


Abstract
Opioid use disorder (OUD) is a major socioeconomic burden. An ideal OUD pharmacotherapy will mitigate the suffering associated with opioid-withdrawal, inhibit the effects of high efficacy opioids, and minimize opioid-cravings while being safe and accessible to a diverse patient population. Although current OUD pharmacotherapies inhibit the euphoric effects of opioids of abuse, the extent to which they safely alleviate withdrawal and opioid-cravings corresponds with their intrinsic µ opioid receptor (MOR) efficacy. In addition to inhibiting the euphoric effects of opioids of abuse, the medium efficacy MOR agonist buprenorphine alleviates withdrawal and opioid-cravings, but its intrinsic MOR efficacy is sufficient such that its utility is limited by abuse and safety liabilities. Although the MOR antagonist naltrexone minimizes euphoria and has no abuse liability, it exacerbates suffering associated with withdrawal and opioid cravings. Therefore, a therapeutic with intrinsic MOR activity between the partial agonist (buprenorphine) and the antagonist (naltrexone) would strike a balance between the benefits and liabilities of these two therapeutics. To address this need, we derived RM1490, an MOR agonist based on a nonmorphinan scaffold that exhibits approximately half the intrinsic MOR efficacy of buprenorphine. In a series of preclinical assays, we compared RM1490 with buprenorphine and naltrexone at doses that achieve therapeutic levels of central nervous system MOR occupancy. RM1490 exhibited a behavioral profile consistent with reduced reward, dependence, and precipitated withdrawal liabilities. RM1490 was also more effective than buprenorphine at reversing the respiratory depressant effects of fentanyl and did not suppress respiration when combined with diazepam.
SIGNIFICANCE STATEMENT In preclinical studies, RM1490 has a physiological and behavioral profile suitable for opioid use disorder maintenance therapy.
Introduction
Opioid use disorder is a major socioeconomic burden that impacts approximately 0.2% of adults (Mark et al., 2001; Gowing et al., 2015; Council of Economic Advisers, 2017). Abuse of high-efficacy opioids is driven by a combination of their positively reinforced euphoric effects and by their negatively reinforced effects. In the latter, opioid use alleviates suffering associated with opioid withdrawal and cravings (Koob and Volkow, 2010). Currently, three medications with high, medium, and low intrinsic µ opioid receptor (MOR) efficacy are used in OUD treatment, namely methadone, buprenorphine and the antagonist naltrexone, respectively. Notably, the intrinsic MOR efficacy of these medications determines the level of detoxification required before they can be safely introduced, their efficacy at alleviating suffering associated with opioid withdrawal and cravings, and, importantly, their respective liabilities.
Before complete detoxification from an opioid of abuse, methadone and buprenorphine can be introduced as replacement therapies to alleviate opioid withdrawal and cravings. In contrast, complete detoxification is required before introduction of naltrexone. Although few deaths occur as a direct result of suffering associated with opioid withdrawal, a large proportion of patients do not complete the detoxification required prior to naltrexone introduction (Preston et al., 1999; Lee et al., 2018).
Upon successful completion of the respective detoxifications and induction into OUD medication–assisted treatment, patients are maintained on doses of methadone, buprenorphine (2–24 mg/day), or naltrexone (50 mg/day). These doses of naltrexone and buprenorphine occupy approximately 90% of MOR in the central nervous system (CNS) and are sufficient to prevent the euphoric effects of opioids of abuse (Lee et al., 1988; Weerts et al., 2008; Greenwald et al., 2014; Mattick et al., 2014). Buprenorphine treatment is sufficient to alleviate symptoms of opioid withdrawal and cravings. Although naltrexone inhibits the euphoric effects of opioids of abuse, it does not alleviate opioid withdrawal or cravings. In fact, naltrexone precipitates opioid withdrawal–like effects that exacerbate patient dropout rates (Krupitsky et al., 2016; Carroll et al., 2018). On the other hand, the moderate level of intrinsic MOR efficacy of buprenorphine is sufficient such that it is abused in some populations (Häkkinen et al., 2012), and drug-drug interactions leading to respiratory depression are observed when buprenorphine is mixed with benzodiazepines (Nielsen et al., 2007). Furthermore, the utility of buprenorphine is limited by a series of metabolites that are high-efficacy MOR agonists (Moody et al., 2011; Häkkinen et al., 2012; Tompkins et al., 2014). Because of the mentioned limitations, physicians must complete both training and undertake to treat a limited number (in 2021 this legislation is under revision) before being approved to prescribe buprenorphine for OUD (Greenwald et al., 2014; Martin et al., 2018).
Because of the aforementioned risks, regulatory challenges, and social stigma associated with buprenorphine treatment, the next stage in OUD therapy involves transitioning either to no medication or to maintenance on the more readily accessible and safer MOR antagonist naltrexone. Critically, both transitions are impeded by the physiologic dependence that develops after long-term exposure to buprenorphine (Nigam et al., 1994; Eissenberg et al., 1996; Cowan, 2007; Paronis and Bergman, 2011) and by the large difference in intrinsic efficacies between buprenorphine and naltrexone, which necessitates a risky period of unmedicated detoxification before introduction of naltrexone (Bentzley et al., 2018).
An ideal OUD pharmacotherapy would suppress withdrawal and cravings for the abused opioid, have limited respiratory liabilities (e.g., impair respiration in normoxic conditions or chemosensory responses to CO2), and, if desired, be amenable to discontinuation. In addition, it would have minimal interactions with other classes of abuseable drugs, particularly benzodiazepines, and would be convenient to administer. Although tamper-proof formulations and modifications of the morphinan scaffold have sought to address the liabilities of buprenorphine and naltrexone, to date there is no clinical strategy that both overcomes their liabilities and facilitates the treatment transition from buprenorphine to a safer, more-accessible therapeutic (Ling et al., 2011). In the present study, we describe an MOR agonist based on a nonmorphinan scaffold that exhibits approximately half the intrinsic MOR efficacy of buprenorphine; we evaluated it in preclinical models to assess its efficacy, rewarding, and respiratory liabilities.
Materials and Methods
Compounds
Buprenorphine hydrochloride, morphine pellets (75 mg each of base), and morphine sulfate were provided by the National Institute on Drug Abuse drug supply program. DAMGO ([D-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin), forskolin, IBMX (3,7-Dihydro-1-methyl-3-(2-methylpropyl)-1H-purine-2,6-dione), naloxone hydrochloride, fentanyl, and diazepam were obtained from Sigma Aldrich (St Louis, MO). Carfentanil was obtained from Cayman Chemical. Ham-F12 and FBS were purchased from Invitrogen (Carlsbad, CA). RM1049 and RM1490 were provided by R2M Pharma (South San Francisco, CA) and synthesized as previously reported (Medina et al., 2019). Naloxone, morphine, and fentanyl were dissolved in 0.9% saline; stock solutions of the other compounds were prepared by dissolving in DMSO and kolliphor and then diluting with 0.9% saline to working concentrations, which contained 1% or less of DMSO and kolliphor. Vehicle control solutions consisted of 0.9% saline, 1% DMSO, and 1% kolliphor. Unless otherwise stated, injections were subcutaneous at 3 to 5 ml/kg.
