



Abstract
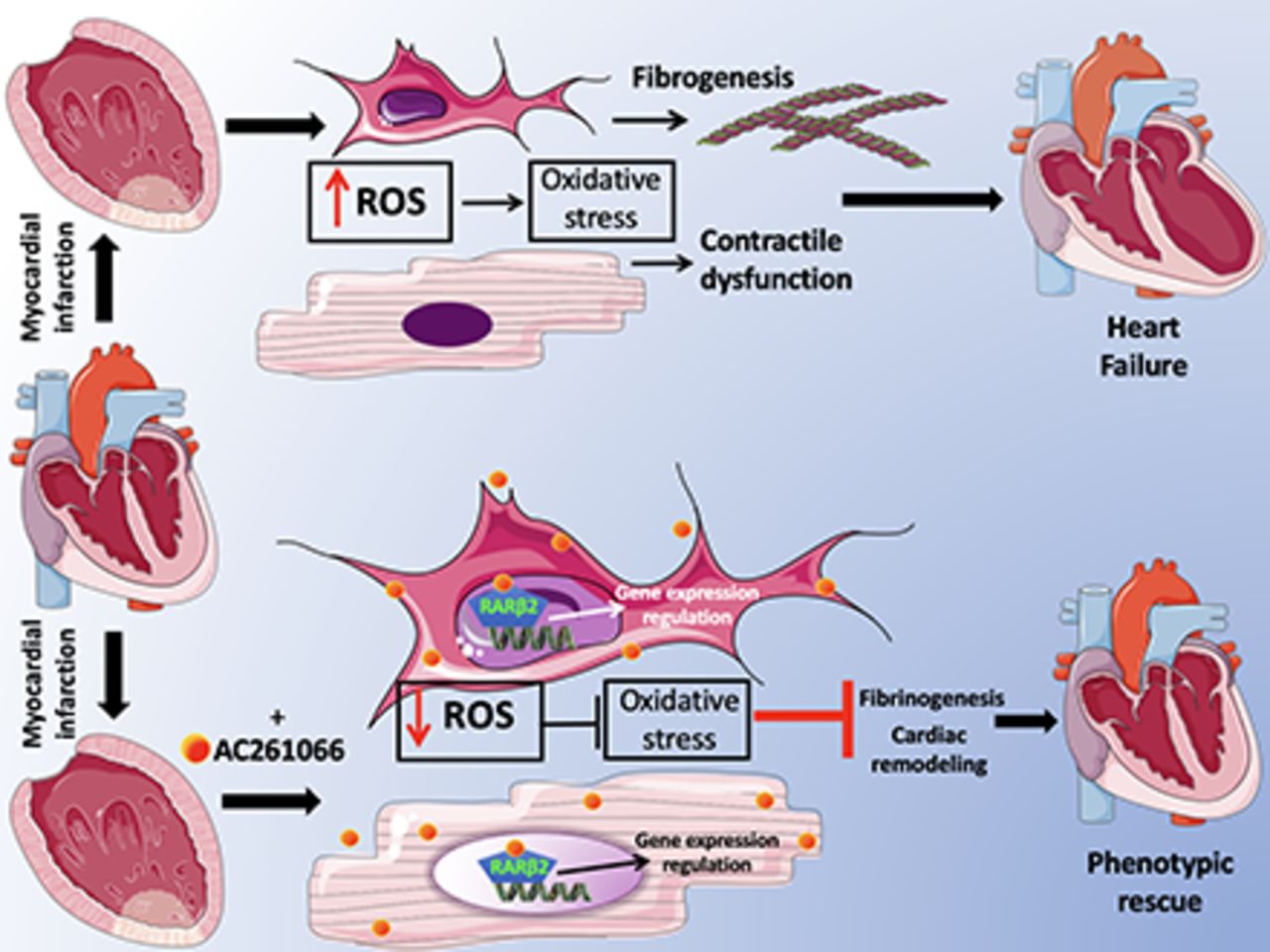
We previously demonstrated that the selective retinoic acid receptor (RAR) β2 agonist AC261066 reduces oxidative stress in an ex vivo murine model of ischemia/reperfusion. We hypothesized that by decreasing oxidative stress and consequent fibrogenesis, AC261066 could attenuate the development of contractile dysfunction in post-ischemic heart failure (HF). We tested this hypothesis in vivo using an established murine model of myocardial infarction (MI), obtained by permanent occlusion of the left anterior descending coronary artery. Treating mice with AC261066 in drinking water significantly attenuated the post-MI deterioration of echocardiographic indices of cardiac function, diminished remodeling, and reduced oxidative stress, as evidenced by a decrease in malondialdehyde level and p38 mitogen-activated protein kinase expression in cardiomyocytes. The effects of AC261066 were also associated with a decrease in interstitial fibrosis, as shown by a marked reduction in collagen deposition and α-smooth muscle actin expression. In cardiac murine fibroblasts subjected to hypoxia, AC261066 reversed hypoxia-induced decreases in superoxide dismutase 2 and angiopoietin-like 4 transcriptional levels as well as the increase in NADPH oxidase 2 mRNA, demonstrating that the post-MI cardioprotective effects of AC261066 are associated with an action at the fibroblast level. Thus, AC261066 alleviates post-MI cardiac dysfunction by modulating a set of genes involved in the oxidant/antioxidant balance. These AC261066 responsive genes diminish interstitial fibrogenesis and remodeling. Since MI is a recognized major cause of HF, our data identify RARβ2 as a potential pharmacological target in the treatment of HF.
