


Abstract
TPN672 [7-(2-(4-(benzothiophen-4-yl) piperazin-1-yl)ethyl)quinolin-2(1H)-one maleate] is a novel antipsychotic candidate with high affinity for serotonin and dopamine receptors that is currently in clinical trial for the treatment of psychiatric disorders. In vitro binding study showed that TPN672 exhibited extremely high affinity for serotonin 1A receptor (5-HT1AR) (Ki = 0.23 nM) and 5-HT2AR (Ki = 2.58 nM) as well as moderate affinity for D3R (Ki = 11.55 nM) and D2R (Ki = 17.91 nM). In vitro functional assays demonstrated that TPN672 acted as a potent 5-HT1AR agonist, D2R/D3R partial agonist, and 5-HT2AR antagonist. TPN672 displayed robust antipsychotic efficacy in rodent models (e.g., blocking phencyclidine-induced hyperactivity), significantly better than aripiprazole, and ameliorated negative symptoms and cognitive deficits in the sociability test, dark avoidance response, Morris water maze test, and novel object recognition test. The results of electrophysiological experiments showed that TPN672 might inhibit the excitability of the glutamate system through activating 5-HT1AR in medial prefrontal cortex, thereby improving cognitive and negative symptoms. Moreover, the safety margin (the ratio of minimum catalepsy-inducing dose to minimum effective dose) of TPN672 was about 10-fold, which was superior to aripiprazole. In conclusion, TPN672 is a promising new drug candidate for the treatment of schizophrenia and has been shown to be more effective in attenuating negative symptoms and cognitive deficits while having lower risk of extrapyramidal symptoms and hyperprolactinemia.
SIGNIFICANCE STATEMENT TPN672 is a promising new drug candidate for the treatment of schizophrenia and has been shown to be more effective in attenuating negative symptoms and cognitive deficits while having a lower risk of extrapyramidal symptoms and hyperprolactinemia. A phase I clinical trial is now under way to test its tolerance, pharmacokinetics, and pharmacodynamic effects in human volunteers. Accordingly, the present results will have significant impact on the development of new antischizophrenia drugs.
Introduction
Schizophrenia is a severe psychiatric disorder that profoundly affects both the individual and society. Schizophrenia is characterized by diverse psychopathology; the core features are positive symptoms (delusions and hallucinations, so-called psychotic symptoms in which contact with reality is lost), negative symptoms (impaired motivation, reduction in spontaneous speech, and social withdrawal), and cognitive impairment (temporary or permanent loss of mental function) (Kahn et al., 2015; Owen et al., 2016; Krogmann et al., 2019). Schizophrenia is characterized by a sequential trajectory that involves four phases: premorbid phase, prodromal phase, psychotic phase, and stable phase (Kelly et al., 2019). The psychotic phase is the formal onset of schizophrenia and is marked by repeated episodes of florid positive symptoms. After the first psychotic break, psychotic symptoms become exacerbations or remissions across patients and through the course of the illness, and finally the disease reaches a stable phase, during which psychotic symptoms are less prominent and negative symptoms and the stable cognitive deficits increasingly predominate (Tandon et al., 2009). The negative and cognitive symptoms are associated with long-term effects on social function.
Antipsychotic drugs remain the main approach for the treatment of schizophrenia, but there are significant unmet medical needs in the current treatments. First, antipsychotic drugs may produce clinical benefits in patients with psychosis through antagonism or partial agonism of the postsynaptic dopamine D2R. EPS are major side effects of first-generation antipsychotics caused by the blockade of D2R in the substantia nigra striatum, which is mainly manifested as abnormal movements such as muscle spasms, rigidity, tremors, restlessness, and involuntary movements (such as tardive dyskinesia) (Casey, 1996). Compared with first-generation antipsychotics, atypical antipsychotics that have antagonist activity on both D2R and 5-HT2AR show fewer side effects related to EPS, but they also have other side effects, such as dyslipidemia, weight gain, diabetes, and prolonged the time from the beginning of the QRS complex to the end of the T wave (QT interval), with no or limited effect on negative symptoms and cognitive dysfunction. Second, high levels of nonadherence, mainly because of side effects such as movement disorders and metabolic and cardiometabolic side effects, cause poor recovery and relapse of symptoms (Galletly et al., 2016). Therefore, the discovery of new antipsychotics that are effective for negative and cognitive symptoms with reduced side effects attracts great attention in both academia and industry.
Recent animal and clinical studies have shown that 5-HT1AR agonists can improve cognitive and negative symptoms of schizophrenia and can reduce EPS caused by antipsychotic drugs. 5-HT2AR antagonists and D3R partial agonists can also reduce EPS caused by dopaminergic dysfunction (Meltzer et al., 2003; Kiss et al., 2008, 2010).
The first Serotonin receptor (5-HTR) subtype to be cloned and characterized by its high affinity for 5-HT was 5-HT1AR (Meltzer et al., 2003). The 5-HT1AR are expressed on the soma, presynaptic, and postsynaptic membrane in hippocampal neurons. 5-HT1AR agonists can regulate 5-HT neuronal activity, thereby maintaining the balance of excitatory and inhibitory transmission. By enhancing the dopaminergic activity in the prefrontal cortex, 5-HT1AR agonism can improve the negative symptoms and cognitive impairment of patients (Meltzer and Sumiyoshi, 2008; Celada et al., 2013) and can also improve the positive symptoms indirectly and alleviate EPS (Newman Tancredi 2010; Shimizu et al., 2010; Celada et al., 2013). 5-HT1AR can regulate GABAergic inhibitory interneurons by regulating glutamatergic excitatory neurons and exerting neuroprotective effects (Llado-Pelfort et al., 2012). Therefore 5-HT1AR may also be an important target for antipsychotics.
