


Abstract
β3-Adrenergic receptor expression is enhanced in the failing heart, but its functional effects are unclear. We tested the hypothesis that a β3-agonist improves left ventricular (LV) performance in heart failure. We examined the chronic effects of a β3-agonist in the angiotensin II (Ang II)–induced cardiomyopathy mouse model. C57BL/6J mice were treated with Ang II alone or Ang II + BRL 37344 (β3-agonist, BRL) for 4 weeks. Systolic blood pressure in conscious mice was significantly elevated in Ang II and Ang II + BRL mice compared with control mice. Heart rate was not different among the three groups. Systolic performance parameters that were measured by echocardiography and an LV catheter were similar among the groups. LV end-diastolic pressure and end-diastolic pressure-volume relationships were higher in Ang II mice compared with control mice. However, the increase in these parameters was prevented in Ang II + BRL mice, which suggested improvement in myocardial stiffness by BRL. Pathologic analysis showed that LV hypertrophy was induced in Ang II mice and failed to be prevented by BRL. However, increased collagen I/III synthesis, cardiac fibrosis, and lung congestion observed in Ang II mice were inhibited by BRL treatment. The cardioprotective benefits of BRL were associated with downregulation of transforming growth factor-β1 expression and phosphorylated-Smad2/3. Chronic infusion of a β3-agonist has a beneficial effect on LV diastolic function independent of blood pressure in the Ang II–induced cardiomyopathy mouse model.
Significance Statement Chronic infusion of a β3-adrenergic receptor agonist attenuates cardiac fibrosis and improves diastolic dysfunction independently of blood pressure in an angiotensin II–induced hypertensive mouse model. This drug might be an effective treatment of heart failure with preserved ejection fraction.
Introduction
Heart failure with preserved ejection fraction (HFpEF) comprises approximately half of the cases of congestive heart failure (HF) and has a poor prognosis, despite preserved systolic function (Senni et al., 1998). The survival rate has increased over time in patients with HF with reduced ejection fraction (HFrEF) but not in patients with HFpEF (Owan et al., 2006). Recently, sacubitril/valsartan, which is an angiotensin receptor-neprilysin inhibitor, was found to improve survival and reduce the rate of hospitalization for HF compared with enalapril in patients with HFrEF (Morrow et al., 2019). However, an optimal therapeutic strategy for patients with HFpEF has not been established. The mechanism of HFpEF is considered to be different from that of HFrEF. Therefore, investigation of the mechanisms and therapeutic strategies for HFpEF is important.
β-Adrenergic stimulation is one of the most important mechanisms for modulation of cardiac function (Wallukat, 2002). In addition to β1- and β2-adrenergic receptors (ARs), a third β-AR called β3-AR has been characterized in humans and several species, such as rat, mouse, sheep, and dog myocardium (Gauthier et al., 1996; Zhao et al., 2007). β3-AR plays an important role in modulating cardiovascular function in HF (Gauthier et al., 1998; Pott et al., 2006). In the chronically failing heart, the inotropic response is diminished with elevated circulating catecholamines, partly by downregulation and desensitization of β1-AR (Bristow et al., 1982; Engelhardt et al., 1996). Conversely, recent evidence suggests that β3-AR is upregulated in the failing heart in human and animal models (Cheng et al., 2001; Moniotte et al., 2001; Treskatsch et al., 2014). In contrast to β1/β2-AR, β3-AR stimulation induces a negative inotropic effect through activation of a nitric oxide synthase pathway (Gauthier et al., 1998; Brixius et al., 2006; Pott et al., 2006; Belge et al., 2014; Schena and Caplan, 2019). The role of β3-AR in modulating heart function is controversial. In a chronic β3-AR deficiency model, a cardioprotective effect through improving sarcoplasmic reticulum Ca2+ uptake was reported (Ziskoven et al., 2007). However, adverse left ventricular (LV) remodeling was observed in mice lacking β3-AR with pressure overload (Moens et al., 2009). Another study that used mice with cardiac overexpression of β3-AR that were subjected to neurohormone-induced cardiomyopathy suggested protection (Belge et al., 