Cell Culture and In Vitro Assays
MOR cAMP assays: CHO Tag-lite human MOR stable cell line from Cisbio (NCBI accession number: NM_000914.3; Bedford, MA) were seeded and grown to approximately 80% confluence in Ham F-12 with 10% FBS, 50 U/ml penicillin, 50 µg/ml streptomycin, 2 mM Hepes, and 1 mg/ml geneticin (Invitrogen, Carlsbad, CA). Cells were then harvested using accutase (Corning, Corning, NY), centrifuged at 1300 RPM for 5 minutes, and plated at 5000 cells per 5 μL per well in a 5× dilution of stimulation buffer consisting of the HTRF cAMP Gi kit, water, and IBMX at 0.5 mM (Cisbio, Bedford, MA) in white HTRF low-volume 384-well plates (Cisbio, Bedford, MA). Plates were then incubated at 37°C in 5% CO2 for 10 minutes. For the agonist assay, forskolin was added to a final concentration of 4 μM. For the antagonist assay, forskolin (4 μM) and DAMGO at 90% of its effective concentratoin (EC90) were added. Test compounds were dissolved in DMSO and water and then serially diluted to working concentrations such that the concentration of DMSO was less than 0.1%. Diluted test compounds were added at 2.5 μL per well, and plates were incubated at 37°C and 5% CO2 for 15 minutes and then at room temperature for 15 minutes. Next 5 μl/well of cAMP Eu-cryptate and 5 μl/well of anti–cAMP-d2 (both diluted 1:20 in lysis buffer) were added, and plates were incubated at room temperature for 1 hour. After incubation, plates were read in a Synergy Neo2 multimode reader (Biotek, Winooski, VT). Plate reader settings were set to time-resolved fluorescence with excitation at 330 nm and emissions of 620 nm and 665 nm. Emission fluorescence was normalized (665/620nm signal × 1000). For the agonist assay, data were normalized using the maximal DAMGO response. Measurements were performed in triplicate, and the dose response was fit using nonlinear regression.
MOR GTPγS assays were conducted at Multispan Inc. (Hayward, CA) in membranes obtained from stably transfected CHO cell expressing the MOR receptor (CHO-K1 Multispan Inc., C1350-1a) using SPA beads (Perkin Elmer, RPNQ0001).
Animals
Male Sprague-Dawley rats (225–300 g) from Charles River were used for all experiments. Unless otherwise stated, animals were acclimatized to the local holding facility for a minimum of 3 days after arrival from Charles River. Animals were maintained on a 12-hour light cycle. All experiments were approved by the Institutional Animal Care and Use Committee in accordance with the animal care standards set forth by National Institutes of Health.
Behavior
All behavioral experiments were performed with the experimenter blinded to the treatment condition.
Acetic Acid–Induced Writhing
Animals were acclimatized to the recording chamber for 2 days for 15 minutes per day. The recording chamber consisted of 2 webcams and a custom-built plexiglass chamber 38”×38” ×18” height that was further enclosed in a sound-attenuating box from Med associates Inc. (Fairfax, VT). As schematized in Fig. 1F on the day after acclimatization, either saline or the antagonist naloxone 0.3 mg/kg was injected subcutaneously, and then 5 minutes later the test compound was injected subcutaneously; 30 minutes later, 2% acetic acid was injected intraperitoneally. After the intraperitoneal injection, animals were placed into the recording chamber. Videos were scored in 5-minute bins at 5–15 minutes after the intraperitoneal injection by an experimenter blinded to the treatment conditions.

RM1490 is an MOR agonist that reduces acetic acid–induced writhing. (A) Chemical structures of naltrexone, RM1490, buprenorphine, and RM1049. In (B–D) are representative dose-response curves: in B for activation of cAMP in CHO cells expressing human MOR by test compounds as single agents, in C for inhibition of cAMP activated by the high-efficacy MOR agonist DAMGO EC90, and in D for activation of GTPγS in CHO cells membranes expressing human MOR. (E) Corresponding EC50, IC50, and maximum efficacy values (n = 3 experiments per treatment and 3 replicates per experiment). (F and G) The timeline and summary data for changes in writhing after intraperitoneal injection of 2% acetic acid; compared with animals injected with vehicle, agonists buprenorphine (0.3 mg/kg) and RM1490 (3 mg/kg) dose-dependently reduce the number of writhes [mean diff: 3.183, 95% CI (0.4046 to 5.961) P = 0.015, and mean diff: 2.833, 95% CI (0.05456 to 5.611), P = 0.04, respectively], whereas the antagonist naloxone increases the number of writhes [mean diff: −3.267, 95% CI (−6.045 to −0.4890), P = 0.013] and inhibits the effect of both agonists. [n = 8–10 per group, *P < 0.05, ns P > 0.05 vs. vehicle post hoc test; ANOVA F (10, 157) = 5.948, P = 0.0001]. Bup, buprenorphine; NA, not available; ns, not significant.
Hot-Plate
Animals were acclimatized to the hot-plates from IITC (Woodlands, CA) that were switched off (room temperature) for 10 minutes per day for 2 days. On the following day, hot-plates were switched on and set to 52°C. To determine the baseline (seconds): vehicle was injected, and 15 minutes later the animal was placed on the hot-plate. An experimenter blinded to the treatment condition monitored the animal for responses, including a lick of a hind paw, shake of a hind paw, and stomping of the hind paws; after either two of the listed occurred or if the animal vocalized, jumped (all four feet), or reached the nominal cutoff time of 30 seconds without exhibiting a detectable response, the animal was immediately removed from the hot-plate, returned to its home cage, and the time was recorded. Testing was repeated at 45, 90, and 120 minutes after the injection. All experiments were recorded on video, and a subset of experiments were scored by a second blinded observer. Criteria were selected based on preliminary experiments and ensured maximum consistency between scores. To determine the test score (seconds), the next day the procedure was repeated after injection of test compound: Animals were tested at 45, 80, 120, and 180 minutes after injection (timeline schematic Fig. 2D). If an animal did not respond to touch upon being picked up for testing, its righting reflex was tested; animals that did not right in 5 seconds were assigned the cutoff score of 30 seconds and not tested at that time point. To test inhibition of the antinociceptive effects of agonists with high intrinsic MOR efficacy (Fig. 1, C and E), test compounds were mixed together and injected subcutaneously (Fig. 2G). Percentage maximum possible effect (%MPE) was calculated as: 100*[(test − baseline)/(30 seconds − baseline)], wherein 30 seconds is the cutoff time. Inhibition of antinociception was calculated as: %MPE − 100%. Average effect in summary bar graphs was calculated as the average of test scores between 45 and 120 minutes.