D3R has been considered as a potential target for antipsychotic agents (Gross and Drescher, 2012; Sun et al., 2016). D3R are less abundant than their D2R counterparts in the brain and are principally located in mesolimbic regions such as the ventral striatum, islands of Calleja, nucleus accumbens, globus pallidus, and thalamus, but they are also found in the frontal and other cortical regions as well as the cerebellum (Watson et al., 2012). Mice genetically deficient in the D3R show increased cognitive flexibility in the attentional set-shifting task and improved retention in a passive avoidance test (Micale et al., 2010; Corponi et al., 2019). D3R antagonists demonstrated procognitive effects in experimental learning paradigms (Millan et al., 2007; Gross et al., 2013) and inhibited haloperidol-induced catalepsy (Gyertyan and Saghy, 2007). These findings suggested that D3R antagonists might have some potential against negative symptoms and reduced EPS potential. Cariprazine, an antipsychotic drug with dopamine D3R partial agonism, with better improvement of cognitive and negative symptoms effects in mice, has been successfully approved by the Food and Drug Administration (Kiss et al., 2010; Caccia et al., 2013). We hypothesized that a mixed D3R/D2R partial agonist could be an effective antipsychotic that would be free of EPS and have beneficial effects on cognition and negative symptoms (Kiss et al., 2010; Gyertyan et al., 2011).
Several atypical antipsychotic drugs, such as ziprasidone, lurasidone, aripiprazole, brexpiprazole, and cariprazine, have affinity for the 5-HT1AR, but their efficacy and side effects are different, presumably as a result of their balanced differences in 5-HT1AR/5-HT2AR/D2R/D3R affinity. The best 5-HT1AR/5-HT2AR/D2R/D3R affinity balance needs to be studied to improve the impact on cognitive dysfunction and reduce negative symptoms, EPS, and metabolic side effects. Currently, the discovered atypical antipsychotic drugs have a lower affinity for 5-HT1AR than D2R and D3R. Therefore, it may be of great significance to design and evaluate compounds with higher affinity for 5-HT1AR than D2R and comparable affinity for 5-HT2AR and D3R to D2R.
Our study examined in vitro and in vivo activities of a novel antipsychotic candidate TPN672. TPN672 (Fig. 1) (patent application: WO2019242717), 7-(2-(4-(benzothiophen-4-yl) piperazin-1-yl)ethyl)quinolin-2(1H)-one maleate, showed a powerful 5-HT1AR agonistic effect and has the unique characteristics of D2R/D3R partial agonistic and 5-HT2AR antagonizing activity. Compared with other antipsychotics, TPN672 showed weaker EPS adverse effects but significantly improved positive symptoms, negative symptoms, and cognitive impairments in phencyclidine (PCP)-induced animal models of schizophrenia.

Chemical structure of TPN672.
Materials and Methods
Animals
Male/female ICR mice (20–25 g) and male Sprague-Dawley rats (180–220 g) were used in this study. Experimental animals were housed in a thermostatically controlled room at 23 ± 2°C and 55%–65% relative humidity on a 12-hour dark/light cycle (lights on from 6:00 to 18:00). All procedures on animals were approved by the Institutional Animal Care and Use Committee of Shanghai Institute of Materia Medica or China Pharmaceutical University.
Drugs
TPN672, brexpiprazole, and PCP were synthesized by Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Aripiprazole (ARI) and risperidone (RIS) were purchased from Jiangsu Nhwa Pharmaceutical Co. Ltd. WAY100635 was purchased from Sigma-Aldrich. Canine serum prolactin ELISA kit was purchased from Abcam. All other chemicals are analytical level and were obtained from commercial sources. In most experiments, TPN672 was used as the maleate salt. For in vivo experiments, PCP was dissolved in saline and injected intraperitoneally at a dose of 5 mg/kg of body weight for rats and 7 mg/kg for mice, whereas TPN672, aripiprazole, and risperidone were suspended with 0.5% carboxymethyl cellulose-Na and administrated intragastrically at a volume of 10 ml/kg body weight.
Radioligand-Receptor Binding Assays
Human receptor binding assays were performed under the incubation conditions shown in Table 1.
Summary of binding assay conditions for human receptor assay in vitro
8-OH-DPAT, 8-hydroxy-2-dipropylaminotetralin
Radioligand-receptor binding assays were used to identify the affinity of TPN672, aripiprazole, risperidone, and brexpiprazole for G protein–coupled receptors, and the protocols were performed according to previous reports, with some modifications (Song et al., 2012). The reaction mixture containing 20–30 μg membrane receptor protein and 1 to 2 nM 3H-labeled ligand was incubated at 30°C for 50 minutes, and then the reaction was stopped in ice-cold water. The reaction mixture was transferred to the Millipore sample collector and filtered by glass microfiber filters (Whatman). The filter paper was rinsed with 50 mM Tris-HCl buffer (pH 7.4) and dried up. Radioactivity was quantified by Tri-Carb 2910TR (PerkinElmer LAS Ltd., Beaconsfield, UK) with a lipophilic scintillation solution. IC50 values were analyzed by nonlinear regression using Prism 8.0.2. Ki values were calculated according to the Cheng-Prusoff equation: Ki = IC50/(1 + S/Kd), where S is the radioligand concentration and Kd is the dissociation constant (Ishibashi et al., 2010).
Receptor Functional Activity
Receptor functional activity was performed under the incubation conditions shown in Table 2.