2014). RR + SS-(±)-4-(2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl)phenoxyacetic acid (BRL) 37344 is possibly the most widely used of all of the β3-AR agonists in humans and mice (Schena and Caplan, 2019). In acute experiments in sheep, there was no difference between the effects of BRL on stroke work and cardiac output in the chronic ischemic failing heart and the normal heart. There was also a negative relationship between the dose of BRL and the end-systolic pressure-volume relation in the normal heart. However, there was a significant shift in the relationship between the dose of BRL and the end-systolic pressure-volume relation in a positive direction after induction of the failing heart in this previous study (Bundgaard et al., 2010). Chronic subcutaneous administration with BRL can significantly reduce the occurrence of ventricular tachycardia (Zhou et al., 2008). Niu et al. (2012) and Niu et al. (2014) showed that chronic infusion of BRL had substantial cardioprotective effects, preventing cardiac hypertrophy, cardiac fibrosis, and systolic dysfunction in the failing heart. However, the physiologic role of β3-AR stimulation and whether chronic infusion of a β3-agonist improves hemodynamic assessment of cardiac function in the HF model remain unknown.
The angiotensin II (Ang II)–infused mouse is a well established model through blood pressure (BP)-dependent and BP-independent mechanisms of hypertensive cardiomyopathy. Chronic pressure overload initially induces compensated LV hypertrophy (LVH) but finally causes cardiac fibrosis and damage to myocytes, which lead to HF (Mehta and Griendling, 2007; Xu et al., 2008; Yagi et al., 2008; Peng et al., 2011; Valero-Munoz et al., 2017; Brenes-Castro et al., 2018). A β1-AR blocker prevented an increase in cardiac cytokines, such as transforming growth factor (TGF)-β1, in a cardiomyopathy model with continuous Ang II infusion (Becher et al., 2012). Therefore, the current study was undertaken to test the hypothesis that a β3-agonist improves cardiac remodeling and LV diastolic function in the Ang II–induced cardiomyopathy mouse model.
Materials and Methods
Animals.
In total, 22 C57BL/6J male mice (8 to 9 weeks old; CLEA Japan, Tokyo, Japan) were included in this study. The mice were kept on a constant 12-hour light/dark cycle and provided standard mouse chow and water ad libitum.
The 22 mice were randomly divided into three groups and treated over a 4-week period. The study groups consisted of the control group (n = 7), the Ang II–treated group (1.4 mg/kg per day; Sigma-Aldrich Japan, Tokyo, Japan) (n = 8), and the Ang II + BRL–treated group (2.4 mg/kg per day; Sigma-Aldrich Japan) (n = 8). Niu et al. (2012) examined the effects of chronic BRL infusion in a transverse aortic constriction (TAC) mouse model of HF. The dose of BRL used in this study was decided as previously described. Ang II and BRL were administered using an osmotic minipump (Alzet model 2004; Alza Corp., Mountain View, CA). One mouse in the Ang II + BRL group died from intrathoracic bleeding on the 4th day. BP and echocardiographic data before implantation of the pumps of this mouse were similar to other mice. All mice, except for this mouse, survived over the 4-week Ang II administration period. Therefore, the Ang II + BRL group finally comprised seven mice.
The care and treatment of experimental animals were in accordance with institutional guidelines. This study was approved by the Animal Care and Use Committee of Nippon Medical School.
BP and Heart Rate.
Before implantation of the osmotic minipumps and at the end of this study, systolic BP and heart rate were measured in a conscious state in all mice using the indirect tail-cuff method (BP-98A; Softron, Tokyo, Japan). The experiments were conducted in a quiet location, where mice remained for 30 minutes before experiments began. The conscious mice were individually restrained in a mouse holder at an ambient temperature of 37–38°C for 10 minutes, and an occlusion cuff was placed at the proximal part of the tail. Measurements of BP and heart rate values were made more than three times and averaged. Signals from the cuff pressure transducer were recorded continuously on the monitor of a computer. Mice were acclimated to restraint and tail-cuff inflation for at least 3 days before conduction of experiments.