SIGNIFICANCE STATEMENT A previous report showed that the selective retinoic acid receptor (RAR) β2 agonist AC261066 reduces oxidative stress in an ex vivo murine model of ischemia/reperfusion. This study shows that AC261066 attenuates the development of contractile dysfunction and maladaptive remodeling in post-ischemic heart failure (HF) by modulating a set of genes involved in oxidant/antioxidant balance. Since myocardial infarction is a recognized major cause of HF, these data identify RARβ2 as a potential pharmacological target in the treatment of HF.
Introduction
Myocardial fibrosis is a key feature of heart failure (HF), contributing significantly to contractile dysfunction (Weber et al., 2013; Leask, 2015; Gourdie et al., 2016; Moore-Morris et al., 2016; Travers et al., 2016; Humeres and Frangogiannis, 2019; Nagaraju et al., 2019; Wang et al., 2020b). An increased production of reactive oxygen species (ROS) is a recognized critical cause of myocardial fibrosis (Burgoyne et al., 2012; Purnomo et al., 2013; Wang et al., 2017). Vitamin A (retinol), retinoic acid, and related metabolites, collectively known as retinoids, exert several effects on heart development and play a major role in cardiac regeneration in zebrafish models (L.J. Gudas, article in press). We recently showed that a selective retinoic acid receptor (RAR) β2 agonist, AC261066, was cardioprotective, leading to a diminution of norepinephrine spillover and significantly alleviating reperfusion arrhythmias in obese (i.e., high fat diet–fed) and genetically hypercholesterolemic (i.e., Apolipoprotein E (ApoE) knockout ) mice. These cardioprotective changes were associated with a reduction in oxidative stress (Marino et al., 2018).
On these grounds, our hypothesis is that AC261066 can attenuate the development of myocardial contractile failure in HF by decreasing oxidative stress and consequent fibrotic response. To test this hypothesis, we used an established murine model of ischemic HF, obtained by permanent occlusion of the left anterior descending (LAD) coronary artery, in which cardiac function is influenced by left ventricular maladaptive remodeling (Lutgens et al., 1999). Permanent LAD occlusion typically results in HF 3–4 weeks postsurgery and is widely used as a model of ventricular remodeling after myocardial infarction (MI), especially because it reproduces human HF with reduced ejection fraction (van den Borne et al., 2009; Houser et al., 2012; Lara-Pezzi et al., 2015; Eaton et al., 2016; Okuhara et al., 2019).
We report here that oral treatment of mice with the RARβ2 agonist AC261066, after induction of permanent LAD coronary artery occlusion, attenuated the decline in left ventricular ejection fraction (LVEF), decreased the development of oxidative stress in cardiomyocytes, and reduced interstitial fibrosis. Based on further experiments directly conducted in cardiac murine fibroblasts subjected to hypoxia, we propose that the cardioprotective effects of AC261066 originate from actions on both cardiomyocytes and fibroblasts.
Materials and Methods
In Vivo Studies
MI was obtained via ligation of the LAD coronary artery, performed as we previously reported (Santulli et al., 2015; Kushnir et al., 2018). Briefly, male mice (3 months old) were anesthetized with isoflurane (4% for induction, 2% for maintenance), the heart was exposed via a left thoracotomy, and a permanent knot was tied around the LAD coronary artery. After surgery, mice were randomized to receive AC261066 (3 mg/100 ml) in drinking water or vehicle.