Summary of receptor function activity test conditions in vitro
Ultra Lance cAMP assay was used to test four compounds on D1R, D4R, and serotonin 1B receptor with antagonist mode and D2R and 5-HT1AR with agonist and antagonist mode. The compound was transferred to the assay plate by echo liquid handler, and then cells were collected with stimulation buffer. In total, 10 µl of the cell solution was transferred to the assay plate, centrifuged at 600 rpm for 3 minutes, and incubated for 60 minutes at room temperature, and then 5 μl of 4× Eu-cAMP tracer solution and 5 μl ULight-anti-cAMP solution were added to the assay plate, centrifuged at 600 rpm for 3 minutes, and incubated for 60 minutes at room temperature. Finally, the plate was read on EnVision. Readout parameters were as follows: excitation, 320 nm or 340 nm; emission, 615 nm/665 nm; delay time, 20 microseconds; window time, 200 microseconds.
Fluorescence luminescence system (FLIPR Tetra 384) calcium assay was used to test four compounds on H1, M1, α1A, 5-HT2A, 5-HT2B, and 5-HT2C antagonist mode. The cells were inoculated in a 384-well plate and incubated for 16–24 hours in a 37°C and 5% CO2 incubator. Then, the cells were taken out, the culture medium was removed, 30 μl calcium 5 dye was added to each well, and it was incubated for 1 hour at 37°C and 5% CO2. Compounds were transferred to the plate with 30 μl assay buffer by Echo. Compounds were added 15 μl per well and incubated for 10 minutes at room temperature. Inducer was added 22.5 μl per well, and the calcium flux signal was measured with FLIPR. Readout parameters were as follows: excitation, 470/495 nm; emission, 515 nm/575 nm; read interval, 1 second; number of reads, 120; number of reads before dispense, 10.
Homogeneous time-resolved fluorescence was used to evaluate the agonist and antagonist activity of human D3R expressed in CHO cells. The cells were suspended in buffer (Hanks’ balanced salt solution for agonist test, Hepes for antagonist test) and then distributed in the microplates with a density of 104 cells per well. Thereafter, NKH 477, an adenylate cyclase activator, was added at a final concentration of 1.5 μM. The reference agonist dopamine was added at a final concentration of 10 nM in the agonist activity test. For basal control measurements of the antagonist activity test, dopamine was omitted from the wells containing 1 µM butaclamol, a standard reference antagonist. After incubation for 30 minutes at 37°C, the cells were lysed, and the fluorescent receptor (D2-labeled cAMP) and fluorescent donor (europium-labeled anti-cAMP antibody) were added. After 60 minutes at room temperature, the fluorescence transfer was measured by a microplate reader (Envison, Perkin Elmer).
Electrophysiology
Concentric stimulation electrodes were placed on the second layer of the PrL brain area of the prefrontal cortex, and the whole-cell patch-clamp recording technique was used to record the AMPA receptor caused by the current stimulation on the pyramidal neurons in the fifth layer mediated evoked excitatory postsynaptic current (eEPSC). The changes of eEPSC were measured after perfusion of TPN672 (100 nM) with external fluid, and whether WAY100635 can affect the effect of TPN672 on eEPSC was measured. Then double-pulse stimulation was used to further verify the effect of TPN672 on 5-HT1AR–mediated changes in glutamate transmission.
PCP-Induced Hyperactivity in Mice and Rats
Locomotor activity in rats was measured using activity boxes (box for mice: 25 × 25 × 40 cm; box for rats: 45 × 45 × 60 cm) equipped with video tracking software (EthoVision XT, Noldus). TPN672, risperidone, aripiprazole, or vehicle was administered orally 1 hour before injection of PCP (7 mg/kg, s.c., for mice and 5 mg/kg, s.c., for rats). The locomotor activity was measured immediately after PCP injection and recorded for 1 hour.
Dark Avoidance Experiment
The dark escape box (dimensions: 46 × 19.5 × 20 cm) was subdivided into two compartments by a hurdle (1 mm in width, 3 cm in height). There is a channel with a diameter of 5.0 cm between the two chambers and a copper grid at the bottom of the box.
Mice were injected subcutaneously with PCP 7.5 mg/kg or normal saline for 7 consecutive days, and 24 hours after the last PCP administration, the animals were given the corresponding dose of drug or vehicle by gavage for 5 consecutive days. The animals were trained 30 minutes after the last dose. Before training, the head of the mouse was put into the bright room with the back of the hole, adapted to the environment for 2 minutes, and then the 0.5 mA was passed to the dark room’s copper grid. After entering the dark room, the mouse would escape to the bright room after being shocked. After 24 hours, the memory test of the mouse was performed. The time when the mouse entered the dark room for the first time (the dark avoidance incubation period) and the number of times the mouse entered the dark room (the number of errors) were recorded. If the mouse had not entered the dark room within 5 minutes, the incubation period was counted as 300 seconds. The experimental observation indexes were the escape latency and the number of errors of the mice.
Morris Water Maze
The Morris water maze consisted of a circular pool (120 cm in diameter) that was filled with water (25 ± 1°C). The mice were trained in the Morris water maze over four daily sessions (S1, S2, S3, S4). An escape platform (9 cm in diameter) was located in the center of the second quadrant 1 cm below the water surface. Black ink was added to the pool to reduce the visibility of the platform. Video tracking was conducted with a video camera focused on the full diameter of the pool. Navigation parameters were analyzed by using the video tracking system (EthoVision XT, Noldus). The four sessions were performed on consecutive days between 9:00 and 12:00.
The water maze experiment lasts 5 days. The first 4 days are the positioning navigation experiment (also the training phase), and the 5th day (that is, 24 hours after the end of the last training) was the space exploration experiment (also the test phase). After the positioning navigation experiment, the platform in the water labyrinth experimental device was removed, and the space exploration experiment was conducted 24 hours after the last training. For each daily trial, the mice were taken from the home cage and placed in one of the four randomly determined locations in the water maze with their heads facing the center of the water maze. A trial was started when the mouse was released from one of three randomly chosen start positions. After the rat found and climbed onto the platform, the trial was stopped and the escape latency was recorded. The maximum trial length was 90 seconds. In the test phase, a quadrant was selected randomly and the mice of each group were put into the water to make them swim freely for 90 seconds. Escape latency (seconds) of the mice to find the hidden platform was evaluated during each trial. If the mouse had not climbed onto the platform in 90 seconds, the experimenter guided the mouse to the platform by hand and recorded the escape latency of 90 seconds.