Echocardiography.
Echocardiography was performed using a high-frequency 15-MHz linear scanner (Sequoia, ACUSON; Siemens Medical Solutions, Munich, Germany) on a warmed pad to maintain the body temperature of the mouse at a constant 37°C. Mice were anesthetized with sodium pentobarbital (50 mg/kg) that was injected intraperitoneally. M-mode echocardiography was performed to measure the following parameters from the M-mode image of the LV. Interventricular septal thickness (IVST), posterior wall thickness (PWT), and LV diastolic dimension were measured. The LV ejection fraction (LVEF) as an index of systolic function was calculated by the cubed method as follows: LVEF = [(LVDd)3 − (LVDs)3]/(LVDd)3, where LVD indicates LV diameter, d indicates diastolic, and s indicates systolic. LV mass was calculated using the two-dimensional area-length method as described previously (Collins et al., 2001). Pulsed Doppler measurements of LV diastolic inflow, peak velocities of early and late diastolic filling, and their ratio (E/A) as an index of diastolic function were measured.
Hemodynamic Measurements.
At 4 weeks after Ang II or Ang II + BRL administration, cardiac catheterization was performed with intraperitoneal injection of sodium pentobarbital (50 mg/kg), and the mice were intubated. A 1.2-F catheter-tip micromanometer (Scisense Inc., London, Ontario, Canada) was inserted into the LV cavity via the right carotid artery (Perrino et al., 2006). Absolute volume was calibrated, and steady-state pressure and volume measurements were digitally recorded. Heart rate, LV end-systolic pressure, LV end-diastolic pressure (LVEDP), peak positive LV rate of pressure development (dP/dt), peak negative LV dP/dt (min LV dP/dt), time constant relaxation (τ), stroke volume, and cardiac output were calculated (Steendijk and Baan, 2000).
LV pressure-volume relations were measured via transient preload reduction by occlusion of the abdominal vena cava. A total of 10–20 consecutive cardiac cycles were obtained over 2 seconds, from which the end-systolic pressure-volume relation slope and end-diastolic pressure-volume relation slope were derived.
After the data were recorded under steady state and preload was reduced by abdominal vena cava occlusion, parallel conductance was obtained by injecting 10 μl of 30% hypertonic saline into the central venous line.
One mouse in the control group and two mice in the Ang II group were not able to have hemodynamics measured because of failure of catheter insertion into the LV cavity. Therefore, the control group comprised six mice, and the Ang II group comprised six mice.
Pathologic Analyses.
After hemodynamic measurements were performed, mice were anesthetized with peritoneal injection of sodium pentobarbital (200–300 mg/kg) and euthanized, and the heart and lungs of each mouse were excised. The heart was dissected to remove the atria and right ventricular free wall, and the LV weight was measured. The weight of the LV was standardized to body weight and used as an index of ventricular hypertrophy. Lung weight was measured before air drying to evaluate pulmonary congestion.
The LV was cut into two sections of the apex and base. The base was fixed in 10% formalin and imbedded in paraffin. Fixed tissues were sectioned at 4-μm thickness and stained with hematoxylin and eosin and Masson’s trichrome. Pathologic images were captured using an Olympus AX80 microscope (Olympus, Tokyo, Japan) and an Olympus DP50 digital video camera system (Olympus) and were analyzed with the ImageJ software program (National Institutes of Health, Bethesda, MD) (Chang et al., 2009).
The cross-sectional area of myocytes was measured from hematoxylin and eosin sections. Suitable cross-sections were defined as those having nearly circular capillary outlines and circular-to-oval myocyte sections (Senbonmatsu et al., 2000). No correction for oblique sectioning was made. The outline of 100–200 myocytes was traced, and the mean area was calculated for the LV in each mouse. The group mean was calculated for each region and group.
