice sample and 300 μl for MC4R KO mice sample). Extracted triglyceride content was measured using the Triglyceride-E test (432-40201; Fujifilm Wako Pure Chemical Industries, Osaka, Japan).
The hepatic collagen levels were measured using a commercially available kit (QZBTOTCOL1; Quickzyme Biosciences, Leiden, Netherlands) according to manufacturer’s instructions.
The percentage of hepatic Sirius red–positive area was measured as follows. Liver samples fixed with 10% neutral buffered formalin were embedded in paraffin. Liver histology was assessed in 4-μm–thick sections stained with H&E and Sirius red. The Sirius red–positive areas were quantified using four randomly selected fields per liver sample by using the WinROOF software (Mitani Co., Tokyo, Japan). The pathologist was blinded to the animal information until the completion of the measurement.
Hepatic mRNA levels were measured as follows. Total RNA was isolated from the liver samples that were stored in RNA later by using a Lipid Tissue Mini kit (Qiagen, Tokyo, Japan). Reverse-transcription reactions were performed using a SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific, Tokyo, Japan), according to manufacturer’s instructions. Gene expression was quantified by TaqMan real-time PCR (ABI7900; Thermo Fisher Scientific, Tokyo, Japan) using Platinum qPCR SuperMix-UDG (Thermo Fisher Scientific, Tokyo, Japan) and primers/Taqman probe sets (Thermo Fisher Scientific, Tokyo, Japan). The following primer-probe sets were used: monocyte chemoattractant protein-1 (Ccl2; MCP-1, Mm00441242_m1), adhesion G protein-coupled receptor E1 (F4/80, Mm00802529_m1), collagen type 1 α1 (Col1α1; Mm00801666_g1), collagen type 1 α2 (Col1α2; Mm00483888_m1), α-smooth muscle actin (Acta2; αSMA, Mm00725412_s1), transforming growth factor-β1 (Tgfβ1; TGF-β1, Mm01178820_m1), acetyl-CoA carboxylase α (Acaca; ACC1, Mm01304257_m1), acetyl-CoA carboxylase β (Acacb; ACC2, Mm01204671_m1), fatty acid synthase (Fasn; Mm00662319_m1), stearoyl-CoA desaturase 1 (Scd1; Mm00772290_m1), and ribosomal protein lateral stalk subunit P0 (36B4, Mm00725448_s1). The relative gene expression was calculated using the ΔΔCt method and normalized to 36B4 expression.
Measurement of Cellular or Hepatic M-CoA
Tissue samples were homogenized in 6% perchloric acid containing malonyl-13C3-CoA (Sigma-Aldrich, St. Louis, MO) as an internal standard. After centrifugation, the supernatant was subjected to solid-phase extraction by using an Oasis HLB Extraction Cartridge (Waters, Milford, MA). The analyte was eluted with acetonitrile and supplemented with dibutylammonium acetate (Tokyo Chemical Industries, Tokyo, Japan), which was followed by rinsing of the column with ultrapure water. The eluate was dried under a stream of nitrogen, and the residue was reconstituted in 100 μl of ultrapure water. An aliquot of 10 μl was injected into the liquid chromatography–tandem mass spectrometry system, which consisted of a Shimadzu LC-20AD HPLC system (Shimadzu, Kyoto, Japan) and an API5000 or API5500 mass spectrometer (AB Sciex, Foster City, CA). The analytical column was a CAPCELL CORE C18 column (2.7 μm, 2.1 × 50 mm; Shiseido, Kanagawa, Japan) used at 40°C. The mobile phases consisted of (A) 50 mM ammonium carbonate/ammonium hydroxide (pH 9) supplemented with dibutylammonium acetate and (B) acetonitrile. The analyte was eluted using a gradient of 95% solvent A/5% solvent B to 5% solvent A/95% solvent B. The flow rate of the mobile phase was 0.3 ml/min. Detection was performed using multiple-reaction monitoring in the positive ionization mode [selected reaction monitoring m/z = 854.1 → 347.1 or 854.1 → 303.3 for M-CoA and m/z = 857.2 → 350.2 for malonyl-13C3-CoA (internal standard)]. Analyst software (version 1.6.2, AB Sciex) was used for data acquisition and processing. The concentration of compounds in each sample was back-calculated using a calibration curve generated from a series of calibration standards.