Mice were injected with PCP (7.5 mg/kg) or saline intraperitoneally for 10 consecutive days. After the last injection of PCP, animals were given intragastrically the corresponding dose of drug or vehicle once a day for 7 consecutive days. The Morris water maze experiment started 24 hours after the last dose.
Novel Object Recognition
The procedure was modified according to Bevins and Besheer (2006). The task was performed in a 40 × 50 × 50 cm chamber. All animals were given a habituation session during which they were allowed to freely explore the environment for 10 minutes. No objects were placed in the chamber during the habituation trial. At 24 hours after habituation, training was conducted by placing individual rats into a chamber for 3 minutes in which two identical objects (object A1 and A2) were positioned in two adjacent corners, 10 cm away from the walls. In a short-term memory test performed 1 hour after training, the mice explored the chamber for 3 minutes in the presence of one novel and one familiar object. Two triangular cones (3 cm at the bottom and 8 cm in height) were used as familiar objects, and one cylinder (3 cm in diameter and 8 cm in height) was used as a novel object. All objects presented had similar textures, colors, and sizes. The time animals spent on observing, licking, sniffing, or touching the object was recorded as object interaction. Standing, sitting, or leaning on the object is not recorded. The time spent on exploring new and old objects was recorded separately. Sprague-Dawley rats were intraperitoneally injected with PCP (5 mg/kg) for 14 consecutive days, after which the animals were washed out for 3 days and were then given the corresponding dose of drug or vehicle by gavage for 7 consecutive days (days 18–24) prior to the testing day. Object recognition in this test was reflected by discrimination index [(time with novel object − time with familiar object)/(time with novel object + time with familiar object)] (Denninger et al., 2019).
Social Interaction Test
The social interaction test was performed in an open-field box measuring 40 × 40 × 50 cm. Above the arena was a lamp (3.5 lux) that illuminated the open field. A video camera was placed above the arena to record activity in the open field. Each rat was acclimatized to the arena for a period of 10 minutes the day before testing. On the day of testing, rats were again paired with unfamiliar weight-matched conspecifics that had received the same treatment and were placed in the arena for 10 minutes. In this experiment, the time rats spent on positive social behaviors, such as combing their hairs, chasing each other’s tail, crossing each other’s body, and mutual licking, was observed and recorded.
Rats were injected with PCP (5 mg/kg) or saline intraperitoneally for 14 consecutive days. After the last injection of PCP, the animals were washed for 7 days, after which the animals were given the corresponding dose of drug or vehicle by gavage once a day for 7 consecutive days. The social experiment started 24 hours after the last dose.
Catalepsy Test
Male and female ICR mice were used in this test. Animals were given intragastrically the corresponding dose of drug or vehicle. After 1, 2, and 4 hours of administration, the forepaws of mice were placed on a stainless steel bar. If this position was kept for 20 seconds or longer, catalepsy would be judged to be positive.
Prolactin
Female ICR mice were treated with a single dose of TPN672, risperidone, and aripiprazole for the toxicity evaluation, and blood was collected 1 hour after administration. Serum was prepared by centrifugation at 4°C, 1600g, and the prolactin concentration in the serum was detected with the mice serum prolactin ELISA kit.
Statistical Analysis
For in vitro assays, the concentration-response curves, Ki, IC50, EC50, and Emax values were calculated by nonlinear regression analysis using GraphPad Prism 8.0.2. For in vivo and electrophysiological experiments, data were analyzed using GraphPad Prism 8.0.2 and were presented as means ± S.E.M. The data were analyzed statistically by one-way ANOVA and Dunnett’s post hoc test (A, B) or two-way ANOVA and Dunnett’s post hoc test. Student’s t test (for comparisons between two groups) was used in the social interaction test between the vehicle-treated and model groups and the electrophysiological experiments. P < 0.05 was set as statistical significance.
Results
In Vitro Pharmacology
To determine the molecular targets that mediate the response to TPN672, the compounds were tested against several known molecular targets (G protein–coupled receptors). Table 2 summarizes the affinity values, and of particular interest are the relatively high affinities of TPN672 for 5-HT1A (Ki = 0.23 nM), which is 1624.3- and 30.2-fold to that of risperidone and aripiprazole, respectively, and it had higher selectivity for 5-HT1A related to D2, with a ratio of 77.8 (determined by the affinity value of 5-HT1AR divided by that of D2R), than risperidone (ratio < 0.01), aripiprazole (ratio = 0.59), brexpiprazole (ratio = 2.5), and cariprazine (ratio = 0.23) (Table 3). It is also interesting that TPN672 showed a good balance between D2R and D3R, whose Ki values are 17.91 and 11.55 nM, respectively. The follow-up functional profile testing showed that TPN672 exhibited a broad range of activities at several receptors, including D2R, D3R, 5-HT1A, and 5-HT2AR (Fig. 2; Table 4). The most notable activity of TPN672 was its agonistic activity to 5-HT1AR (maximum efficacy = 100%, EC50 = 0.28 nM), which is far more potent than that of aripiprazole (EC50 = 159 nM) and brexpiprazole (EC50 = 66.8 nM). The maximum agonistic and antagonistic activities of TPN672 for D2R were 27.52% and 61.71%, respectively; for D3R, they were 59.0% and 19.3%, respectively. Its maximal antagonistic activity for 5-HT2AR was 99.6%.