LV interstitial fibrosis and perivascular fibrosis were quantitatively analyzed from Masson’s trichrome–stained sections. The ratio of interstitial fibrosis to the total LV area was calculated from four to five randomly selected microscopic fields from each of the sections. Perivascular fibrosis was assessed by calculating the ratio of the area of collagen-stained material to the luminal area and was averaged from three vascular cross-sections from each mouse.
Western Blot Analyses.
Mouse LV tissues were homogenized in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP40) with protease inhibitor cocktail. Homogenates were centrifuged at 10,000g for 10 minutes at 4°C. The supernatant was transferred to new tubes. The protein concentration was determined by the bicinchoninic acid protein assay (Thermo Fisher Scientific, Rockford, IL). An equal amount (20 µg) of protein sample from mouse hearts was loaded per lane in a 10% polyacrylamide gel, which was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membranes were blocked with 25% skim milk for 1.5 hours and then incubated with 1:1000 diluted primary antibodies against TGF-β1 (ab66043; Abcam, Cambridge, England), Smad2/3 (3102; Cell Signaling Technology, Danvers, MA), phospho-Smad2(Ser465/467)/Smad3(Ser423/425) (8828; Cell Signaling Technology), collagen type I (ab34710; Abcam), collagen type III (ab7778; Abcam), and glyceraldehyde-3-phosphate dehydrogenase (G9545-25UL; Sigma-Aldrich, St. Louis, MO) at 4°C overnight. Membranes were washed with PBS with Tween 20 and incubated with anti-rabbit secondary antibodies that were diluted 1:1000 with 25% skim milk for 2 hours at 25°C. Membranes were incubated with SuperSignal West Dura Extended Duration Substrate (Thermo-Pierce, Waltham, MA), and immunoreactive bands were visualized using the ChemiDoc MP Imaging System (Bio-Rad Laboratories, Hercules, CA). Densitometry was performed using Photoshop software (Adobe, San Jose, CA).
Statistical Analyses.
All data are shown as the means ± S.D. Group data were compared using one-way analysis of variance with Tukey’s post hoc test for multiple comparisons. The relationship between lung/body weight and the end-diastolic pressure-volume relation slope was evaluated using Spearman’s rank test. A value of P < 0.05 was considered to be the minimal level of significance. All statistical analyses were performed using Dr. SPSS II (IBM, Armonk, NY).
Results
BP and Heart Rate.
The effects of Ang II and Ang II + BRL treatment in conscious mice on systolic BP and heart rate are shown in Fig. 1. Before starting treatment, there was no significant difference in BP or heart rate among the three groups (data not shown). After 4 weeks, systolic BP was significantly higher in Ang II and Ang II + BRL mice compared with control mice (both P < 0.001), but there was no difference between Ang II and Ang II + BRL mice by the tail-cuff method. Heart rate was not significantly different among the three groups.

Systolic BP (A) and heart rate (B) of the three groups. General findings measured in the conscious state using the indirect tail-cuff method are shown. White bars indicate control mice. Solid bars indicate Ang II mice. Gray bars indicate Ang II + BRL37344 mice. Data shown are means ± S.D. from seven to eight samples per group. *P < 0.001 vs. control.
Echocardiography.
Echocardiographic data are shown in Fig. 2. Echocardiographic analysis showed that the IVST, PWT, and LV mass were significantly higher in Ang II and Ang II + BRL mice compared with control mice (both P < 0.05). Other echocardiographic parameters, including LVEF and E/A, were similar among the three groups.

Echocardiographic findings of the three groups. Bar graphs show the effects of Ang II + BRL37344 on IVST (A), PWT (B), LVDd (C), ejection fraction (EF) (D), LV mass (E), E/A (F), heart rate (HR) (G), and motion mode image (H) in mice under pentobarbital anesthesia. Data shown are means ± S.D. from seven to eight samples per group. *P < 0.05 vs. control. bpm, beat per minutes.