Statistical Analysis
All data in the graph are represented as mean ± S.D. Statistical analyses were performed in EXSUS ver.8.0 in combination with SAS ver. 9.3 or GraphPad PRISM software. To evaluate the effects of compound-1 in vivo, we analyzed the statistical differences between vehicle and drug treatment in C57BL/6J mice or MC4R KO mice using Student’s t test, Aspin-Welch test, Dunnett’s test, and Steel’s test. To confirm the establishment of the disease state, we analyzed the statistical differences between WT mice and vehicle by using Student’s t test or Aspin-Welch test. For all tests, P values of <0.05 were considered statistically significant. The correlations between hepatic M-CoA content and hepatic TG as well as hepatic M-CoA content and hepatic collagen were analyzed using Pearson’s correlation coefficient.
Results
Inhibition of Mouse ACC1 and ACC2
The inhibitory activities of compound-1 and compound-2 (dual ACC1/2 inhibitor) (Kamata et al., 2012) on recombinant mouse ACC1 and ACC2 enzymes were evaluated. Compound-1 inhibited recombinant human ACC1 and ACC2 at IC50 values of 0.58 nM and >10,000 nM, respectively (Mizojiri et al., 2018), as well as mouse ACC1 and ACC2 with IC50 values of 1.9 nM and >10,000 nM, respectively (Table 1). Conversely, compound-2 inhibited mouse ACC1 and ACC2 with IC50 values of 6.0 nM and 6.4 nM, respectively. These results indicate that compound-1 is selective for ACC1 inhibition, whereas compound-2 inhibits both ACC1 and ACC2.
Inhibitory activity of compound-1 and compound-2 on recombinant mouse ACC1 and ACC2 proteins
Effect of Compound-1 on M-CoA Content and DNL in HepG2 Cells
To assess the effect of compound-1 on M-CoA content and DNL in cultured cells, we investigated the effect of compound-1 on M-CoA production and [14C] acetate incorporation into fatty acids in HepG2 cells. Compound-1 decreased the M-CoA content in cells with IC50 of 16.0 nM (Fig. 1A) and inhibited [14C] acetate incorporation into fatty acids with IC50 of 12.7 nM (Fig. 1B) in a dose-dependent manner. Additionally, the unbound fraction of compound-1 in the medium was 0.09. The estimated unbound compound-1–based IC50 when compound-1 inhibited M-CoA production and [14C] acetate incorporation was 1.44 nM and 1.14 nM, respectively.

Effect of compound-1 on M-CoA content and fatty acid synthesis in HepG2 cells. (A) Effect of compound-1 on M-CoA content in HepG2 cells. (B) Effect of compound-1 on [14C] acetate incorporation into fatty acids in HepG2 cells. Data are presented as the mean ± S.D. (n = 3)
Effect of Compound-1 on Hepatic M-CoA Content and DNL in C57BL/6J Mice
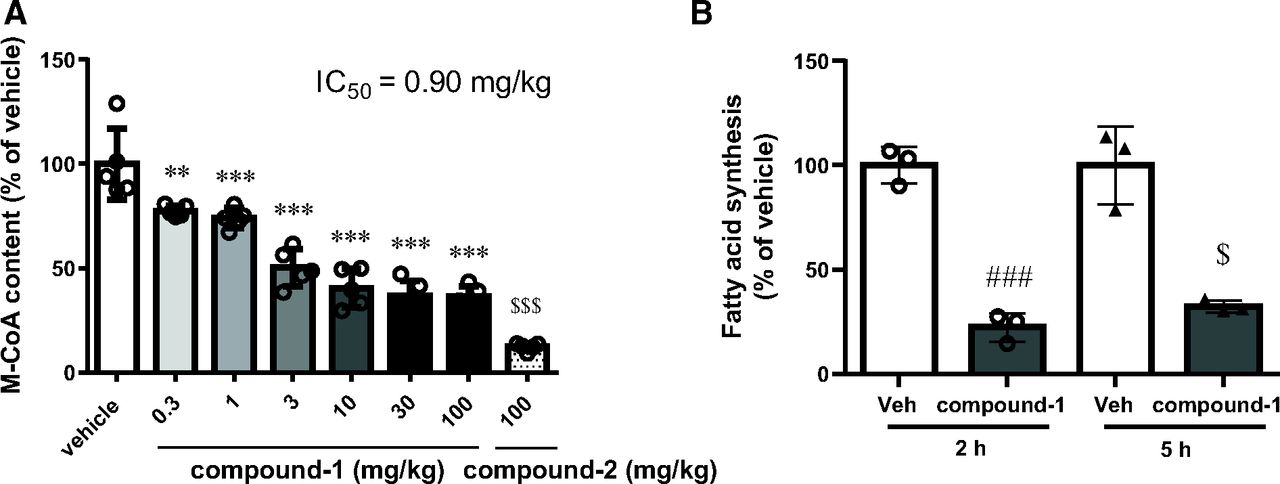