Binding affinities for cloned human receptor in vitro (mean ± S.E.M., n = 3)
Data represent Ki obtained from three experiments performed in duplicate for each concentration and expressed as mean values (nonlinear regression analysis).

Effects of TPN672 on D2R, D3R, 5-HT1AR, and 5-HT2AR compared with antipsychotic drugs aripiprazole, risperidone, and brexpiprazole. (A) D2R agonist. (B) D2R antagonist. (C) D3R agonist. (D) D3R antagonist. (E)5-HT1AR agonist. (F) 5-HT2AR antagonist. The effects are determined by cAMP production or intracellular Ca2+ concentrations. The concentrations of test compounds range from 0.03 nM to 100 μM. Data are presented as means ± S.E.M. (n = 3) and show percentage rate of baseline.
Functional effects of TPN672 on human receptor in vitro (IC50/EC50, nM)
Data represent IC50 or EC50 (nonlinear regression analysis) obtained from a single experiment performed in triplicate for each concentration.
Electrophysiology
We performed whole-cell patch-clamp recording in brain slice containing the PrL and used concentric bipolar electrode to stimulate fibers containing glutamatergic afferents to the PrL (Fig. 3A). EPSCs were evoked by local electrical stimulation in the presence of GABAA receptor antagonist picrotoxin and NMDA receptor antagonist d-2-Amino-5-phosphonovalerate. As illustrated in Fig. 3, B and C, the tested sample PrL neuron was clamped at −70 mV, and the recorded eEPSCs were totally blocked by AMPA receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline, indicating that the recorded eEPSCs are mediated by AMPA receptor. Then, we examined the effect of TPN672 on eEPSCs in PrL to address whether it could modulate glutamate transmission. The results showed that TPN672 (10 μM) induced a significant decrease in the amplitude of eEPSCs to 67.51 ± 3.97% of the control (n = 10, P < 0.001, paired t test), and this inhibitory effect of TPN672 on glutamatergic eEPSCs could be washed out (Fig. 3, D and E). To confirm whether the glutamatergic transmission decreased via a presynaptic mechanism, we further examined the effect of TPN672 on the paired-pulse ratio (PPR). As shown in Fig. 3F, stimuli elicited a pair of eEPSCs, with the second eEPSC amplitude larger than the first one. Application of TPN672 reduced the peak amplitude of both EPSCs. However, TPN672 significantly enhanced the paired-pulse facilitation of eEPSCs from 1.13 ± 0.15 to 1.35 ± 0.15 (n = 5, P < 0.05, paired t test; Fig. 3G), strongly suggesting that a presynaptic mechanism mediates the inhibition of eEPSCs induced by TPN672 in prefrontal cortex. Finally, bath application of WAY100635 (3 µM), a 5-HT1AR receptor antagonist, significantly blocked the inhibition of TPN672 on eEPSCs (n > 5, P > 0.05, paired t test; Fig. 3, H and I).

Activation of 5-HT1AR inhibits the glutamatergic transmission in the mPFC pyramidal neurons. (A) The image shows the placement of stimulating electrode and the recorded mPFC neuron on a coronal brain slice. (B and C) Raw traces show the AMPA receptor–mediated eEPSC on mPFC neuron in the presence of pertussis toxin. Note that the fast evoked EPSC was totally blocked in the presence of AMPA/KA receptor antagonist NBQX. (D) Bath application of TPN672, a highly potent artificial synthetic 5-HT1AR agonist, decreases the amplitude of eEPSCs, and this effect could be washed out. (E) Bar graphs show the effect of TPN672 on the AMPA-mediated eEPSC. (F) TPN672-induced decrease in amplitude of the eEPSCs was associated with an increase in the PPR. (G) The plots and group data of the mean PPR obtained in the absence and presence of TPN672. (H and I) The inhibitory effect of TPN672 on eEPSCs was blocked by selective 5-HT1AR antagonist WAY100635 (n = 5). *P < 0.05 and ***P < 0.001 vs. control, Student’s t test. D‐APV,D-2-amino-5-phosphonovalerate. Ctrl, control. NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline. mPFC, medial prefrontal cortex.
Effects of TPN672 on PCP-Induced Hyperactivity in Mice and Rats
Acute treatment of PCP can induce robust hyperactivity in rodents, similar to the positive symptoms of patients with schizophrenia, and is used as a routine assay in preclinical studies and is widely used to screen novel compounds with antipsychotic effects (Moffat et al., 2017; Dedic et al., 2019).
In the open-field test in mice, results of the two-way ANOVA showed that drug treatment was the only significant factor [P < 0.001, F(6,124)= 8.41], and there were no significant differences among sex [P = 0.7327, F(1,124) = 0.1171]. TPN672 significantly inhibited the increase in spontaneous movement distance of animals induced by PCP in a dose-dependent manner (Fig. 4). Mice receiving 0.1–0.3 mg/kg TPN672 (0.1 and 0.3 mg/kg, P = 0.0140 and P = 0.0001, respectively) showed effects similar to animals receiving 1 mg/kg aripiprazole (P = 0.0134) or 0.1 mg/kg risperidone (P = 0.0001) (two-way ANOVA and Dunnett’s post hoc test). Rats administered 3–10 mg/kg TPN672 showed similar or better effects than animals administered 10 mg/kg aripiprazole (3 and 10 mg/kg TPN672, P = 0.0037 and P < 0.0001, respectively; 10 mg/kg aripiprazole, P = 0.0179) (one-way ANOVA test with Dunnett’s post hoc test).

Single acute treatment with TPN672, ARI, and RIS inhibited the hyperactivity induced by PCP. (A) Inhibition of hyperactivity in rats (n = 12 to 13 males per group). (B) Inhibition of hyperactivity in mice (n = pooled analysis of 10 males and 9 to 10 females per group). Data are presented as means ± S.E.M.; ###P < 0.001 vs. vehicle, two-way ANOVA and Dunnett’s post hoc test; *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle + PCP, one-way ANOVA test with Dunnett’s post hoc test. Veh, vehicle.