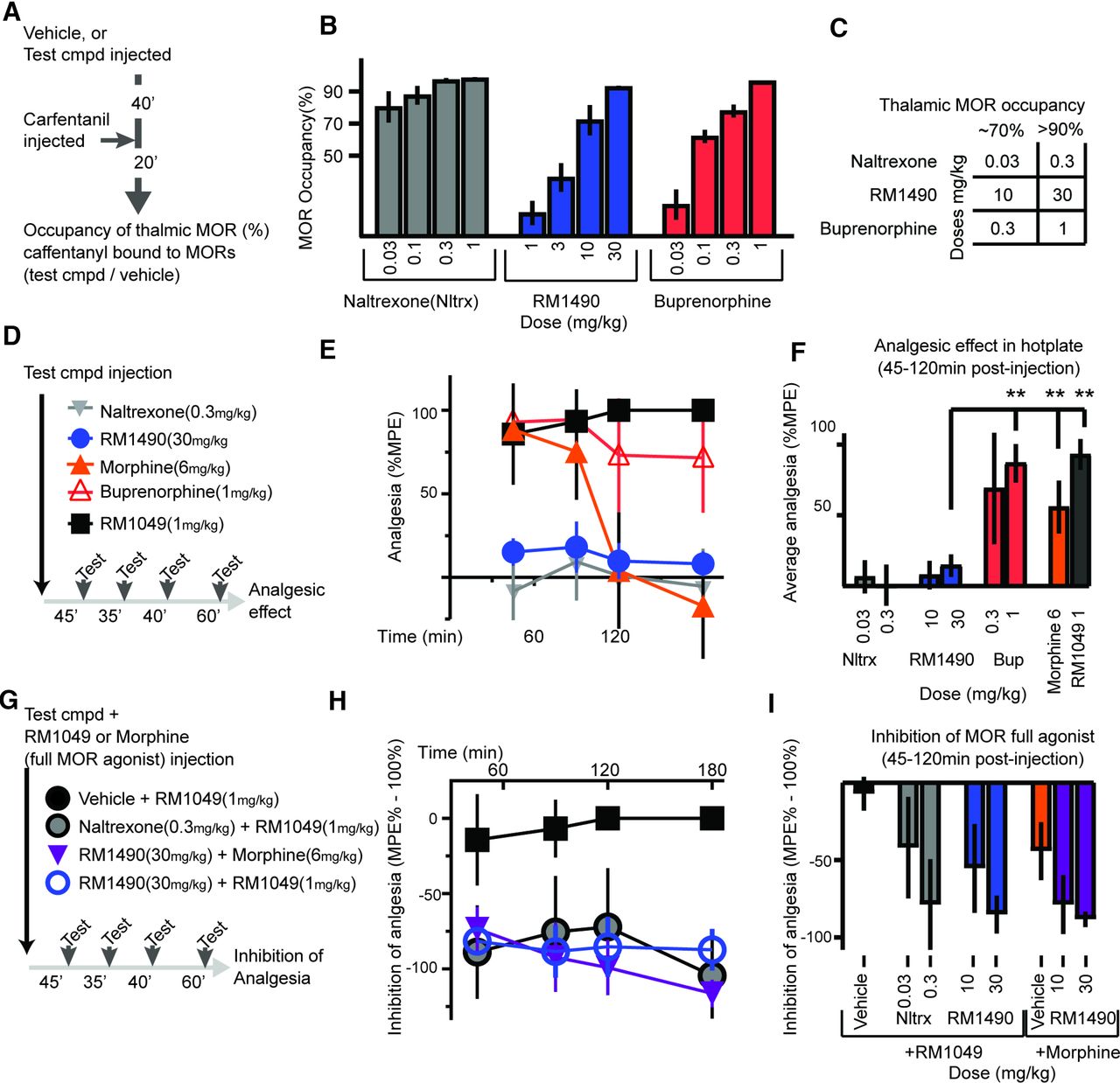
RM1490 dose-dependently binds brain MOR and reduces the antinociceptive effects of high-efficacy MOR agonists in the hot-plate test. (A and B) Timeline of experiment to assay MOR occupancy in the thalamus and corresponding summary data for 4 doses per compound (n = 3–4 per dose). (C) Table of doses for each compound corresponding to approximately 70% and 90% occupancies that are used for experiments. (D–I) Timelines and corresponding summary data for 2 sets of hot-plate experiments. In (D–F), the test compounds were administered as single agents to determine their antinociceptive effect in the hot-plate test. Buprenorphine (1mg/kg) [mean diff: 72.37, 95% CI (54.74 to 90.00)], morphine (6 mg/kg) [mean diff: 41.44, 95% CI (23.04 to 59.84)], and RM1049 (1 mg/kg) [mean diff: 78.58, 95% CI (61.54 to 95.61)] induced more antinociception than RM1490 (30 mg/kg) [n = 8–16 per group, ANOVA F (7, 70) = 33.88 P < 0.0001; **P < 0.001 Dunnett’s post hoc test]. (G–I) Coinjection of a test compound (vehicle, naltrexone, or RM1490) and a high-efficacy MOR agonist (RM1049 or morphine), revealed that RM1490 inhibited the antinociceptive effect of the high-efficacy MOR agonists ([vehicle + RM1049 (1 mg/kg) vs. RM1490 (30 mg/kg) + RM1049 (1 mg/kg): mean diff: −78.08, 95% CI (−115.5 to −40.70), P < 0.001] and [vehicle + morphine (6 mg/kg) vs. RM1490 (30 mg/kg) + morphine (6 mg/kg): mean diff: −44.05, 95% CI (−84.24 to −3.865), P = 0.02)]; [n = 8–16 per group, ANOVA F (7, 47) = 12, P < 0.0001]). Bup, buprenorphine; cmpd, compound; Nltrx, naltrexone.
Receptor Occupancy
Experiments were performed at Melior Inc. using procedures similar to those described by Need et al. (2007). The timeline of the in vivo phase is schematized in Fig. 2A: Vehicle or test compounds were injected subcutaneously, and 40 minutes later the tracer (carfentanil) was injected intravenously through a lateral tail vein. After 20 minutes, animals were sacrificed, and cerebellar and thalamic tissue samples were collected and processed for liquid chromatography tandem mass spectrometry.
For liquid chromatography tandem mass spectrometry analysis, tissue samples were homogenized 1:4 (v/v) with water: 2-propanol: DMSO (50:30:20 v/v/v). Homogenates were then centrifuged, and the supernatant was collected for analysis. Tandem mass spectrometry detection was done with an Applied Biosystems Sciex API 4000 instrument in positive-ion electrospray ionization mode that was calibrated using samples from the vehicle group spiked with test compound. To calculate occupancy, the concentration of carfentanil detected in the thalamus (MOR-containing region) was normalized to levels in the cerebellum (MOR-null region), and percentage receptor occupancy was calculated as: 100*{1 − [ratiot − 1)/(ratioc − 1)]} = % Occupancy, wherein ratiot and ratioc refer to the average thalamus to cerebellar ratio of vehicle-treated and test compound–treated animals, respectively.
Dose Selection Based on Receptor Occupancy
Food and Drug Administration (FDA) guidance recommends that the minimally effective daily dose of buprenorphine between 4 and 24 mg be empirically selected (FDA, 2018). This dose range occupies 64% to 96% of MOR in the CNS, respectively (Greenwald et al., 2014). In contrast to buprenorphine, naltrexone is prescribed at doses that occupy approximately 95% of CNS MOR (Lee et al., 1988; Weerts et al., 2008). Therefore, we benchmarked RM1490 versus naltrexone and buprenorphine in this occupancy range (65% to 90%).