Hemodynamic Measurements.
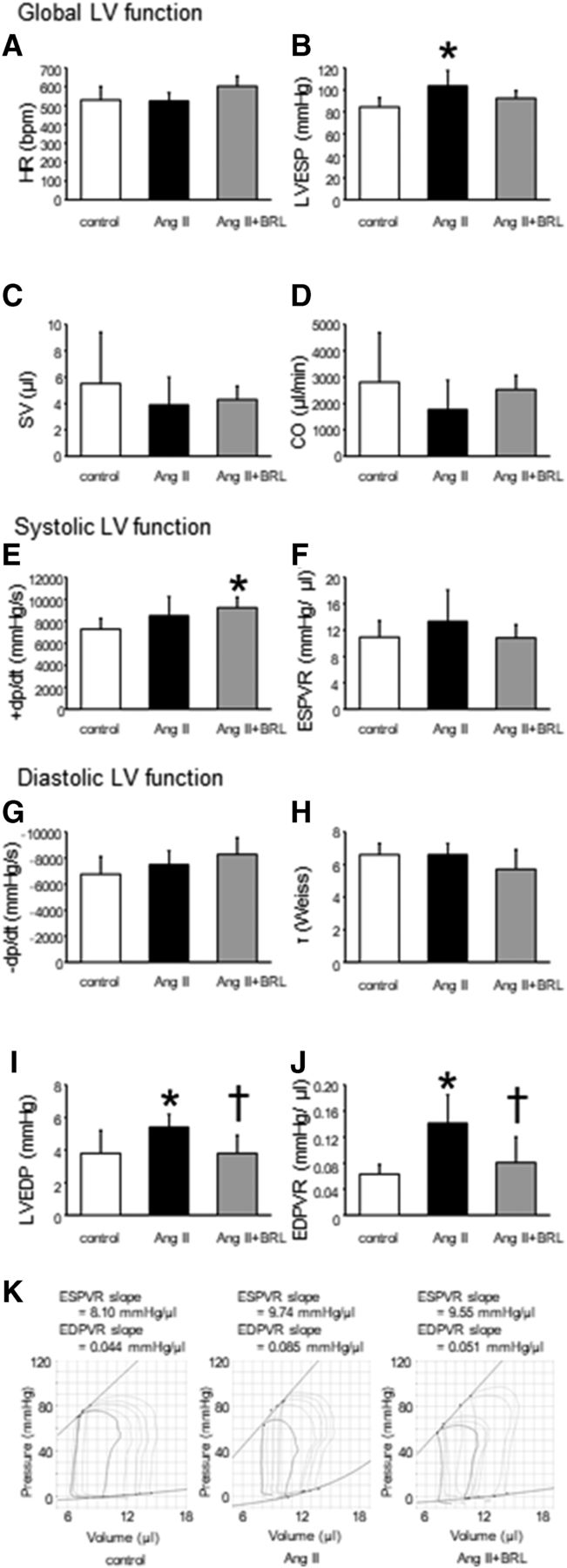
Figure 3 shows the main hemodynamics parameters. LV end-systolic pressure was significantly higher in Ang II mice (P < 0.05), but not in Ang II + BRL mice, compared with control mice under anesthesia. In steady-state analysis, stroke volume and cardiac output were not different among the three groups.