M-CoA is formed from acetyl-CoA by ACC, and the hepatic M-CoA level could be used as a pharmacodynamic marker of ACC inhibition (Glien et al., 2011; Harriman et al., 2016). Therefore, we assessed the ability of compound-1 to suppress hepatic M-CoA content in C57BL/6J mice, which were fed normal chow. At 2 hours after a single dose, compound-1 significantly and dose-dependently decreased hepatic M-CoA content from the lowest tested dose of 0.3 mg/kg. The efficacy of compound-1 tended to plateau at approximately 30 mg/kg, and a maximum reduction of −64% was noted at 100 mg/kg (Fig. 2A). Conversely, compound-2 (100 mg/kg, to achieve >85% inhibition), an ACC1/2 dual inhibitor, reduced hepatic M-CoA by 88% compared with vehicle (Fig. 2A). To analyze the correlation between compound-1 blood exposure and hepatic M-CoA reduction, we measured the plasma concentration of compound-1 at 2 hours after dose. We already confirmed that the plasma and hepatic concentrations of compound-1 were similar in a separate time-course study. Thus, the plasma concentration can be used as the index for analyzing the pharmacokinetics and pharmacodynamics (unpublished data). The IC50 of hepatic M-CoA reduction was 1.88 µg/ml (0.90 mg/kg) of plasma concentration in C57BL/6J mice (Supplemental Table 1). The exploratory in vitro study suggested high plasma protein binding of compound-1 to mice plasma, and the estimated unbound compound-1–based IC50 was <5 nM. We also confirmed that compound-1 (10 mg/kg) did not change the M-CoA content in muscles in which ACC2 is predominantly expressed 1 hour after a single dose (unpublished data).

Effects of single oral dose of compound-1 in C57BL/6J mice. (A) Effect of compound-1 and compound-2 on hepatic malonyl-CoA in normal-diet–fed C57BL/6J mice 2 hours after a single oral dose. Data are presented as the mean ± S.D. (n = 5). $$$P < 0.001 vs. vehicle by Aspin-Welch test; **P < 0.01 and ***P < 0.001 vs. vehicle by Dunnett’s test. (B) Effect of compound-1 (10 mg/kg) on [14C] acetate incorporation into fatty acids in WD-fed C57BL/6J mice 2 and 5 hours after dosing. Data are presented as the mean ± S.D. (n = 3). ###P < 0.001 vs. vehicle by Student’s t test; $P < 0.05 vs. vehicle by Aspin-Welch test. Veh, vehicle.
To assess DNL inhibition by compound-1 in vivo, we evaluated the inhibition of [14C] acetate incorporation into fatty acids in mice that were fed WD. Compound-1 (10 mg/kg) inhibited hepatic fatty acid synthesis in WD-fed C57BL/6J mice by 77.6% and 67.8% at 2 and 5 hours after oral administration, respectively (Fig. 2B).
Based on these pharmacodynamic results, we selected the doses of 3, 10, and 30 mg/kg for the repeated dose study to investigate the anti-NAFLD/anti-NASH effect of compound-1 in WD-fed MC4R KO mice.
Compound-1 Improves Hepatic Steatosis and Fibrosis in WD-Fed MC4R KO Mice
To evaluate the effects of compound-1 on NAFLD/NASH, we orally administered compound-1 to WD-fed MC4R KO mice once daily for 8 weeks at doses of 3, 10, and 30 mg/kg.