Conditioned Place Preference Using Opioid Naïve Rats
The recording chamber consisted of 2 webcams and a custom-built plexiglass chamber 38”×38” ×18” that was further enclosed in a sound-attenuating box from Med Associates Inc. (Fairfax, Vermont). Animals were tracked using the video from the camera positioned above the chamber using software (Viewer2 from Biobserve GmbH). The timeline for determining place preference in opioid-naïve animals is schematized in Fig. 3A; to determine the baseline time during the pretest, a divider with a cutout door (5” ×5” ×4”) was used to divide the chamber into two sides of equal dimensions. Each side of the chamber had distinct visual and tactile cues, including floors of different consistency (smooth vs. bumpy) and visual cues (2-cm stripes vertical vs. horizontal). The time that animals spent freely exploring each side of the chamber was measured for 20 minutes (1200 seconds). To reduce the effect of intrinsic biases, animals that spent >900 seconds on one side of the chamber were excluded from the study. Conditioning began the following day, with subcutaneous injection of vehicle in the morning (between 8 and 11 AM) and test compound 4 hours later in the afternoon (between 12 and 3 PM). After each injection, animals were confined to one side of the chamber for 45 minutes and then returned to their home cage. The injections and conditioning procedure were repeated two additional times at 48-hour intervals. Two days after the last injection, the divider with the cutout was placed in the chamber, and the animals were again allowed to explore both sides of the chamber for 20 minutes (1200 seconds); the time spent on the side of the chamber associated with the test compound was determined. Preference score percent was calculated as: 100*[(test − baseline)/(600)]. Morphine at 6 mg/kg s.c. has been shown to induce place preference (Bardo et al., 1995; Tao et al., 2006).

RM1490 induces less conditioned place preference than buprenorphine and inhibits morphine-induced CPP. (A) Timeline and (B) summary data for CPP in opioid-naïve animals showing that RM1490 does not induce CPP in opioid-naïve animals. At doses that occupy >90% of CNS MOR: buprenorphine (1 mg/kg) vs. RM1490 (30 mg/kg) [mean diff: −39.15, 95% CI (−77.53 to −0.7696), P = 0.04; n = 6–7 per group, ANOVA F (2, 15) = 5.120, P = 0.02; *P < 0.05]. (C and D) Timeline and summary data for morphine-induced CPP in opioid-naïve animals. Note that morphine-induced CPP is inhibited by preinjection of RM1490 and naltrexone [mean diff: 26.73, 95% CI (6.673 to 46.78), P = 0.009, and mean diff: 42.46, 95% CI (22.40 to 62.51), P = 0.001, respectively; n = 8 per group, ANOVA F (2, 21) = 12.87, P = 0.0002; **P < 0.01]. Bup, buprenorphine; Nltrx, naltrexone; ns, not significant.
Conditioned Place Preference and Avoidance after Morphine, Buprenorphine of RM1490
Baseline time spent on either side of the conditioned place preference (CPP) chamber was determined during the pretest as described above and as schematized in Fig. 4C. Next animals were implanted subcutaneously with either two 75-mg pellets or a 2ML1 osmotic minipump from ALZET (Durect, Cupertino, CA). Minipumps contained either 1 or 3 mg/ml of buprenorphine HCl or 10 mg/ml of RM1490. Pumps were removed 5–6 days after implantation. Twenty-four hours later, vehicle was injected subcutaneously in the morning, and test compound was administered in the afternoon. The injections and conditioning procedure were repeated two additional times at 24-hour intervals. Preference was determined 48 hours later as above in the CPP procedure in opioid-naïve rats.

RM1490 precipitates fewer somatic and behavioral signs of withdrawal than naltrexone during detoxification from buprenorphine, and naltrexone does not precipitate signs of withdrawal from RM1490. (A) Timeline for removal of minipumps, injection of test compound after pump removal, and scoring of precipitated somatic signs of withdrawal. (B) Summary data showing that during detoxification from buprenorphine, RM1490 unlike naltrexone does not precipitate somatic signs of withdrawal [mean diff: −9.200, 95% CI (−13.38 to −5.021), P < 0.0001]. Furthermore, after 5 days of exposure to RM1490, naltrexone does not precipitate robust somatic signs of withdrawal [mean diff: 7.10, 95% CI (2.199 to 12.00), P = 0.001]. (C) Timeline for removal of minipumps and conditioning in CPP chambers with test compounds. (D) From the left, summary data for conditioning during detoxification from buprenorphine reveal that naltrexone induced place aversion, RM1490 did not affect place preference, and buprenorphine induced place preference (compared with RM1490 (30 mg/kg): naltrexone [mean diff: −81.73, 95% CI (−133.9 to −29.58), P = 0.001] and buprenorphine (1 mg/kg): [mean diff−52.67, 95% CI (−104.8 to −0.5111), P = 0.047] (n = 5–10 per group, ANOVA F (3, 21) = 14.10, P < 0.0001; P < 0.0001; **P < 0.01, *P < 0.05). On the right side of the graph for conditioning, after removal of minipumps containing vehicle or RM1490 (10 mg/ml), naltrexone does not induce place aversion (vehicle minipump removed [mean diff: −78.70, 95% CI (−144.2 to −13.25), P = 0.02] and RM1490 minipump removed [mean diff: −84.27, 95% CI (−149.7 to −18.81) P = 0.013]; (n = 5–10 per group, ANOVA F(2, 12) = 6.493, P = 0.0123; #P < 0.05). Bup, buprenorphine; Nltrx, naltrexone.
Scoring of Somatic Signs of Withdrawal
Somatic signs were scored and assigned weighted scores as previously described (Gellert and Holtzman, 1978; Schulteis et al., 1994). During the session, animals were monitored during two 5-minute epochs at 2 and 12 minutes after injection. The following behaviors were counted and assigned weighted scores based on the number of occurrences: wet dog shakes (<3 score = 2; >3 score = 4) and escape attempts (jumping with four feet off ground (2–4, 5–9, >10, score = 1, 2, 3, respectively). Any occurrence of teeth chattering/excessive facial grooming (score = 2), abdominal constriction (abdominal twitches; score = 2), pronounced swallowing movements (score = 2), abnormal posture (indicative of visceral discomfort; score = 3), ptosis (score = 2), erection or ejaculation (score = 3), chromodacryorrhea (porphyrin on face; score = 5), profuse salivation (score = 7), and vocalization on handling (score = 3) were noted and given weighted scores. After the session, fecal pellets and diarrhea were counted (pellets > 15/diarrhea: score = 2), and any changes in weight (score = 1 per 1% loss) were noted and scored. A global weighted score was then calculated per animal per session.
Open Field
Individual animals were injected with test compound 30 minutes prior to being placed in to an open-field arena: (a custom-built plexiglass chamber 38”×38”×18”height that was further enclosed in a sound-attenuating box from Med associates Inc. (Fairfax, VT)). Animals were tracked using the video from the camera positioned above the chamber using software (Viewer2 from Biobserve GmbH). Timeline schematic is in Fig. 6D. The dose of diazepam (3 mg/kg) has been shown to induce place preference in rats (Gray et al., 1999).
Pharmacokinetics: Collection and Analysis of Plasma Samples
To determine plasma exposures in animals implanted with ALZET minipumps: animals were lightly anesthetized with isoflurane, and approximately 0.75 ml of blood was collected from a tail vein and mixed with 0.1M EDTA. Samples were centrifuged at 4°C for 15 minutes. The supernatant was stored at −80°C until analysis. Samples were analyzed by liquid chromatography–mass spectrometry at Quintara discovery (Hayward, CA). The pharmacokinetics of RM1490 after acute injection was determined using cannulated rats at BioDuro Inc. (San Diego, CA).