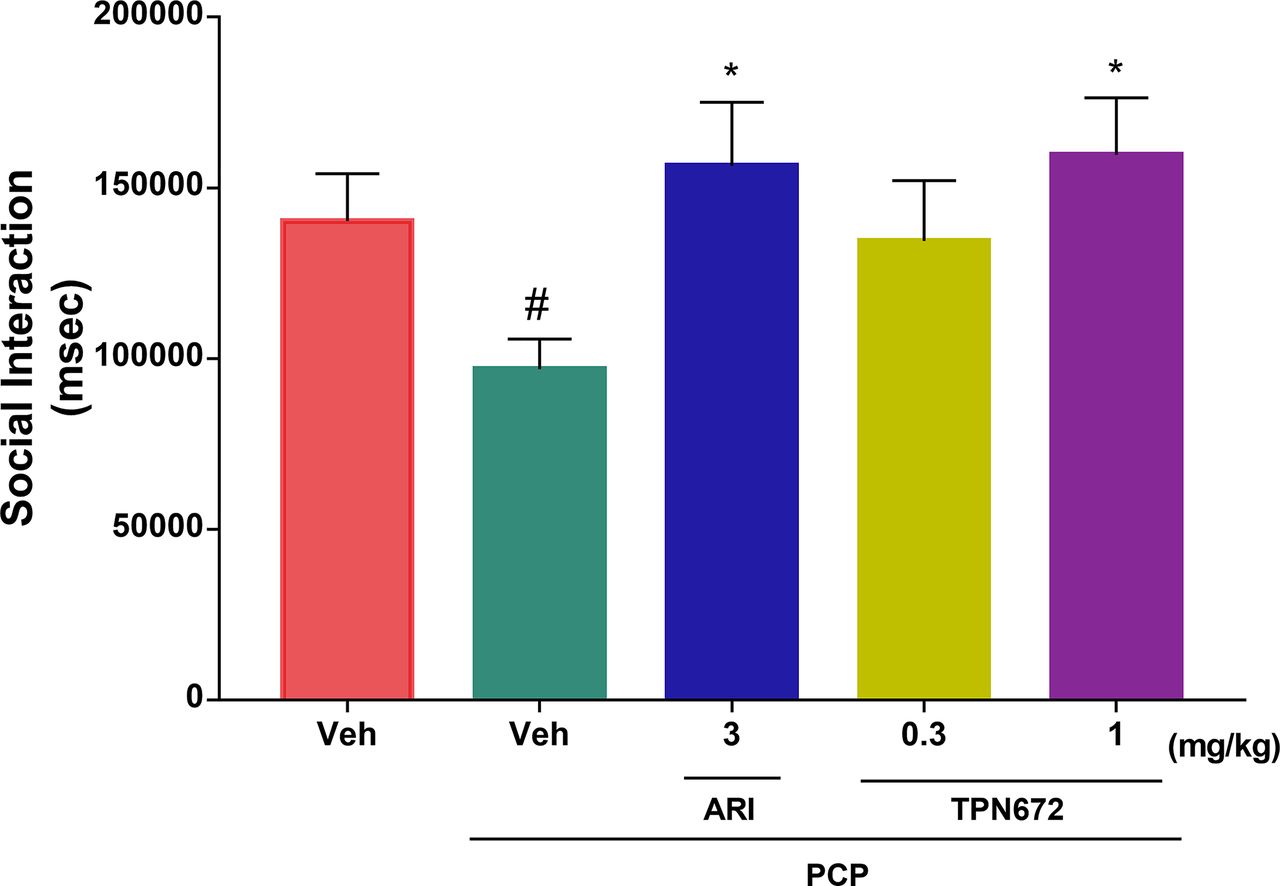
Effect of TPN672 on Social Interaction Behavior of Model Rats
PCP at a dose of 5 mg/kg significantly decreased the total time spent in social interaction (P = 0.0489 vs. vehicle). TPN672 improved the social deficits caused by PCP (Fig. 5). Post hoc analysis of one-way analysis variance for multiple comparisons showed that administration of 1 mg/kg TPN672 significantly improved the social deficits in the PCP rat model compared with the untreated group (P = 0.0305). The positive control, aripiprazole, at doses of 3 mg/kg also significantly improved the social deficits caused by PCP (P = 0.042) (Fig. 5).

Effects of TPN672 and aripiprazole on PCP-induced social deficits in rats (n = 12–14 males per group). Data are presented as means ± S.E.M.; #P < 0.05 vs. vehicle, Student’s t test; *P < 0.05 vs. vehicle + PCP, one-way ANOVA test with Dunnett’s post hoc test. Veh, vehicle.
Effects of TPN672 on Cognitive Deficits
To examine potential therapeutic effects of TPN672 on cognitive deficits commonly associated with schizophrenia, we performed three experiments. In the passive avoidance task, we observed that PCP reduced passive avoidance (PA) error latency (P < 0.0001 vs. vehicle) and drastically reduced times of PA error (P = 0.0002 vs. vehicle), suggesting impaired cognition. TPN672 (0.025 and 0.5 mg/kg) induced a dose-dependent inhibition of PA error latency (P = 0.0129 and P < 0.0001, respectively) and PA error times (P = 0.0022 and P < 0.0001, respectively), similar to aripiprazole (0.4 mg/kg, P < 0.0001 for error latency and P < 0.0001 for times of PA error) (Fig. 6, A and B).