The liver weight and hepatic TG content in WD-fed MC4R KO mice were significantly higher than those in WT mice (Fig. 3, A and B). The total liver weights in compound-1–treated groups (3, 10, and 30 mg/kg) were reduced by 9.1%, 23.8%, and 57.1%, respectively, compared with that in the vehicle-treated group (Fig. 3A). Additionally, consistent with the improvement of the appearance of fatty liver (unpublished data), compound-1 significantly reduced the total hepatic TG at doses of 10 and 30 mg/kg (Fig. 3B). These results indicate that compound-1 improves liver hypertrophy and steatosis.

Effect of chronic compound-1 on liver weight, TG content, BW, and FI in WD-fed MC4R KO mice. (A) Liver weight, (B) hepatic TG (mg/tissue), (C) BW change at day 56, and (D) cumulative FI at 56 days. Data are presented as the mean ± S.D. (n = 5–8). ##P < 0.01 vs. WT by Student’s t test; $$$P < 0.001 vs. WT by Aspin-Welch test; ***P < 0.001 vs. vehicle by Dunnett’s test; ¶¶P < 0.01 vs. vehicle by Steel’s test.
All animals in the compound-1–treated group (30 mg/kg) showed abnormal findings, such as dryness of the tail, loss of fur around the chest, and flushing of the face on day 56 (unpublished data). No other abnormalities were observed in terms of anatomy and appearance. Only at 30 mg/kg did compound-1 treatment significantly decrease BW without reducing the cumulative FI (Fig. 3, C and D); however, the BW reduction was not correlated with the severity of skin toxicity.
The plasma parameter levels were evaluated before and after the 8-week treatment of compound-1 (Table 2). At the start of drug treatment, plasma AST and ALT, which are hepatic enzymes that could be used as markers of hepatocellular injury (Zechini et al., 2004), and TC levels were higher in WD-fed MC4R KO mice than in WT mice. In contrast, plasma TG levels were lower in WD-fed MC4R KO mice than in WT mice. These data suggested that WD-fed MC4R KO mice exhibited hepatocellular injury and lipid metabolism disruption.
Effect of chronic compound-1 on plasma parameters in WD-fed MC4R KO mice
Compound-1 treatment markedly decreased plasma AST, ALT, and TC levels in a dose-dependent manner, whereas the treatment increased plasma TG concentrations compared with those in the vehicle-treated group. However, plasma TG levels in the compound-1–treated mice never reached statistically significant increased values over those observed in the WT mice.
The plasma concentration and pharmacokinetic parameters of compound-1 before and after the final dose are shown in Supplemental Table 2.
To evaluate the effects of compound-1 on markers of inflammation and fibrosis, we measured the mRNA expression of MCP-1 and F4/80 as markers of inflammation and that of Col1α1, Col1α2, αSMA, and TGF-β1 as markers of fibrosis activity in the liver. Additionally, we evaluated the expression of lipogenesis-related genes, including ACC1 and ACC2, in the liver. Compared with WT mice, WD-fed MC4R KO mice treated with vehicle showed remarkable elevation in the expression levels of MCP-1, F4/80, Col1α1, Col1α2, αSMA, and TGF-β1 (Fig. 4), suggesting progression to NASH with macrophage infiltration induced by MCP-1, which are hypothesized to be induced by hepatic steatosis, was observed in this mouse model. Additionally, we confirmed that the expression of ACC1, Fasn, and Scd1 was increased in this model (Supplemental Fig. 1); these are consistent with the findings of previous studies (Itoh et al., 2011; Konuma et al., 2015; Shiba et al., 2018). Compound-1 significantly decreased the gene expression levels of MCP-1, F4/80, Col1α1, Col1α2, αSMA, and TGF-β1 (Fig. 4), whereas it increased those of ACC1, ACC2, Fasn, and Scd1 (Supplemental Fig. 1).

Effect of chronic compound-1 on hepatic mRNA expression in WD-fed MC4R KO mice. Gene expression of (A) MCP-1, (B) F4/80, (C) Col1α1, (D) Col1α2, (E) αSMA, and (F) TGF-β1. Data are presented as the mean ± S.D. (n = 5–8). #P < 0.05 and ###P < 0.001 vs. WT by Student’s t test; $$P < 0.001 vs. WT by Aspin-Welch test; *P < 0.05 vs. vehicle by Dunnett’s test; ¶P < 0.05 and ¶¶P < 0.01 vs. vehicle by Steel’s test.