Whole-Body Plethysmography
Whole body plethysmograph chambers, hardware, and recording software from DSI (St. Paul, MT) were used to record unrestrained and unanesthetized respiration, with a negative bias flow of 2.5 liters per minute. Chambers were enclosed in a sound-attenuating boxes from Med Associates Inc. (Fairfax, VT). Animals were acclimatized to the chamber for 3 days for 40 minutes per day. After acclimatization and for experiments in normal air, animals were placed into the chamber and recorded for 30 minutes; next, chambers were opened, and test compound was injected. Animals were then returned to their respective chambers, and respiration was recorded for 1–2 hours (Fig. 5A). For experiments to monitor the hypercapnic ventilatory response (HCVR) respiratory response to 10% CO2, animals were injected with drug 20 minutes before being placed into the chamber. This time point was selected based upon preliminary experiments. After test-compound injection, animals were placed into the chamber, and baseline respiration was recorded for 30 minutes (Fig. 5G). Next, the negative bias flow was switched off, and a hypercapnic gas mixture of 10% CO2, 20% O2, and 70% N2 was fed into the chamber at approximately 2 psi. After 2 minutes, the bias flow was restarted; 6 minutes later the hypercapnic gas mixture was switched off, and respiration was monitored for 15 minutes. Data were normalized by dividing by the average minute volume observed during baseline.

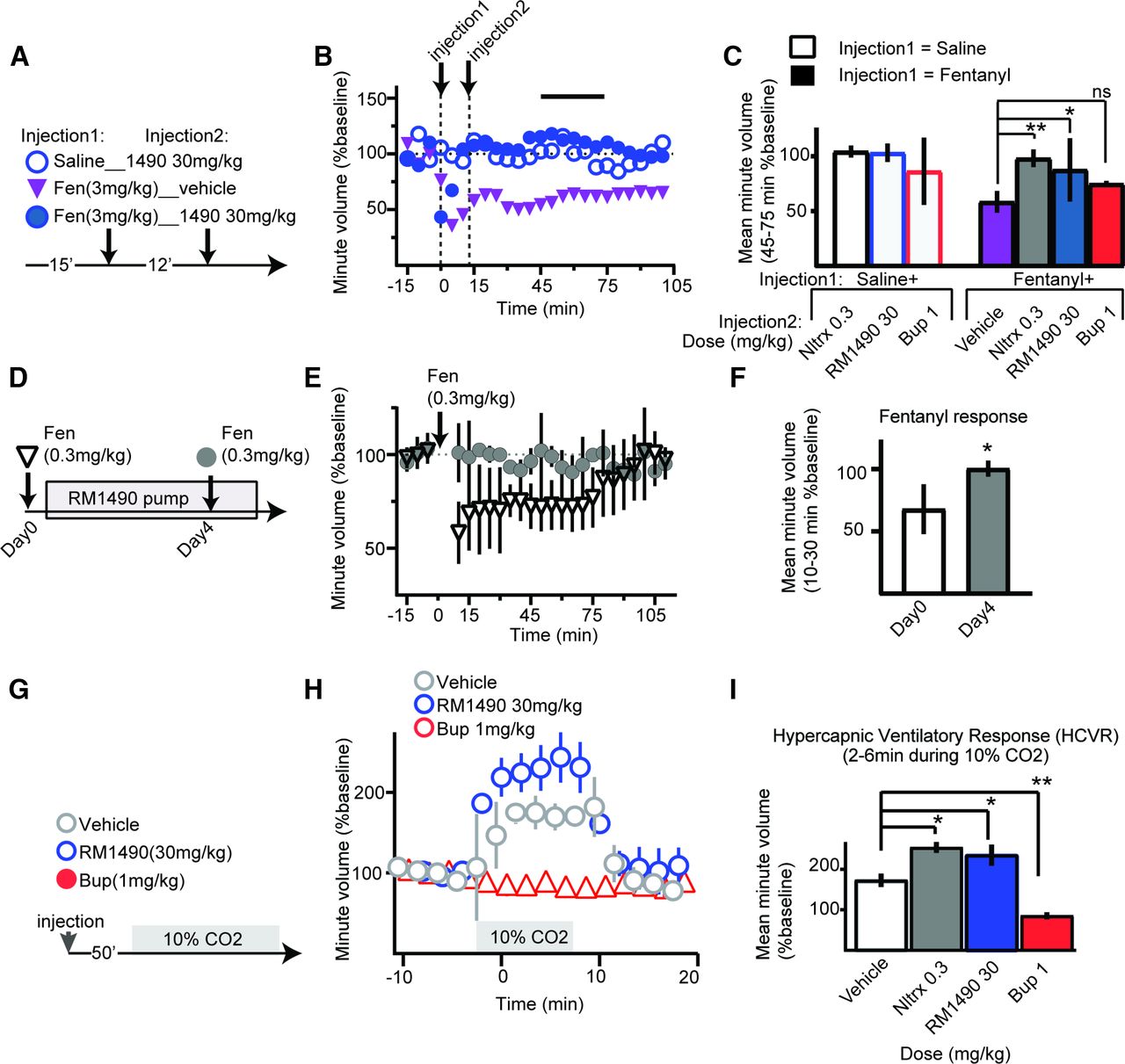
RM1490 has limited respiratory liabilities and rescues fentanyl-induced respiratory depression. (A–C) Timeline, representative example, and summary data for effect of test compounds after injection of either vehicle or fentanyl. The line horizontal in (B) indicates the 30-minute period used to calculate the average minute volume shown in (C). Note that only buprenorphine failed to rescue fentanyl-induced respiratory depression [fentanyl (3 mg/kg) + saline 58.4 ± 9.8% vs. fentanyl (3 mg/kg) + buprenorphine (1 mg/kg) 75 ± 2.6%; mean diff −16.49, 95% CI (−44.30 to 11.32), P = 0.32, ANOVA]. (D–F) Timeline and summary data for fentanyl-induced respiratory depression and occlusion of fentanyl-induced respiratory depression by subchronic RM1490 (10 mg/ml) in a subcutaneous minipump (*P < 0.05, two-tailed unpaired t test). (G–I) Timeline and summary data for effect of test compounds on minute volume in 10% CO2 (hypercapnic conditions); summary data indicate that compared with vehicle treatment naltrexone, RM1490 increased the HCVR [mean diff: −81.15, 95% CI (−112.2 to −50.09), P < 0.0001 and mean diff: −62.19, 95% CI (−93.25 to −31.13), P = 0.0004, respectively], and in contrast buprenorphine suppressed the HCVR [mean diff: 87.57, 95% CI (56.51 to 118.6), P < 0.0001; n = 5 per group, ANOVA F (3, 13) = 77.01, P < 0.0001; *P < 0.05, **P < 0.01]. Bup, buprenorphine; Fen, fentanyl; Nltrx, naltrexone; ns, not significant.
Experimental Design and Statistical Analysis
Statistics were performed using Prism (GraphPad Software Inc, San Diego, CA). Unless otherwise stated, for between subjects, comparison data were analyzed using one-way ANOVA and Dunnett’s multiple comparisons post hoc t test and are presented as mean ± S.D., with the corresponding mean difference (mean diff) and 95% confidence interval (95% CI) in the figure legends and Results section.