Effects of TPN672 on cognitive deficits induced by PCP. (A and B) Passive avoidance experiment (N = 20 females per group), one-way ANOVA test with Dunnett’s post hoc test. (C) Morris water maze test (N = 10 males and 10 females per group), two-way ANOVA and Dunnett’s post hoc test. (D) Novel object recognition test (N = 8–12 males per group), one-way ANOVA test with Dunnett’s post hoc test. Data are presented as means ± S.E.M.; #P < 0.05, ###P < 0.001 vs. vehicle, one-way ANOVA test with Dunnett’s post hoc test; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle + PCP. Veh, vehicle.
The Morris water maze test has been widely used to examine spatial learning and memory in rodents. Results of the two-way ANOVA showed that drug treatment was the only significant factor [P = 0.0002, F(5,93) = 5.329], and there were no significant differences among sex [P = 0.9419, F(1,93) = 0.0053]. The latency to locate the platform was obviously reduced in mice treated with 0.05 or 0.1 mg/kg TPN672 in the test phase (day 4) of the Morris water maze (P = 0.0001 and P = 0.0003, respectively). TPN672 showed comparable effects at 0.05 and 0.1 mg/kg to that of aripiprazole treatment at 0.4 mg/kg. The behavioral data demonstrated that TPN672 effectively alleviated PCP-induced impairments in spatial learning and memory (0.05–0.1 mg/kg, oral; Fig. 6C).
Novel object recognition was used to directly examine the effects of TPN672 on cognitive deficits in PCP-treated rats. In the 3-minute test experiment, rats treated with vehicle showed significant new object recognition damage by spending approximately equal time exploring a familiar object and a novel object. We found that TPN672 (0.3 mg/kg, oral) exhibited comparable effects to that of aripiprazole (3 mg/kg, oral) (Fig. 6D). Both treatments increased the discrimination index (3 mg/kg ARI, P = 0.0133; 0.3 mg/kg TPN672, P = 0.0079), one indicator of recovered cognitive functions.
Adverse Reactions Related to D2R Antagonism
Catalepsy
The minimum dose of TPN672 that induced catalepsy in mice is 1 mg/kg. The dose ratios of cataleptic activity against the inhibitory effect on dopaminergic stimulant-induced behavior and PA tests were 10 for TPN672 and 1 for aripiprazole and risperidone (Table 5).
EPS liability of TPN672 and other antipsychotics
Hyperprolactinemia
D2R antagonism of schizophrenia drugs has the risk of inducing hyperprolactinemia in animals. Mice were given TPN672, risperidone, and aripiprazole orally as a single dose. We found that TPN672 (0.3 mg/kg, oral) and aripiprazole (3 mg/kg, oral) showed no significant difference in serum prolactin level compared with the vehicle-treated mice (Fig. 7), whereas risperidone (0.1 mg/kg) induced a significant increase (P = 0.0099 vs. vehicle) (Fig. 7).

Effect of acute administration of TPN672, ARI, and RIS on the serum prolactin content of female mice. N = 5 per group. Data are presented as means ± S.E.M.; *P < 0.05 vs. vehicle, one-way ANOVA test with Dunnett’s post hoc test. Veh, vehicle.
Discussion
Our results demonstrate that TPN672 is a novel antipsychotic drug with unique characteristics of 5-HT1AR agonism, D2R/D3R partial agonism, and 5-HT2AR antagonism.
Common adverse events of schizophrenia drugs include headache, insomnia and restlessness, hyperprolactinemia, and weight gain. Preclinical and clinical studies suggested that the insufficient signal transduction of D1R in the prefrontal lobe results in cognitive impairment (Goldman-Rakic et al., 2004). The affinity of TPN672 for D1R was roughly equivalent to D2R and D3R, but the antagonistic activity for D2R and D3R was 26-fold and 17-fold for D1R, which suggested that TPN672 had weak D1R antagonistic activity and may have low propensity for causing cognitive impairment. For in vitro functional activity assays, TPN672 had weak antagonistic activity on D4R (related to the positive symptoms of schizophrenia), D1R (related to cognitive impairment), α1A receptor (related to orthostatic hypotension), H1 receptor (related to sedation and weight gain), or cholinergic M1 receptor (related to cognitive impairment) (Table 3) (Kroeze et al., 2003; Das, 2016). In vivo animal experiment results indicate a low risk of EPS and hyperprolactinemia after TPN672 treatment. Research results indicate that TPN672 is expected to be safe and tolerable in clinical practice.
The results of efficacy evaluation suggest that TPN672 not only treats positive symptoms of schizophrenia but also improves negative symptoms and cognitive impairments in the PCP-induced schizophrenia model.
Distinct from other antipsychotics, TPN672 in vitro exhibits powerful affinity to 5-HT1AR and similar affinities to D3R (Ki = 11.55 nM) and D2R (Ki = 17.91 nM) (Table 2).
In addition, TPN672 dose-dependently reduced the PCP-induced hyperactivity in mice and rats, suggesting the potential efficacy of TPN672 on the positive symptoms in schizophrenia. The antagonistic activities of TPN672 on D2R and D3R can inhibit the overactivation of dopaminergic neurons. Moreover, 5-HT1AR agonistic activity can reduce glutamate release, thereby decreasing dopamine release in the midbrain and ameliorating the positive symptoms of patients.
At present, negative symptoms and cognitive disorders of schizophrenia cannot be effectively managed, which hinders patients with schizophrenia from returning to normal life (Kahn R. S., et al, 2015). Subchronic administration of PCP is a widely employed animal model to replicate negative symptoms and cognitive impairments of patients with schizophrenia in antipsychotic research. We used this approach to induce schizophrenia-like behaviors in rodents and performed a sociability test to assess the effects of TPN672 on negative symptoms. Meanwhile, we used the passive avoidance task, the water maze experiment, and the novel object recognition experiment to evaluate the reversal of cognitive impairment by TPN672. Our results show that TPN672 significantly improved negative symptoms. In the passive avoidance task, the water maze task, and the novel object recognition test, TPN672 treatment was effective for cognition improvement.