To assess cardiac function and remodeling after surgery, animals underwent a transthoracic ultrasound procedure, as we previously reported (Santulli et al., 2012; Santulli et al., 2015). Briefly, echocardiography was performed using a small-animal high-resolution imaging system (Vevo2100, FUJIFILM VisualSonics, Toronto, Canada) at the Albert Einstein College of Medicine. The mice were anesthetized by isoflurane inhalation (induction: 4%) and maintained by mask ventilation (isoflurane 1.5%). Fur was removed from the chest by application of a depilatory cream (Veet, Reckitt Benckiser, Slough, UK) to gain a clear image. The mice were placed in a shallow left lateral decubitus position on a heated platform (Santulli et al., 2012). All measurements were averaged on at least 10 consecutive cardiac cycles per experiment. Vevo LAB software (FUJIFILM VisualSonics) was used to acquire images and to assess cardiac morphology and function. Animals were maintained in a specific pathogen-free facility at Albert Einstein College of Medicine in New York City, NY, and the protocol for surgery and echocardiography in rodents was approved by the Einstein Animal Care Committee (00001302, PI: G. Santulli ) in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care international guidelines. All animals were of the C57/BL6J background strain. All experiments were performed in a blinded fashion. Serum concentration of troponin I was measured 24 hours postsurgery using a commercially available immunoassay kit (Lifespan Bioscience, Seattle, WA) to confirm that the infarct size achieved by surgery was similar among groups, as described by Santulli et al. (2015).
Ex Vivo and In Vitro Experiments
The level of malondialdehyde (MDA) in the left ventricle was measured by using a lipid peroxidation assay kit (Abcam, Cambridge, UK), as previously described (Loche et al., 2018).
Histology
For histopathological analyses, hearts were fixed for 4 hours in freshly diluted 4% paraformaldehyde at 4°C and subjected to a graded series of alcohol dehydrations before being embedded in paraffin (Sorriento et al., 2010; Santulli et al., 2012), and serial 5-μm sections were cut from five mice per group. For immunohistochemical analysis, sections were deparaffinized and rehydrated in graded ethanol concentrations and distilled water. Slides were immersed in diluted, citrate-based antigen unmasking solution (H-3300; Vector Laboratories, Burlingame, CA) to unmask antigen using a pressure cooker. Then the slides were treated with 3% hydrogen peroxide prepared in methanol to quench endogenous peroxidase activity. After blocking with phosphate-buffered saline containing 10% goat serum (for rabbit primary antibodies), the sections were incubated with the following primary antibodies overnight at 4°C: rabbit anti–monocyte-chemoattractant protein 1 (MCP-1) (1:100; 2029, Cell Signaling Technology, Danvers, MA), rabbit anti–α-smooth muscle actin (α-SMA) (1:100; 19245, Cell Signaling Technology), and rabbit anti–phospho-p38 (1:100; 4631, Cell Signaling Technology). After incubation with the primary antibodies, the slides were treated with secondary antibodies supplied in the SuperPicture HRP Polymer Conjugate (87-8963; Life Technologies, Thermo Fisher Scientific, Carlsbad, CA). Antibody signals were visualized by peroxidase reaction using 3,3′-diaminobenzidine as a chromogen. As a negative control, tissue sections were incubated in the absence of primary antibody to ensure specificity of the primary antibody. At least eight noncontiguous areas from the anterior and posterior portions of each section were photographed for analysis, and three sections from five different mice were measured. For the evaluation of cardiac fibrosis, slides were subjected to Picrosirius red staining (s2365, Poly Scientific R&D Corp, Bay Shore, NY), as previously described (Yuan et al., 2014); images were captured with a Zeiss Axio Z1 inverted microscope. Images were quantified using ImageJ.
Cell Culture
Primary isolated murine adult cardiac fibroblasts, obtained by enzymatic digestion using type II collagenase (Worthington, Lakewood, NJ) and Protease Type XIV (Millipore Sigma, Burlington, MA) according to standard protocols, as described and validated (Kong et al., 2018; Morelli et al., 2019; Wang et al., 2020b), were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum at 5% CO2 (37°C), unless otherwise stated.
For the hypoxia assays, fibroblasts were treated with the following medium containing (in mM) 116 NaCl, 26.2 NaHCO3, 5.4 KCl, 1.8 CaCl2, 1 NaH2PO4, 0.8 MgSO4, 0.01 glycine, and 0.001 (% w/v) phenol red; before the addition to the cells, this medium was saturated for 10 minutes at 1 atm? with 95% N2 and 5% CO2 mixture. The cells in the described medium were incubated for 6 hours in an anaerobic chamber (hypoxia chamber) filled with the same gas mixture, at 37°C, as described (Gambardella et al., 2018; Amgalan et al., 2020).
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) experiments were carried out as previously described and validated (Santulli et al., 2014; Lombardi et al., 2017; Matarese et al., 2020). Briefly, gene expression was determined through means of an AbiPRISM 7300 fast real-time cycler using the power SYBR Green real-time PCR master mix kit and quantified by built-in SYBR Green Analysis (Gambardella et al., 2018; Mone et al., 2021); samples were measured in triplicates, and results were confirmed by at least three independent experiments; the relative amount of specific mRNA was normalized to β-actin. Sequences of oligonucleotide primers (Merck KGaA, Darmstadt, Germany) for gene analysis are indicated in Supplemental Table 1.