Results
RM1490 Is a Brain-Penetrant Neutral MOR Agonist with Limited Analgesic Activity
The potency and efficacy of RM1490 and RM1049 at the MOR were compared with naltrexone and buprenorphine in CHO membranes and cells expressing human MOR. As summarized in Fig. 1E, RM1490 had a high potency but significantly lower efficacy than buprenorphine at MOR in both the GTPγS and cAMP assays: values as EC50 ± S.D. (maximum effect : %Emax) in GTPγS assay were RM1490 (3.4 ± 0.4 (27)) versus buprenorphine (1.4 ± 0.7 (57)) and in cAMP assay were RM1490 (4.4 ± 3.5 (18)) versus buprenorphine (4.0 ± 2.3 (58)). In contrast, RM1049 was a high-affinity and high-efficacy agonist in the GTPγS assay at (0.4 ± 0.1 (91)) and in the cAMP assay at (0.3 ± 0.1 (103)) (Fig. 1, C–E). In a competition assay, RM1490 inhibited the cAMP response to DAMGO EC90 to a similar extent to naltrexone: RM1490 (26.38 ± 10.5 (90)) versus naltrexone (8.4 ± 1.9 (100)), whereas RM1049 had no inhibitory effect (Fig. 1, D–E). After subcutaneous injection, the half-life of RM1490 1 mg/kg was 2.04 ± 0.6 hours (Supplemental Fig. 1).
To assay the in vivo MOR agonist activity of RM1490, we used the acetic acid–induced writhing test. In this test, acetic acid was injected intraperitoneally, causing stimulation of visceral nociceptors and writhing; buprenorphine reduced writhing, whereas naloxone increased writhing (Fig. 1D). Similar to buprenorphine, RM1490 reduced writhing. Importantly, the effects of buprenorphine and RM1490 were antagonized by pretreatment with naloxone (Fig. 1D).
To determine whether RM1490 crosses the blood-brain barrier and binds to MOR, we used a receptor occupancy assay. As schematized in Fig. 2A, preinjection of either naltrexone (0.03 and 0.3 mg/kg), RM1490 (10 and 3 mg/kg), or buprenorphine (0.3 and 1 mg/kg) reduced carfentanil binding to MOR in the thalamus by greater than 70% and 90%, respectively (Fig. 2, B and C).
Next, we sought to compare the antinociceptive effect of RM1490 in the hot-plate test to naltrexone and buprenorphine. As schematized in Fig. 2D, we injected doses of each compound that achieved approximately 70% or greater occupancy of thalamic MOR (Fig. 2C). Only buprenorphine and the full-efficacy MOR agonist RM1049 elicited robust antinociception (Fig. 2, E and F). However, RM1490, when coinjected with the full-efficacy MOR-agonist RM1049 or with an analgesic dose of morphine, dose-dependently inhibited the antinociceptive effects of the full agonists (Fig. 2, G–I).
RM1490 Has Limited Rewarding, Aversive, and Physical Dependence Liabilities
To investigate the reward associated with RM1490, we conditioned opioid-naïve animals with doses of buprenorphine, naltrexone, and RM1490 that achieved approximately 70% and 90% occupancy of thalamic MOR as described in Fig. 2. Only buprenorphine induced a change in place preference (Fig. 3B). Next, we investigated whether RM1490 inhibited place preference induced by a high-efficacy MOR agonist. Initially, we confirmed that robust place preference was induced by morphine (6 mg/kg) and then determined that this effect was inhibited to a similar extent by preinjection of naltrexone (0.3 mg/kg) or RM1490 (30 mg/kg; ANOVA; mean diff: −15.73, 95% CI (−37.06 to 5.597), P = 0.17 Tukey’s) (Fig. 3, C and D). Given that similar levels of place preference were induced by buprenorphine (1 mg/kg) and morphine (6 mg/kg) [(48% vs. 61%) (Fig. 3B vs. Fig. 2D), P = 0.27, unpaired t test], an inhibition experiment was not possible with these two compounds.
To investigate whether RM1490 precipitated somatic signs of buprenorphine withdrawal, animals were implanted with subcutaneous minipumps continuously releasing buprenorphine (Fig. 4A). Five days after pump implantation, the average plasma levels of animals implanted with buprenorphine minipumps (4.2 ± 0.8 ng/ml) were similar to plasma levels (5.7 ± 0.4 ng/ml) required to achieve MOR occupancy of approximately 70% (Fig. 2). On day 5, the minipumps were removed. Subsequently, we determined that the global somatic score of precipitated buprenorphine withdrawal was significantly more pronounced after naltrexone (0.3 mg/kg) versus RM1490 (30 mg/kg) (mean diff: −9.200, 95% CI (−13.38 to −5.021); ANOVA P < 0.001) (Fig. 4B). Therefore, the intrinsic MOR efficacy of RM1490 is not sufficient to precipitate somatic signs of buprenorphine withdrawal.
Next, the same timeline in Fig. 4A was used in experiments to determine whether naltrexone precipitates somatic signs of withdrawal from RM1490. Animals were implanted with minipumps containing either vehicle or RM1490 at 10 mg/ml. The dose of RM1490 was selected as it achieved approximately 70% MOR occupancy in the thalamus (67 ± 8%, n = 4 animals), which matches the occupancy estimated after 5 days of exposure to buprenorphine (1 mg/ml minipump). In contrast to the naltrexone-precipitated somatic signs seen after buprenorphine detoxification, naltrexone failed to precipitate somatic signs after 5 days of exposure to vehicle or RM1490 (Fig. 4B).
Having investigated the somatic signs of RM1490 withdrawal, we sought to investigate the behavioral impact. To this end, we used the protocol schematized in Fig. 4C. After removal of the minipumps on day 5, animals were conditioned over the next 3 days with naltrexone, RM1490, or buprenorphine in the CPP chambers. Naltrexone at 0.3 mg/kg (the dose that achieves 90% MOR occupancy) induced greater place aversion in buprenorphine treated animals compared with opioid-naïve animals (−79.1 ± 16% vs. 0.1 ± 25%; P = 0.003) (Figs. 3B and 4D) and compared with animals that had been implanted with vehicle pumps (−0.4 ± 30%, P < 0.001) (Fig. 4D). In contrast, RM1490 (30 mg/kg) had no effect on place preference either in animals undergoing buprenorphine detoxification (2.6 ± 44%) or in opioid-naïve animals (8 ± 37%) (Figs. 4D and 3B, respectively). Furthermore, buprenorphine at doses that occupied approximately 70% and 90% of MOR in the thalamus induced robust place preference (Fig. 4D). Thus, in animals undergoing buprenorphine-detoxification, RM1490 was less aversive than naltrexone and less rewarding than buprenorphine. RM1490 (30 mg/kg) also failed to induce aversion during detoxification from morphine, unlike naltrexone that induced place aversion [RM1490 (30 mg/kg): −3 ± 34%, naltrexone (0.3 mg/kg: −31 ± 37%; Supplemental Fig. 3)]. In keeping with the neutral somatic symptom and CPP profile produced by RM1490 administration, naltrexone conditioning for 3 days also failed to induce a change in place preference in animals after removal of RM1490 pumps (Fig. 4D).
Since CPP conditioning entails pairing of the injection of saline and test compound with the respective sides of the chamber, a potential interpretation of the lack of effect of RM1490 on place preference in the three CPP assays undertaken (Figs. 3 and 4) is that its effects are comparable with those of saline. However, when these CPP results are considered collectively with the ability of RM1490 to inhibit morphine-induced place preference (Fig. 3D) and the results of the experiments in Figs. 1 and 2 (buprenorphine-like reduction of writhing, accompanied by inhibition of the antinociceptive effect of two full MOR agonists in the hot-plate test), a more parsimonious interpretation of the analgesia and CPP data is that at 90% MOR occupancy, the intrinsic MOR efficacy of RM1490 is neither rewarding nor aversive.