We measured the hepatic M-CoA content as a pharmacodynamic marker at 20 hours after the final dose in WD-fed MC4R KO mice. Compound-1 dose-dependently reduced hepatic M-CoA content, and the maximum efficacy was observed in the 30-mg/kg group, which was 62% lower than that in the vehicle-treated group (Fig. 5).

Effect of chronic compound-1 on total hepatic M-CoA content in WD-fed MC4R KO mice. Data are presented as the mean ± S.D. (n = 5–8). ***P < 0.001 vs. vehicle by Dunnett’s test.
Additionally, we investigated the antifibrotic effect of compound-1 by conducting biochemical and histologic analyses in the liver. Hepatic collagen content and Sirius red–positive area were significantly higher in WD-fed MC4R KO mice than in WT mice (Fig. 6, A, B, and C). Compound-1 significantly decreased hepatic collagen (mg/liver) at doses of 10 and 30 mg/kg and Sirius red–positive area at doses of 3 and 10 mg/kg. Notably, hepatic M-CoA reduction was correlated with the decrease in hepatic TG and collagen (Fig. 6, D and E; R2 = 0.5716, ****P < 0.0001, and R2 = 0.5472, ****P < 0.0001).

Effect of chronic compound-1 on liver fibrosis in WD-fed MC4R KO mice. (A) Total hepatic collagen content, (B) Sirius red–positive area, (C) representative images of liver sections stained with Sirius red (magnification 10×, scale bar = 100 µm), (D) correlation between hepatic M-CoA and hepatic TG, and (E) correlation between hepatic M-CoA and hepatic collagen. Data are presented as the mean ± S.D. (n = 5–8). ###P < 0.001 vs. WT by Student’s t test; $$$P < 0.001 vs. WT by Aspin-Welch test; *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle by Dunnett’s test.
Discussion
Hepatic DNL is markedly higher in patients with NAFLD than in healthy individuals (Donnelly et al., 2005; Fabbrini et al., 2008; Lambert et al., 2014), and ACC1—the rate-limiting enzyme for hepatic DNL—is upregulated in patients with NAFLD (Kohjima et al., 2007), which suggests that abnormal ACC1 activity contributes to the pathogenesis of fatty liver, a key feature of NAFLD. In the current study, compound-1, a novel selective ACC1 inhibitor, was found to influence hepatic M-CoA concentrations, which in turn reduced hepatic lipid accumulation and prevented hepatic fibrosis in WD-fed MC4R KO mice.
First, we determined that compound-1 is highly selective at the enzyme level for mouse ACC1 and not ACC2, thus providing a candidate molecule for further studies on the biological function of ACC1. We also confirmed the effect of compound-1 on the pharmacodynamic marker M-CoA in a liver cell model indicating that ACC1 inhibition is sufficient to reduce intracellular M-CoA concentrations. The cellular M-CoA production and subsequent fatty acid synthesis were dose-dependently suppressed by compound-1 treatment. Therefore, compound-1 inhibits M-CoA content and fatty acid synthesis in hepatocytes by selectively blocking ACC1 and could represent a potential therapeutic strategy for improving hepatic steatosis.
To confirm that the reduced in vitro DNL translates in vivo, we administered a selective ACC1 inhibitor to C57BL/6J mice. Hepatic M-CoA concentrations negatively correlated with the plasma concentration of compound-1 and fatty acid synthesis, which were measured by the incorporation of acetate into lipids, were significantly suppressed by compound-1 treatment (10 mg/kg; Fig. 2; Supplemental Table 1). Additionally, we confirmed the ACC1 selectivity for compound-1 in vivo by comparing its effect on the reduction in M-CoA levels in the liver and muscles. These results support that compound-1 inhibits DNL via ACC1 inhibition in vivo and corroborates the in vitro results.