Chemicals and Reagents
All chemicals were purchased from Millipore Sigma unless otherwise stated. AC261066 was purchased from Bio-Techne Corporation (4046; Minneapolis, MN).
Statistical Analysis of Data
All results are presented as means ± S.D. or means ± S.E.M., as indicated in the figure legends. Statistical analysis was performed using Prism 8.0 software (GraphPad, San Diego, CA). All experiments were repeated at least three times. For comparisons of two groups, the unpaired two-tailed t test using (when appropriate) Welch’s correction for unequal variances was performed. For comparisons of multiple groups, one-way ANOVA was performed followed by Tukey-Kramer’s multiple comparison test. Significant differences were established at a value of P < 0.05.
Results
The RARβ2 Agonist AC261066 Reduces the Post-MI Decline in Cardiac Function and Attenuates Remodeling
Having previously established that the RARβ2 agonist AC261066 displays cardioprotective effects by decreasing oxidative stress (Marino et al., 2018), we hypothesized that the antioxidant effects of AC261066 might also mitigate the development of myocardial contractile failure in an in vivo murine model of post-MI heart failure. Accordingly, we tested if AC261066 could attenuate the decline in LVEF observed post-MI induced by permanent ligation of the LAD coronary artery (Noll et al., 2020). Three-month-old wild-type male C57BL/6 mice underwent MI surgery as described (Santulli et al., 2015). Mice then remained on their original chow diet and drinking water containing 0.1% DMSO or water containing 3.0 mg AC261066/100 ml in 0.1% DMSO/water for 4 weeks. Ultrasound data were collected at days 0 (baseline), 7, 14, and 23 after permanent LAD ligation. We found that 23 days after LAD ligation, when an overt heart failure was evident in vehicle-treated mice, the decline in LVEF was significantly less in hearts from mice treated with AC261066 than in vehicle-treated mice (Table 1).
Baseline and postsurgery characteristics of mice treated with vehicle or AC261066
1.23 ± 0.05
Moreover, the post-MI decline in left ventricular fractional shortening (LVFS), another measure of left ventricular (LV ) contractile function, was significantly smaller in hearts from AC261066-treated mice than in vehicle-treated mice (Table 1). Notably, treatment with AC261066 markedly attenuated post-MI LV remodeling, as proven by measuring the LV end-diastolic internal diameter (Table 1). Furthermore, post-MI lung weight was significantly reduced in drug-treated compared with vehicle-treated mice, strongly suggesting that treatment with AC261066 also decreased pulmonary congestion, another heart failure marker (Table 1).
Importantly, there was no difference in troponin I serum levels (measured 24 hours after LAD ligation as previously described (Frobert et al., 2015; Morelli et al., 2019) between AC261066-treated and vehicle-treated mice, indicating that surgically obtained infarcts did not differ between these two groups. Also, heart rates of drug-treated mice did not change compared with vehicle-treated mice (Table 1).
Notably, the diminished decline in contractile function and remodeling was associated with a significant reduction in the staining for MCP-1 [measured by immunohistochemistry (IHC) in sections distant from MI location 4 weeks after surgery] (Fig. 1). Since MCP-1 is a chemokine known to increase in murine MI models and to potentiate LV remodeling (Morimoto and Takahashi, 2007; Frangogiannis, 2015), its decrease supports a cardioprotective effect of RARβ2 activation in the setting of post-MI contractile failure.

The RARβ2 agonist AC261066 reduces the MCP-1 protein level in the hearts after permanent LAD occlusion. Four weeks after LAD occlusion, mouse hearts were fixed, embedded in paraffin, sectioned, and stained with a MCP-1 antibody. Representative pictures from five mice per group. Magnification, 200×; bar, 50 µm. The stained images (7–8 images per mouse) were quantified using FIJI software. Data are means ± S.D. ***P < 0.001 (intensity).
The RARβ2 Agonist AC261066 Reduces the Post-MI Oxidative Stress in Cardiomyocytes
Since oxidative stress is considered a major cause of ischemic cardiac dysfunction (Misra et al., 2009) and we had previously reported that treatment with AC261066 diminishes oxidative stress (Marino et al., 2018), we next investigated whether the protective effects of AC261066 against the post-MI decline in cardiac contractility and remodeling were accompanied by a reduction in oxidative stress. For this, we measured the level of MDA, an established marker of oxidative stress (Ho et al., 2013; Elkabany et al., 2020), in left ventricles obtained after having euthanized the mice, 4 weeks after surgery. We found that MDA, measured by a lipid peroxidation assay, increased by ∼70% (It does not look like 70%) in the mouse hearts 4 weeks post-MI, compared to the sham group, and that AC261066 treatment reduced MDA levels by ∼60% (It does not look like 60%) as compared with untreated post-MI samples (Fig. 2). These results suggest that AC261066 diminished the MI-induced decline in cardiac function and remodeling by attenuating oxidative stress.