Compared with Buprenorphine, RM1490 Has Reduced Respiratory Liabilities
In these studies, we used whole-body plethysmography and three conditions to characterize the respiratory effects of naltrexone, RM1490, and buprenorphine at doses that occupy >90% of MOR in the thalamus. First, in normoxic conditions (room air), we observed that naltrexone and RM1490 do not suppress minute volume and rescue the respiratory depressant effects of fentanyl (Fig. 5, A–C). Although buprenorphine does not cause a significant decrease in respiration (86.3 ± 30.6%), it fails to rescue the respiratory depressant effects of fentanyl (Fig. 5C). Next, we confirmed that when administered subchronically in a subcutaneous minipump that RM1490 occluded fentanyl induced respiratory depression (Fig. 5, D–F). Secondly, under hypercapnic conditions (Fig. 5G)), we observed that pretreatment with naltrexone and RM1490 caused an increase in the HCVR (Fig. 5, G–I), whereas pretreatment with buprenorphine inhibited the HCVR.
To determine whether the effects of RM1490 were reversible, we compared the effects of fentanyl (3 mg/kg) on respiration before and approximately 4 hours after injection of RM1490 (10 mg/kg). There was no difference in the respiratory depressant effect of either fentanyl injection [fentanyl injection 1 (42 ± 13%) and fentanyl injection 2 at 4 hours after injection of RM1490 (52 ± 6%) P = 0.46, n = 4] (Supplemental Fig. 2).
Unlike Buprenorphine, RM1490 Does Not Exacerbate Respiratory Depression when Combined with Diazepam
To determine whether RM1490 combined with diazepam induces respiratory depression, we coinjected diazepam (3 mg/kg) with naltrexone, RM1490, or buprenorphine. Buprenorphine combined with diazepam significantly suppressed respiration (71 ± 21.3% of baseline). In contrast, neither naltrexone (93 ± 9.8%) nor RM1490 (107 ± 13.3%) suppressed respiration when coinjected with diazepam (Fig. 6, A–C). Next, we tested whether diazepam combined with naltrexone, RM1490, or buprenorphine suppressed locomotor activity in the open-field test. Compared with the distance in meters that animals injected with vehicle traveled (1.04 ± 0.39), naltrexone (0.98 ± 0.28), RM1490 (1.16 ± 0.42), buprenorphine (0.72 ± 0.35), and diazepam (0.73 ± 0.64) did not cause a change in locomotor activity when injected alone [F (4, 39) = 1.562, P = 0.2037, ANOVA]. In contrast, buprenorphine coinjected with diazepam produced a significant decrease in locomotor activity compared with animals injected with diazepam only (Fig. 6F).

Unlike buprenorphine, RM1490 does not exacerbate respiratory depression or locomotor activity when combined with diazepam. (A–C) Timeline, representative example, and corresponding summary data for effect of coinjection of diazepam with test compounds. Note that compared with diazepam only, buprenorphine combined with diazepam suppresses respiration [mean diff: 41.32, 95% CI (14.07 to 68.57), P = 0.0037], whereas combination of diazepam and either RM1490 or naltrexone does not [mean diff: 5.612, 95% CI (−24.24 to 35.46), P = 0.9, and mean diff: 19.63, 95% CI (−10.22 to 49.48), P = 0.2, respectively; n = 4–6 per group: ANOVA F (3, 14) = 6.619, P = 0.005; **P < 0.01]. (D–F) Timeline, superimposed representative 20-minute tracks of 5 animals in an open field test, and corresponding summary for the effect of test compounds as single agents or when combined with diazepam. Note that compared with diazepam as a single agent, only buprenorphine coinjected with diazepam decreases distance traveled [mean diff: 0.552, 95% CI (0.440 to 1.064), P = 0.032; n = 6–10: ANOVA F (3, 25) = 2.603, P = 0.0743 *P < 0.5]. Bup, buprenorphine; Nltrx, naltrexone; ns, not significant.
Discussion
Summary of Findings
The aims of an OUD pharmacotherapy are to mitigate signs and symptoms of opioid withdrawal, inhibit the effects of high-efficacy opioids, and minimize opioid cravings while being safe and accessible to a diverse outpatient population. In the present study, we benchmarked the novel MOR agonist RM1490 versus the legacy molecules naltrexone and buprenorphine at levels of MOR occupancy observed in clinical studies. RM1490 is as effective as naltrexone and more effective than buprenorphine at inhibiting the effects of high-efficacy opioids in three assays: inhibition of antinociception in the hot-plate test, inhibition of morphine-induced place preference, and inhibition of fentanyl-induced respiratory depression. Importantly, RM1490 is also less rewarding than buprenorphine and less aversive than naltrexone in CPP assays. Based on these preclinical results, RM1490 should inhibit the acute euphoric and respiratory effects of opioids of abuse, minimize symptoms of opioid withdrawal, and reduce opioid craving while having a lower abuse liability than buprenorphine.
RM1490 In Vivo MOR Efficacy and CNS Occupancy
Both the peripherally restricted MOR-agonist loperamide and centrally penetrant partial-agonist buprenorphine reduce writhing in the acetic acid test (Emerich et al., 1998; Labuz et al., 2007). In contrast, the antagonist naloxone increases writhing (Kokka and Fairhurst, 1977; Huang et al., 2001). RM1490 reduced writhing, indicating that its MOR intrinsic efficacy is sufficient to elicit visceral analgesia.
Antinociception in the hot-plate test correlates with both the intrinsic MOR efficacy of the agonist (Schmauss et al., 1982; Zimet et al., 1986; Gades et al., 2000), and with occupancy of thalamic MOR (Takai et al., 2018). RM1490 was not antinociceptive in the hot-plate test but reduced the effects of higher-efficacy agonists. Combined with the finding of dose-dependent occupancy of thalamic MOR by RM1490 (Fig. 2), RM1490 crosses the blood-brain barrier and dose-dependently occupies CNS MOR. Notably, RM1490 doses used in behavioral assays achieved or exceeded therapeutic MOR occupancy levels observed in clinical studies (Weerts et al., 2008; Greenwald et al., 2014).
RM1490 Comparison with Buprenorphine
Concerns associated with buprenorphine therapy include: 1) a series of active metabolites that are full MOR agonists (Moody et al., 2011; Häkkinen et al., 2012; Tompkins et al., 2014), 2) subversion for recreational purposes or to relive symptoms of dependence (Nigam et al., 1994; Eissenberg et al., 1996; Tompkins et al., 2014; Derbel et al., 2016), and 3) respiratory depression either in patients with compromised respiratory function or when buprenorphine is combined with benzodiazepines (FDA, 2012, 2018; Schuman-Olivier et al., 2013; Jones and McAninch, 2015; Dowell et al., 2016; Hirschtritt et al., 2017). Critically, preclinical models can be used to assess these high-risk circumstances.
Consistent with previous reports, we observed that buprenorphine and morphine induced comparable levels of place preference in opioid-naïve animals (Tzschentke, 2007). In contrast, RM1490 and naltrexone did not affect place preference in the same test (Fig. 3), indicating that RM1490 is less rewarding than buprenorphine.
The physical dependence liabilities of buprenorphine are detectable in antagonist-precipitated withdrawal assays. In these assays, the ability of previously ineffective doses of antagonist to precipitate somatic signs of withdrawal and aversion are measured during the acute stages of buprenorphine detoxification (Dum and Herz, 1981; Berthold and Moerschbaecher, 1988; Nigam et al., 1994; Eissenberg et al., 1996; Cowan, 2007; Paronis and Bergman, 2011; Cohier et al., 2014). We observed that doses of the antagonist naltrexone that did not induce aversion in opioid-naïve animals precipitated both somatic signs of withdrawal and place aversion during buprenorphine detoxification. In contrast, naltrexone failed to induce either place aversion or somatic signs after 5 days of exposure to RM1490 (Fig. 4). Based on these preclinical models, the physical dependence liability of RM1490 would be predicted to be substantially lower than that of buprenorphine.