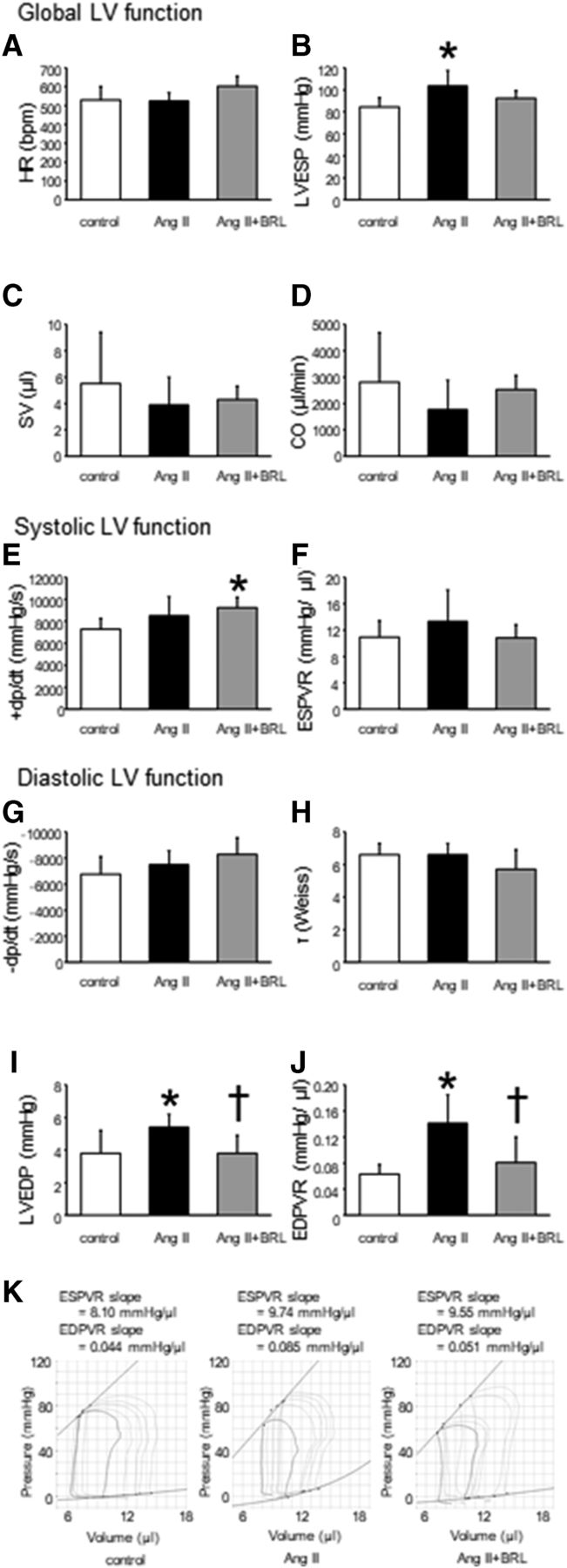
Hemodynamic findings of the three groups. Bar graphs show the effects of Ang II + BRL37344 on heart rate (HR) (A), LV end-systolic pressure (LVESP) (B), stroke volume (SV) (C), cardiac output (CO) (D), peak positive LV dP/dt (+dp/dt) (E), end-systolic pressure-volume relationship (ESPVR) (F), peak negative LV dP/dt (−dp/dt) (G), time constant relaxation (τ) (H), LVEDP (I), and end-diastolic pressure-volume relationship (EDPVR) (J) in mice under pentobarbital anesthesia. Representative systolic and diastolic pressure-volume loops during a preload reduction by occlusion of the abdominal vena cava. Upper lines indicate the resulting end-systolic pressure-volume relationship, and lower lines indicate end-diastolic pressure-volume relationship (K). Data shown are means ± S.D. from six to seven samples per group. *P < 0.05 vs. control, †P < 0.05 vs. Ang II.
The end-systolic pressure-volume relation slope, which is an index of sensitive systolic function, was similar among the three groups. LVEDP and the end-diastolic pressure-volume relation slope, which is an index of diastolic function, were significantly higher in Ang II mice (5.4 ± 0.8 mm Hg and 0.141 ± 0.044 mm Hg/μl, respectively) compared with control mice (3.8 ± 1.4 mm Hg and 0.063 ± 0.015 mm Hg/μl, respectively, P < 0.05). However, the increase in these parameters was prevented in Ang II + BRL mice (3.8 ± 1.1 mm Hg and 0.081 ± 0.039 mm Hg/μl, respectively, P < 0.05), suggesting improvement in myocardial stiffness.