The RARβ2 agonist AC261066 limits the increase in oxidative stress induced by MI in mouse hearts. Four weeks after LAD occlusion, MDA was measured in cardiac tissue with a Lipid Peroxidation Assay Kit (Abcam, ab118970). Data are means ± S.E.M. of triplicate experiments. *P < 0.05 versus sham; #P < 0.05 versus vehicle.
In agreement with this notion, we found that the increase in phospho-p38 level, another marker of oxidative stress (Ma et al., 1999), measured by IHC in sections distant from MI location, was reduced by ∼25% (P < 0.01) 4 weeks post-MI in the hearts of mice treated with AC261066 as compared with untreated post-MI samples (Fig. 3).

The RARβ2 agonist AC261066 reduces the phospho-p38 (p-p38) protein level in the hearts after permanent LAD occlusion. Four weeks after LAD occlusion, mouse hearts were fixed, embedded in paraffin, sectioned, and stained with a phospho-p38 (p-p38) antibody. Representative pictures from five mice per group. Magnification, 200×; bar, 50 µm. The stained images (seven images per mouse) were quantified using FIJI software. Data are means ± S.D. **P < 0.01.
The RARβ2 Agonist AC261066 Reduces the Post-MI Development of Cardiac Fibrosis
Inasmuch as remodeling is a major consequence of the development of cardiac fibrosis (Shih et al., 2011; Humeres and Frangogiannis, 2019) and fibrosis contributes to a long-term disruption of heart function (Tallquist and Molkentin, 2017; Tallquist, 2018), we next investigated whether the anti-remodeling effects of AC261066 after LAD ligation were associated with a decrease in interstitial fibrosis. For this, we measured collagen deposition in heart sections distant from MI location 4 weeks after permanent LAD coronary artery occlusion. We found that MI induced significant interstitial fibrosis (Fig. 4), measured by Picrosirius red staining. Remarkably, in the hearts from mice treated with AC261066, collagen depositio was reduced by ∼50% (P < 0.05) as compared with hearts from vehicle-treated mice (Fig. 4). This finding suggests that AC261066 inhibits post-MI remodeling and contractile dysfunction by limiting excessive fibrogenesis.

The RARβ2 agonist AC261066 limits cardiac fibrosis in mice after permanent LAD coronary artery occlusion. Four weeks after LAD occlusion, mouse hearts were fixed, embedded in paraffin, sectioned, and stained (Picrosirius red). Representative pictures from five mice per group. Bar, 100 μm. Collagen deposition was quantified using FIJI software (plugin commands: Edit>Invert) to obtain images in which Picrosirius red–positive fibers appear in light blue (panels on the right). Images were captured with a Zeiss Axio Z1 inverted microscope. Data are means ± S.D. *P < 0.05.
The expression of α-SMA is recognized to denote the activation of fibroblasts and their transformation into myofibroblasts, another marker of fibrogenesis (Meng et al., 2018). Further corroborating our findings that treatment with AC261066 diminished post-MI collagen deposition, we observed that the increased expression of α-SMA, measured by IHC, at 4 weeks post-MI in sections remote from MI location, was reduced by ∼50% (P < 0.0001) by AC261066 (Fig. 5). These results strengthened the concept that AC261066 reduces the post-MI development of cardiac fibrosis.

The RARβ2 agonist AC261066 reduces the α-SMA protein level in the hearts after permanent LAD coronary artery occlusion. Four weeks after LAD occlusion, mouse hearts were fixed, embedded in paraffin, sectioned, and stained with an α-SMA antibody. Representative pictures from five mice per group. Magnification, 200×; bar, 50 μm. α-SMA stained images (6–7 images per mouse) were quantified using FIJI software. Data are means ± S.D. ****P < 0.0001 (percent area).