The respiratory liabilities of buprenorphine are apparent when CO2 levels are elevated or when buprenorphine is coadministered with benzodiazepines. When CO2 levels are ≥5%, both buprenorphine and the high-efficacy MOR agonists (fentanyl) induce about a 50% reduction in respiration (Dahan et al., 2005; van Dorp et al., 2006; Weil et al., 1975). In accordance with this previous work, we observed that buprenorphine had limited effects on respiration in normoxic condition (room air); however, it inhibited the increase in minute volume during exposure to elevated CO2 (Fig. 5). Like naltrexone, RM1490 induced a limited increase in minute volume during exposure to CO2 (Fig. 5). It should be noted that in room air, RM1490 reversed the respiratory depressant effects of fentanyl, whereas buprenorphine failed to induce a significant recovery (Fig. 5). Based on these results, RM1490 should be safer in high-risk populations that either suffer from an impairment in respiratory function or whose respiration has been suppressed by fentanyl or another opioid.
It is estimated that 30%–66% of people using buprenorphine co-use benzodiazepines (Bramness and Kornør, 2007; Nielsen et al., 2007; Lavie et al., 2009; Maurice Preter, 2009; Hwang et al., 2016; Stein et al., 2016; Zhu et al., 2018). Despite widespread recognition of the risks, including respiratory depression and sedation, the rates of coprescription, co-use, and associated overdoses are increasing (Lintzeris and Nielsen, 2010; Schuman-Olivier et al., 2013; Jones and McAninch, 2015; Hirschtritt et al., 2017). In keeping with previous preclinical studies (Gueye et al., 2002; Nielsen and Taylor, 2005; Cohier et al., 2014), co-injection of buprenorphine with diazepam suppressed both respiration (Fig. 6) and decreased activity in the open field test (Fig. 6). In contrast, neither effect was observed upon coinjection of RM1490 with diazepam (Fig. 6). Based on these preclinical models, respiratory liabilities associated with RM1490 and diazepam co-use may be reduced compared with co-use of diazepam with buprenorphine.
RM1490 Comparison with Naltrexone
As a result of its low intrinsic MOR efficacy, naltrexone has no abuse liability and is prescribed at doses that achieve >90% occupancy of CNS MOR. At this high exposure, naltrexone inhibits euphoria and respiratory suppression associated with abused opioids but does not alleviate opioid cravings or withdrawal (Lee et al., 1988; Weerts et al., 2008; Greenwald et al., 2014; Mattick et al., 2014; Carroll et al., 2018; Morgan et al., 2018). Prior to introduction of naltrexone therapy, patients must complete an unpleasant opioid detoxification process to avoid precipitating severe symptoms of withdrawal from either opioids of abuse or replacement therapies (methadone and buprenorphine). Although deaths during naltrexone (induction and treatment) are not likely to occur as a direct result of suffering associated with opioid cravings, opioid withdrawal, or opioid detoxification, each of these factors contributes to the low rates of induction and compliance (Sullivan et al., 2007; Krupitsky et al., 2016; Carroll et al., 2018; Lee et al., 2018; Morgan et al., 2018).
Based on the present study, the intrinsic efficacy of RM1490 is low enough to provide naltrexone-like inhibition of the effects of full-efficacy opioids in three settings: analgesia, reward, and respiratory suppression. However, unlike naltrexone, the intrinsic efficacy of RM1490 is high enough that during buprenorphine detoxification, it does not precipitate somatic signs or aversion. Therefore, a molecule with RM1490-like MOR efficacy should be as effective as buprenorphine and naltrexone at inhibiting the rewarding and respiratory effects of high-efficacy opioids and should be more effective than naltrexone at alleviating symptoms of opioid craving and withdrawal.
Limitations of RM1490 and Study
Our rodent studies indicate that RM1490 strikes a balance between the benefits and liabilities of buprenorphine and naltrexone. Future clinical studies beyond the scope of the current work will be needed to determine whether RM1490 efficacy will be sufficient to relieve opioid cravings and promote compliance. Two additional limitations of the present studies should also be noted. First, compared with buprenorphine and naltrexone, relatively high doses of RM1490 were required to achieve therapeutic levels of CNS MOR occupancy. It is expected that the relatively simple molecular structure of RM1490 versus the morphinan scaffold (that the legacy molecules are based on) will enable development of more potent orally bioavailable molecules. Second, although CPP experiments were used to test reward associated with RM1490 in opioid-naïve and opioid-experienced animals, the self-administration liability of RM1490 or the willingness of animals to work for RM1490 was not measured. Notably, opioid self-administration correlates with the intrinsic efficacy of the molecule, and buprenorphine is self-administered weakly (Wade et al., 2015). Therefore, given the intrinsic MOR efficacy of RM1490 is approximately half that of buprenorphine, it is expected that RM1490 will have a negligible self-administration liability and will be less aversive than naltrexone. However, as mentioned, future studies will be needed to determine whether the intrinsic efficacy of RM1490 is sufficient to promote compliance.
Forward-Looking Statement
Legacy opioid receptor ligands have variable oral bioavailability [e.g., methadone (36%–100%) (Carlos et al., 2002; Eap et al., 2002; Lugo et al., 2005) and codeine (12%–85%) (Persson et al., 1992; Kirchheiner et al., 2007; Zur et al., 2014; Murakami, 2017)]. In addition, they have active circulating metabolites and display both complex pharmacology (due to poor selectivity among the opioid receptors) and poor pharmacokinetic properties. Historically, optimization of drug-like properties has been difficult and limited due to the synthetic complexity of the natural product core. The combination of the behavioral effects of RM1490, its relatively uncrowded molecular structure, and our comprehensive understanding of the RM1490-MOR interaction are expected to lead to orally bioavailable molecules that will be suitable for the maintenance phase of OUD therapy.
Acknowledgments
We thank Melior for assistance in developing and running the receptor occupancy studies. Compounds listed in methods were supplied by the National Institute on Drug Abuse (NIDA) drug supply program.
Authorship Contributions:
Participated in research design: Youngblood, Li, Gehlert, Medina, Schwartz.
Conducted experiments: Youngblood, Li, Medina, Schwartz.
Contributed new reagents or analytic tools: Medina.
Performed data analysis: Li, Medina, Schwartz.
Wrote or contributed to the writing of the manuscript: Gehlert, Medina, Schwartz.
Footnotes
- Received July 13, 2020.
- Accepted May 17, 2021.
This work was supported by National Institutes of Health National Institute of Drug Abuse [Grant 1UG3DA049598-01].
B.Y., D.R.G., J.C.M., and N.S. are employees of and stockholders in Epiodyne Inc. J.C.M. and K.L. are also stockholders and employees of R2M Pharma Inc.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- 95% CI
- 95% confidence interval
- CNS
- central nervous system
- CPP
- conditioned place preference
- FDA
- Food and Drug Administration
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- HCVR
- hypercapnic ventilatory response
- mean diff
- mean difference
- MOR
- μ opioid receptor
- %MPE
- percentage maximum possible effect
- OUD
- opioid use disorder
- SD
- standard deviation
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}