Pathologic Analyses.
Data of morphometric findings are shown in Fig. 4. The LV/body weight ratio was significantly higher in Ang II and Ang II + BRL mice compared with control mice (both P < 0.01), but there was no difference between Ang II and Ang II + BRL mice. The lung/body weight ratio was significantly higher in Ang II mice compared with control mice (P < 0.01). However, the increase in the lung/body weight ratio with Ang II was prevented by BRL treatment. The relationship between lung/body weight and the end-diastolic pressure-volume relation slope is shown in Fig. 4G. An increase in lung/body weight was positively correlated with an increase in the end-diastolic pressure-volume relation slope (P < 0.01, r = 0.63).

Morphometric findings of the three groups. Bar graphs show the effects of Ang II + BRL37344 on body weight (A), LV weight (B), atrium weight (C), lung weight (D), LV/body weight (BW) (E) and lung/BW (F) in mice under pentobarbital anesthesia. Correlations between lung/BW and end-diastolic pressure-volume relationship (EDPVR) (G). Data shown are means ± S.D. from seven to eight samples per group. *P < 0.05 vs. control, **P < 0.01 vs. control.
Chronic Ang II administration resulted in a significantly higher cross-sectional area of cardiac myocytes in Ang II and Ang II + BRL mice compared with control mice (both P < 0.01), but there was no difference between Ang II and Ang II + BRL mice (Fig. 5, A and D). Masson’s trichrome staining showed that interstitial fibrosis and perivascular fibrosis were significantly higher in Ang II mice compared with control mice (both P < 0.01). These increases in the extent of interstitial fibrosis and perivascular fibrosis were significantly inhibited by treatment with BRL (Fig. 5, B, C, E, and F).

Pathologic analysis of the three groups. Bar graph shows cross-sectional area measurements of myocytes (A), LV interstitial fibrosis (B), and LV perivascular fibrosis (C). Representative hematoxylin and eosin–stained myocardial sections showing myocyte sizes (scale bar, 50 μm) (D). Representative Masson’s trichrome–stained myocardial sections showing LV interstitial fibrosis (scale bar, 200 μm) (E) and LV perivascular fibrosis (scale bar, 100 μm) (F). Data shown are means ± S.D. from seven to eight samples per group. *P < 0.01 vs. control, †P < 0.05 vs. Ang II, ††P < 0.01 vs. Ang II. CAS, cross sectional area.
Western Blot Analyses.
Chronic Ang II administration resulted in significantly higher TGF-β1 expression compared with control mice (Fig. 6, P < 0.05). However, this increase was markedly less in Ang II + BRL mice compared with Ang II mice (P < 0.05). To determine the effect of BRL on TGF-β1–mediated intracellular signaling and the consequences associated with cardiac fibrosis, we evaluated levels of Smad2/3, phospho-Smad2/3, and collagen types I and III in myocardium of the mouse models. Consistent with the results of TGF-β1 expression, phosphorylated Smad2/3 and collagen I/III levels in Ang II–treated mouse hearts were markedly elevated compared with those in controls (all P < 0.05). In the presence of BRL, the elevation in phosphorylation levels and amount of protein of these molecules were significantly suppressed in Ang II–treated mouse hearts (all P < 0.05).

Western blots showing the levels of TGF-β1, Smad2/3, phospho-Smad2/3, collagen I, and collagen III. Data shown are means ± S.D. from four samples per group. *P < 0.05 vs. control, †P < 0.05 vs. Ang II. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-Smad, phospho-Smad.