We also evaluated the impact of ACC1 inhibition on steatosis and fibrosis through repeated compound-1 dosing in a genetic and diet-induced WD-fed MC4R KO mouse model. This model shows multiple histopathological features common to patients with NASH, including hepatic steatosis, inflammation, and fibrosis. Furthermore, the expression of liver lipogenesis-related genes, including ACC1, is elevated in this mouse model compared with WT control mice (Itoh et al., 2011; Konuma et al., 2015; Shiba et al., 2018). The reported pathological features in this model were also noted in our WD-fed MC4R KO mice. In contrast to the increased ACC1 expression, expression of ACC2 was unchanged in the livers of model mice compared with that in WT mice (Supplemental Fig. 1). This is consistent with the findings of a previous study on human patients with NAFLD (Kohjima et al., 2007), which suggests that ACC1 might contribute more strongly to NAFLD progression than ACC2.
After 8 weeks of compound-1 treatment, hepatic TG accumulation was attenuated and plasma AST and ALT levels were significantly reduced, indicating that ACC1 inhibition improved hepatic steatosis resulting in ameliorated hepatocellular damage. Furthermore, compound-1 reduced the mRNA expression of genes related to inflammation and collagen synthesis marker levels, hepatic collagen content, and Sirius red–positive area, which suggests that ACC1 inhibition can also reduce inflammation and fibrosis progression either directly by its inhibition in nonhepatocytes or indirectly by reducing the hepatic lipid content (Bates et al., 2020). Indeed, we observed a correlation between reduced M-CoA concentration via ACC1 inhibition and reduced hepatic TG and collagen content. Cumulatively, these results suggest that inhibiting hepatic DNL by selective ACC1 inhibition may represent a viable strategy for treating NAFLD/NASH.
Although the Sirius red–positive area was not reduced after treatment with 30 mg/kg of compound-1, this may not be attributed to the diminished antifibrotic efficacy but rather the remarkable improvement of liver hypertrophy, which increases the total analysis area per analysis unit screen at 30 mg/kg compared with that in the other groups. Indeed, fibrosis gene expression and total hepatic collagen content were dose-dependently decreased after compound-1 treatment. However, further studies are needed to address the discrepancy between the hepatic collagen content and Sirius red–positive area.
BW loss was observed in WD-fed MC4R KO mice treated with 30 mg/kg compound-1 compared with vehicle-treated mice. Although the improvement in hepatic hypertrophy could partially be considered a cause of the BW loss, we hypothesize that compound-dependent toxicities account for the primary cause at the highest dose. Heterozygous ACC1 mutant mice and liver-specific ACC1-deficient mice did not show BW reduction compared with WT mice (Abu-Elheiga et al., 2005; Harada et al., 2007), indicating that the inhibition of ACC1 in the liver is not responsible for the reduced BW. We further evaluated the specificity of compound-1 for 72 panels, including G-protein–coupled receptor, kinase, non–kinase enzyme , ion channel, transporter, and nuclear hormone receptor up to concentrations of 10 μM, and the compound corresponded to only 5-hydroxytryptamine receptor 2B (HTR2B) (IC50 = 3.3 μM; unpublished data). However, this efficacy of HTR2B is not expected to have contributed to the observed in vivo toxicity since the estimated unbound fraction-based Cmax at 30 mg/kg in WD-fed MC4R KO mice was <160 nM, which was >20-fold lower than the in vitro IC50 against HTR2B.
We were unable to discern the impact of weight loss observed with 30 mg/kg compound-1 on the improvement in NASH caused by ACC1 inhibition. Previously we confirmed that 15% calorie restriction resulted in 11.6% reduction in BW without significantly reducing plasma ALT and AST levels or hepatic collagen content in WD-fed MC4R KO mice compared with animals fed ad libitum (unpublished data). However, marked BW loss (>10%) improved fibrosis in patients with NAFLD/NASH (Mummadi et al., 2008, Glass et al., 2015). Therefore, further studies are needed to confirm whether BW loss or ACC1 inhibition was responsible for the anti-NASH effect of compound-1 at 30 mg/kg.