The RARβ2 Agonist AC261066 Increases the Levels of Protective Gene Transcripts in Ischemic Fibroblasts
Since AC261066 decreased collagen deposition and α-SMA expression in post-MI murine hearts, an indication of antifibrotic effects, we next tested whether the antifibrotic effects of AC261066 could possibly derive from a direct protective effect at the fibroblast level. Accordingly, we measured transcript levels by RT-qPCR of three genes, one prooxidant and two protective, in wild-type murine cardiac fibroblasts cultured in normoxic and hypoxic conditions, respectively, in the presence and absence of AC261066. We found that fibroblasts cultured in hypoxic conditions exhibited an increase in NADPH oxidase 2 (NOX-2) mRNA and decreases in superoxide dismutase 2 (SOD2) and angiopoietin-like 4 (ANGPTL4) mRNAs relative to fibroblasts cultured in normoxia (Fig. 6). The addition of AC261066 (100 nM) did not change the mRNA levels of NOX-2, SOD2, and ANGPTL4 cells in normoxia but significantly reversed the increase in mRNA for NOX-2 and the decreases in SOD2 and ANGPTL4 mRNAs in hypoxic conditions (Fig. 6). NOX-2 is a recognized pro-oxidant enzyme (Bedard and Krause, 2007). In contrast, SOD2 diminishes ROS steady-state levels (Wang et al., 2018b), whereas ANGPTL4 increases nitric oxide levels, decreases collagen expression, and fibroblast to myofibroblast differentiation (Chen et al., 2019). Hence, these data suggest that the cardioprotective effects of AC261066 against hypoxia are associated with a direct action at the fibroblast level.

The RARβ2 agonist AC261066 decreases the mRNA levels of one pro-oxidant gene (NOX-2) and increases the mRNA levels of two protective genes (SOD2, ANGPTL4) in cultured hypoxic murine cardiac fibroblasts. mRNA levels of the indicated genes were assessed by RT-qPCR in murine cardiac fibroblasts in normoxic or hypoxic conditions for 12 hours and treated with AC261066 (100 nM, 12 hours) or vehicle. β-actin was used as a normalizing mRNA. Data are means ± S.E.M. of triplicate experiments. *P < 0.05 versus normoxia, #P < 0.05 versus vehicle.
Discussion
Retinoids are defined as vitamin A (retinol), related metabolites, and synthetic agonists (Gudas and Wagner, 2011; Tang and Gudas, 2011). Interestingly, all-trans retinoic acid has been found to decline in HF, and protective effects in HF have been attributed to it (Yang et al., 2021). We focused on RARβ, one of the three retinoic acid receptors, because RARβ plays a role in heart development (Ghyselinck et al., 1998), and humans with mutations in RARβ show cardiac abnormalities, as well as other features often not compatible with life (Chitayat et al., 2007; Srour et al., 2013). Furthermore, RARβ mRNA is highly expressed in both cardiomyocytes and cardiofibroblasts isolated from adult C57BL/6 mice (Bilbija et al., 2014), and RARβ mRNA expression is detected across cardiomyocyte subtypes and regions (Litviňuková et al., 2020).
Having previously established that a selective RARβ2 agonist, AC261066, alleviates reperfusion arrhythmias in an ex vivo ischemia/reperfusion model in obese and hypercholesterolemic mice by decreasing oxidative stress (Marino et al., 2018), we hypothesized that the antioxidant effects of AC261066 could also reduce the development of fibrosis in HF. Given that myocardial infarction is a predominant cause of HF (Houser et al., 2012; Okuhara et al., 2019) and is known to trigger a detrimental fibrotic response (Bugg et al., 2020; Mouton et al., 2021), we chose to test our hypothesis at a systemic, preclinical level in an established clinically relevant murine model of ischemic HF. Ischemic HF is achieved in this model by permanent occlusion of the LAD coronary artery, after which cardiac function is influenced by left ventricular maladaptive remodeling (Lutgens et al., 1999; Noll et al., 2020). Other investigators have successfully used this model to assess cardioprotective effects of ACE inhibitors, loop diuretics, and antianginal medications (Wang et al., 2018a; Ma et al., 2019).
We found that oral treatment with AC261066, initiated right after MI surgery and maintained until euthanasia 4 weeks later, reduced the post-MI decline in cardiac function and attenuated remodeling, as indicated by significantly smaller declines in LVEF and LVFS and a concurrent attenuated dilatation of the LV chamber in AC261066-treated mice 23 days after MI surgery, at a time when HF was fully developed. Treatment with AC261066 also resulted in a post-MI decrease in pulmonary congestion, as indicated by a markedly smaller increase in the post-MI lung/body weight ratio. These cardioprotective changes afforded by AC261066 treatment were not confounded by possible surgical artifacts, since infarct sizes were not different in AC261066-treated and vehicle-treated mice, as demonstrated by equal serum troponin I levels measured the first day after surgery.
Having determined that AC261066 alleviates HF by reducing the decline in contractility and remodeling, we searched for possible mechanisms of action and measured genes that are known to be causally associated with the development of HF. Damage from hypoxia-related ROS often precedes structural remodeling of the heart, including fibrosis (Lloyd-Jones et al., 2002; Sawyer et al., 2002; Giordano, 2005; Zhao et al., 2015; Richter and Kietzmann, 2016; Tallquist and Molkentin, 2017; Luptak et al., 2019; Peoples et al., 2019). Moreover, we had previously reported that AC261066 reduces oxidative stress in ex vivo murine hearts subjected to ischemia/reperfusion (Marino et al., 2018). Accordingly, our working hypothesis was that AC261066 may attenuate MI-induced cardiac dysfunction by regulating a set of genes that reduce the deleterious effects of ROS and decrease interstitial fibrosis.