According to the results of the EPS evaluation experiment, the safety margin of TPN672 is 10 times that of risperidone and aripiprazole, which may translate into a better clinical safety/compliance profile. 5-HT1AR and 5-HT2AR in the prefrontal cortex are mostly expressed in pyramidal neurons. 5-HT1AR agonism and/or 5-HT2AR antagonism may indirectly regulate the balance of excitatory and inhibitory transmission in pyramidal neurons. Studies have found that selective 5-HT1AR agonists can stimulate the release of nigrostriatal dopamine and reverse the extrapyramidal side effects caused by D2R antagonists (Nunez 2008; Shimizu et al., 2010).
The imbalance in striatal dopamine levels can lead to a disorder of prolactin secretion (Vanover et al., 2018). Most antischizophrenia drugs, such as risperidone, have side effects that induce hyperprolactinemia, which can lead to sexual dysfunction or aggravation of negative symptoms (Bostwick et al., 2009; Besnard et al., 2014; Peuskens et al., 2014). Therefore, prolactin levels were measured in the safety evaluation after single dosing of TPN672. TPN672 had no effect on the prolactin levels at a dose of 0.3 mg/kg in mice, which is 3-fold of the minimum effective dose (0.1 mg/kg) in mice, suggesting a low risk of hyperprolactinemia.
The glutamate metabolism pathway is a potential molecular mechanism that affects schizophrenia cognition (Thomas et al., 2017). AMPAR is one of the family of glutamate receptors and is considered a suitable target for drug discovery and development because it plays a key role in synaptic plasticity, which is the foundation of learning and memory, including long-term potentiation and long-term depression (Partin, 2015). AMPAR were implicated in the cellular mechanisms underlying the observed antidepressant effects of subanesthetic doses of ketamine (Malhotra et al., 1996). The results of electrophysiological experiments (Fig. 3) showed that TPN672 may inhibit the glutamatergic transmission mediated by AMPAR between synapses and that its mechanism may be related to the activation of presynaptic 5-HT1AR. TPN672 may inhibit the excitability of the glutamatergic system through the 5-HT1AR, thereby improving cognitive and negative symptoms.
Post-mortem and neuroimaging studies have shown that the 5-HT1AR density in the cortex and amygdala of patients with schizophrenia has changed. Evidence in rodent models suggests that activation of 5-HT1AR can block D2R-induced EPS, regulate dopaminergic neurotransmission in the frontal cortex, have a positive effect on mood, and ameliorate N-methyl-D-aspartate receptor antagonist-induced cognitive and social interaction disorders (Celada et al., 2013). A new drug candidate for schizophrenia treatment, SEP-363856, was reported to be a trace amine–related receptor 1 and 5-HT1AR agonist (Dedic et al., 2019; Koblan et al., 2020). Interestingly, early research shows that the selective agonist of trace amine–related receptor 1, RO5166017, can increase the efficacy of 5-HT1AR partial agonists and change the desensitization rate of 5-HT1A autoreceptor in dorsal raphe nucleus (Revel et al., 2011). Although SEP-363856 does not have D2R occupancy, it still exhibits antipsychotic-like behavioral characteristics, indicating that antischizophrenia drugs do not necessarily have D2R antagonistic effects. This further suggests the important role of 5-HT1AR agonistic activity in the development of antischizophrenia drugs.
TPN672 is a novel antischizophrenia drug with 5-HT1AR > 5-HT2AR > D2R ≈ D3R activity characteristics, showing robust efficacy in PCP-induced positive, negative, and cognitive-impairment animal models. Its strong activation of 5-HT1AR may contribute to higher efficacy and safety profile and overcome the limitations of current antipsychotic drugs and reduce the risk of recurrence. Partial agonism of D2R and agonism of 5-HT1AR combined may lower the risk of side effects such as EPS and hyperprolactinemia synergistically, and they are expected to increase patient compliance. TPN672 is currently being investigated in a phase I clinical trial in China (ClinicalTrials.gov identifier: NCT03931668).
Acknowledgments
The authors thank Zikai Zhou for expert suggestions and discussions.
Authorship Contributions
Participated in research design: Y. Wang, Shen, Z. Wang, L. He.
Conducted experiments: Y. Wang, Wu, Peng, Feng.
Contributed new reagents or analytic tools: Y. Wang, Yang, Abame.
Performed data analysis: Y. Wang, Y. He, Z. Wang, L. He.
Wrote or contributed to the writing of the manuscript: Y. Wang, Z. Wang, L. He.
Footnotes
- Received November 11, 2020.
- Accepted May 4, 2021.
This work was supported by the National Natural Science Foundation of China [Grant 81673434]; Double First-Class University plan [Grant CPU2018GY22]; Special Foundation of Chinese Academy of Sciences for strategic pilot technology [Grants XDA12040105 and XDA12040337]; National Science & Technology Major Project Key New Drug Creation and Manufacturing Program, China [Grant 2018ZX09711002]; and Science and Technology Commission of Shanghai Municipality [Grant 17431900500].
No author has an actual or perceived conflict of interest with the contents of this article.
Abbreviations
- ARI
- aripiprazole
- D1R
- dopamine 1 receptor
- D2R
- dopamine 2 receptor
- D3R
- dopamine 3 receptor
- D4R
- dopamine 4 receptor
- eEPSC
- evoked excitatory postsynaptic current
- EPS
- extrapyramidal symptom
- 5-HT
- serotonin
- 5-HT1AR
- serotonin 1A receptor
- 5-HT2AR
- serotonin 2A receptor
- PA
- passive avoidance
- PCP
- phencyclidine
- PPR
- paired-pulse ratio
- RIS
- risperidone
- S
- session
- Copyright © 2021 The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}