Many preclinical and clinical studies have shown that ACC1/2 dual inhibitors increase plasma TG concentrations, which may be a signaling cascade that upregulates TG re-esterification genes and flux (Kim et al., 2017; Goedeke et al., 2018; Chen et al., 2019; Bergman et al., 2020; Matsumoto et al., 2020). Indeed, compound-1 significantly increased the expression of ACC1, Fasn, and Scd1, which are sterol regulatory element-binding protein 1c target genes. However, compound-1 did not significantly elevate plasma TG concentrations compared with WT mice (Table 2). We also confirmed that repeated compound-1 dosing for 8 weeks did not increase plasma TG levels in the amylin liver NASH diet–fed NASH model, a diet-induced preclinical NAFLD/NASH mouse model, compared with those in lean and vehicle-treated mice (unpublished data). Since serum TG concentrations are not increased by ACC1/2 inhibitors at a dose that partially inhibits hepatic DNL (Bergman et al., 2020), and since the cellular specific hepatic M-CoA level is compensated by ACC2 activity under ACC1 inhibition conditions (Mao et al., 2006; Harada et al., 2007), one possible explanation for this difference in plasma TG elevation between ACC1/2 inhibitors and a selective ACC1 inhibitor might be that the latter is unable to fully suppress hepatic M-CoA production because of the flux via ACC2. In our study, the maximum M-CoA–lowering effect by selective ACC1 inhibition was less than that by ACC1/2 inhibition. Additionally, hepatic ACC2 expression was dose-dependently induced by compound-1 after repeated dosing in WD-fed MC4R KO mice (Supplemental Fig. 1). Although further investigations are required, these data suggest that a selective ACC1 inhibitor could improve hepatic steatosis and fibrosis without causing plasma TG elevation because ACC2 compensates the hepatic M-CoA level under ACC1 inhibition, thereby preventing any secondary feedback, including increased circulating TGs.
In conclusion, to our knowledge, this is the first study to show that a selective ACC1 inhibitor has sufficient efficacy in preclinical models, thus providing a viable approach for treating NAFLD/NASH. The data demonstrate a robust reduction in hepatic steatosis and fibrosis in our preclinical animal models. Additionally, selective ACC1 inhibition mitigates the increased TG concentrations observed with ACC1/2 inhibitors in clinical studies. These findings support the ACC1 inhibitor as a potential novel treatment of NAFLD/NASH.
Acknowledgments
The authors are grateful to Nobuyuki Amano, Yuichiro Amano, Ryo Mizojiri, and Noboru Tsuchimori from the Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, Kanagawa, Japan, and Noriko Uchiyama from Global Drug Safety Research Evaluation, Takeda Pharmaceutical Company Limited, Cambridge, MA, for their guidance and valuable comments. The authors would like to acknowledge all Takeda ACC1 project members, especially Kazue Tsuchimori and Sayaka Nakagawa from the Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, Kanagawa, Japan.
Authorship Contributions
Participated in research design: Tamura, Sugama, Iwasaki, Sasaki, Yasuno, Aoyama, Watanabe, Erion, Yashiro.
Conducted experiments: Tamura, Sugama, Iwasaki, Sasaki, Yasuno, Aoyama, Yashiro.
Performed data analysis: Tamura, Sugama, Iwasaki, Sasaki, Yasuno, Aoyama, Yashiro.
Wrote or contributed to the writing of the manuscript: Tamura, Sugama, Iwasaki, Sasaki, Yasuno, Aoyama, Watanabe, Erion, Yashiro.
Footnotes
- Received June 7, 2021.
- Accepted September 14, 2021.
This work was supported by Takeda Pharmaceutical Company Limited. Among the authors, Y.O.T., S.I., M.S., H.Yasu., and H.Yash. are employees and stockholders of Takeda Pharmaceuticals Inc. J.S., K.A., M.W., and D.M.E. were employees and stockholders of Takeda Pharmaceuticals Inc. at the time of their contribution to the study reported.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACC
- acetyl-CoA carboxylase
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- BW
- body weight
- Col1α1
- collagen type 1 α1
- Col1α2
- collagen type 1 α2
- DMEM
- Dulbecco’s modified Eagle medium
- DNL
- de novo lipogenesis
- Fasn
- fatty acid synthase
- FI
- food intake
- HTR2B
- 5-hydroxytryptamine receptor 2B
- KO
- knockout
- MC
- methylcellulose
- M-CoA
- malonyl-CoA
- MC4R
- melanocortin 4 receptor
- MCP-1
- monocyte chemoattractant protein-1
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- PK
- pharmacokinetic
- Scd1
- stearoyl-CoA desaturase 1
- αSMA
- α-smooth muscle actin
- TC
- total cholesterol
- TG
- triglyceride
- TGF-β1
- transforming growth factor-β1
- WD
- Western diet
- WT
- wild type
- Copyright © 2021 The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}