We focused first on the possibility that a protective effect of AC261066 against post-MI contractile failure might be associated with intracardiac changes in MCP-1, a cardiomyocyte-produced chemokine known to increase in murine MI models and to potentiate detrimental LV remodeling (Morimoto and Takahashi, 2007; Frangogiannis, 2015). Oxidative stress increases MCP-1 secretion from cardiomyocytes (Hohensinner et al., 2006). Furthermore, MCP-1 null mice, or mice with a deletion of the MCP-1 receptor, have less post-MI LV remodeling, less LV dilatation, and less impairment of LV function (Xia and Frangogiannis, 2007). Indeed, we found that in mice treated with AC261066, the diminished decline in contractile function and remodeling was associated with a marked reduction in the staining for MCP-1 in heart sections distant from MI location. This finding supports the concept that a decline in MCP-1 likely mediates, at least in part, the cardioprotective effect of RARβ2 activation in the setting of post-MI contractile failure.
p38 mitogen-activated protein kinase (MAPK) is a relevant pathway for LV remodeling and dysfunction and post-ischemic HF progression (Liu et al., 2005; Marber et al., 2011; Yokota and Wang, 2016). In fact, oxidative stress leads to increased MAPK phosphorylation in cardiac cells (Bogoyevitch et al., 1996; Seko et al., 1997; Yin et al., 1997; Clerk et al., 1998). Moreover, cardiomyocyte-specific activation of p38 MAPK (phosphorylation of p38) in transgenic mice is associated with LV remodeling with interstitial fibrosis, myocyte hypertrophy, and systolic and diastolic dysfunction (Streicher et al., 2010). Furthermore, p38 MAPK inhibition improves cardiac function and decreases post-MI LV remodeling (Ma et al., 1999). Based on these findings, we surmised that the protective effects of AC261066, which we observed in post-MI HF, might involve a reduced p38 MAPK activity. Indeed, our data clearly demonstrate a major reduction in the level of of phospho-p38 and number of phosphor-p38-positive cells in cardiac sections taken remotely from MI location. This observation strongly suggests that treatment with AC261066 in the setting of MI-induced HF improved LV remodeling and myocardial function by attenuating p38 MAPK activation by oxidative stress. Notably, treatment with AC261066 not only reduced phospho-p38 level but also markedly decreased MDA formation, a characteristic marker of oxidative stress (Luo et al., 2014). Thus, we postulate that treatment with AC261066 decreases oxidative stress and, in turn, reduces the activation of p38 MAPK pathway, thereby alleviating LV remodeling and contractile dysfunction in post-MI HF.
Oxidative stress and increased ROS production prevail in the MI setting, and ROS are known to stimulate interstitial fibrogenesis (Richter and Kietzmann, 2016; Nakada et al., 2017), ultimately leading to maladaptive remodeling and contractile dysfunction (Gourdie et al., 2016; Tallquist and Molkentin, 2017; Tallquist, 2018; Humeres and Frangogiannis, 2019). We show here that treatment with AC261066 decreases ROS production in post-MI hearts and mitigates remodeling and contractile dysfunction.
Since retinoids can reduce fibrosis in kidney disease (Sierra-Mondragon et al., 2019) and reduce cardiac fibrosis (Wang et al., 2020a), we questioned whether the cardioprotective effects of AC261066 involve an antifibrotic step. Indeed, we found that treatment with AC261066 markedly decreased collagen deposition, the hallmark of fibrogenesis (Prabhu and Frangogiannis, 2016), in areas remote from the infarct. Fibrogenesis is initiated by fibroblast transformation into active myofibroblasts, exemplified by an increased expression of α-SMA (Weber, 1997; Ma et al., 2018). We found that treatment with AC261066 diminished post-MI α-SMA expression, further confirming that AC261066 mitigates remodeling and contractile dysfunction in part by diminishing fibroblast activation, thus impeding maladaptive interstitial fibrosis.
As these findings suggested a possible direct effect of AC261066 at the fibroblast level, we tested this hypothesis in cultured murine fibroblasts in normoxic and hypoxic conditions. Accordingly, by RT-qPCR we measured transcript levels of NOX-2, a recognized pro-oxidant enzyme (Bedard and Krause, 2007); SOD2, known to diminish ROS steady-state levels (Wang et al., 2018b); and ANGPTL4, which has been proven to decrease collagen expression and fibroblast activation (Chen et al., 2019). We found that fibroblasts cultured in hypoxic conditions exhibited an increase in NOX-2 and decreases in SOD2 and ANGPTL4 mRNAs relative to fibroblasts cultured in normoxia. Although AC261066 had little effect on normoxic fibroblasts, it partially reversed changes in transcript levels in hypoxic conditions. Thus, our data support the notion that the cardioprotective effects of AC261066 in the MI setting are associated in part with a direct action at the fibroblast level. This is a novel finding; although NOX-2 had been previously implicated in post-MI cardiac remodeling and contractile dysfunction (Looi et al., 2008), this action had been attributed solely to an effect on cardiomyocytes (Sirker et al., 2016). Although the experimental and clinical potential of NOX-2 inhibition in the post-MI setting has been previously recognized (Bauersachs et al., 2001; Hayashidani et al., 2002; Qin et al., 2007), and diastolic dysfunction and NOX-2 increases have been associated with a loss of RARα signaling (Zhu et al., 2016), our results identify for the first time both cardiomyocytes and fibroblasts as targets of the protective role of a selective RARβ2 agonist in ischemic cardiac dysfunction.
In conclusion (Fig. 7), we propose that AC261066 ultimately alleviates post-MI cardiac dysfunction by sequentially activating RARβ2 in cardiomyocytes and fibroblasts, then regulating a set of genes responsible for decreasing oxidative stress and ROS production. These AC261066 responsive genes then diminish interstitial fibrogenesis and remodeling. Since MI is a recognized major cause of HF, our data identify RARβ2 as a likely pharmacological target in the treatment of contractile dysfunction associated with HF. In future experiments, it could also be of interest to explore the actions of AC261066 in ameliorating the cardiotoxicity associated with some cancer drugs.

Proposed mechanistic sequence for the cardioprotective effects of AC261066.
Strengths and Limitations of Our Study
Our investigation is based on a clinically relevant experimental model of HF with reduced ejection fraction in humans (Houser et al., 2012; Bacmeister et al., 2019). Although several therapeutic lines have been followed to inhibit fibrogenesis in the setting of HF, such as beta-blockers (Ravindranathan et al., 1984; Liu et al., 1997), renin-angiotensin-aldosterone system antagonists (McDonald et al., 1994; Schieffer et al., 1994; Liu et al., 1997), and statins (Chen and Mehta, 2006; Tang et al., 2011), our findings identify a novel, first-in-class drug for the treatment of HF based in part on oxidative stress reduction in cardiomyocytes, and also in cardiac fibroblasts. At this point, it remains to be confirmed that AC261066 acts via RARβ2 by performing similar experiments in mice in which RARβ2 is specifically deleted in cardiomyocytes and/or cardiac fibroblasts. These experiments are beyond the scope of this publication.
Acknowledgments
We thank the Gudas and Santulli laboratory members and Dr. John Wagner for reading the manuscript.
Authorship Contributions
Participated in research design: Tang, Gambardella, Santulli, Gudas, Levi.
Conducted experiments: Tang, Gambardella, Jankauskas, Wang, Santulli.
Performed data analysis: Tang, Gambardella, Jankauskas, Santulli.
Wrote or contributed to the writing of the manuscript: Tang, Gambardella, Santulli, Gudas, Levi.
Footnotes
- Received June 22, 2021.
- Accepted August 10, 2021.
↵1 X.-H.T. and J.G. contributed equally to this work as co-first authors.
This research was supported by the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) [Grant R01-DK113088 to L.J.G.] and Weill Cornell funds (to R.L.). The Santulli Laboratory was supported in part by the NIH NIDDK [Grants R01-DK123259, R01-DK033823, R00-DK107895], National Heart, Lung, and Blood Institute [Grants R01-HL146691, R01-HL159062, T32-HL144456], and National Institute on Aging [Grant R56-AG066431] (all to G.S.); by the Irma T. Hirschl and Monique Weill-Caulier Trusts (to G.S.), and by the American Heart Association [Grants AHA-20POST35211151 to J.G. and AHA-21POST836407 to S.S.J.].
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- α-SMA
- α-smooth muscle actin
- ANGPTL4
- angiopoietin-like 4
- HF
- heart failure
- IHC
- immunohistochemistry
- LAD
- left anterior descending
- LV
- left ventricular
- LVEF
- left ventricular ejection fraction
- LVFS
- left ventricular fractional shortening
- MAPK
- mitogen-activated protein kinase
- MCP-1
- monocyte-chemoattractant protein 1
- MDA
- malondialdehyde
- MI
- myocardial infarction
- NOX-2
- NADPH oxidase 2
- RAR
- retinoic acid receptor
- ROS
- reactive oxygen species
- RT-qPCR
- reverse transcription quantitative polymerase chain reaction
- SOD2
- superoxide dismutase 2
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}