Discussion
The purpose of this study was to evaluate the effect of chronic administration of a β3-AR agonist on cardiac remodeling, function, and progression to HFpEF in Ang II–induced cardiomyopathy in mice. Long-term administration of BRL in these mice did not attenuate LVH, but BRL improved myocardial stiffness and reduced lung congestion. These beneficial effects of BRL were independent of a decrease in BP and were associated with attenuating cardiac fibrosis. Downregulation of TGF-β1 expression might be related to these cardioprotective effects.
β3-AR is upregulated in the failing heart in human and animal studies, and the functional response to β3-AR stimulation is increased (Cheng et al., 2001; Moniotte et al., 2001). In addition, accumulating evidence supports a major role for β3-AR in the animal HF model. Cardiac β3-AR protects against fibrosis in response to hemodynamic stress by regulating nitric oxide and oxidant stress-dependent paracrine signaling to fibroblasts (Hermida et al., 2018). Cardiac-specific overexpression of the β3-AR transgene attenuates development of cardiac myocyte hypertrophy and fibrosis under continuous isoproterenol and Ang II stimuli (Belge et al., 2014). β3-AR agonist therapy eliminates an increase in indices of oxidative stress (Karimi Galougahi et al., 2015) and attenuates LV dilatation, systolic dysfunction, and cardiac hypertrophy in a TAC model (Niu et al., 2012).
Ang II mediates cardiac fibrosis via a TGF-β1–dependent mechanism. Ang II inactivates nitric oxide and increases TGF-β1 expression in cardiac fibroblasts (Mehta and Griendling, 2007). The TGF-β1 signaling pathway is thought to be a powerful mediator of cardiac fibrosis (Schnee and Hsueh, 2000; Rosenkranz, 2004). Cardiac fibrosis is accepted as one of the main determinants of cardiac remodeling. Prevention of the fibrotic process is one of the main targets to reverse cardiac remodeling and improve prognosis. Downregulation of TGF-β1 expression suppresses cardiac fibrosis (Kuwahara et al., 2002). TGF-β1 is the main profibrotic cytokine, which leads to Smad2/3 phosphorylation and transport to the nucleus and then to effects in cardiac fibrotic conditions. Excessive production of collagen types I and III proteins may be responsible for myocardial interstitial collagen deposition, disorder, and even heart damage. The TGF-β1/Smad3 signaling pathway finally produces fibrotic mediators such as collagens (Bujak et al., 2007). A previous study reported that enhanced expression of β3-AR in cardiac myocytes prevented cardiac fibrosis via a TGF-β1–dependent pathway (Belge et al., 2014). A novel and significant finding in the present study was that Ang II–induced cardiac TGF-β1 expression and activation of Smad signaling were significantly attenuated by treatment with a β3-agonist. We speculate that the TGF-β1/Smad signaling pathway was involved in one of the mechanisms of decreasing collagen production and cardiac fibrosis independently of a decrease in BP in the present study.
A previous study reported attenuation of LVH by BRL infusion therapy using a mouse model of pressure overload induced by TAC (Niu et al., 2012). However, a reduction in hypertrophy of cardiac myocytes by β3-AR agonist therapy was not observed in the present study. The pressure overload HF model uses either Ang II infusion or TAC. Ang II infusion and the TAC mouse model show diastolic dysfunction and pulmonary congestion, but these two models differ in systolic function and LV structure. The Ang II–infused mouse develops concentric and dilated hypertrophy and has preserved systolic function. However, TAC-induced pressure overload in mice leads to concentric to eccentric hypertrophy and is preserved to reduced systolic function (Valero-Munoz et al., 2017; Brenes-Castro et al., 2018). Differences between these pressure overload HF models might be associated with failure to prevent LVH in our study. LVH under Ang II infusion is due to BP-dependent and BP-independent factors (Valero-Munoz et al., 2017). In the present study, systolic BP in the conscious condition was not different between Ang II and Ang II + BRL mice. Therefore, BRL infusion therapy might be less effective in preventing LVH in the absence of BP control. The relationship of the BP-independent mechanism of LV hypertrophy and β3-AR agonist therapy needs further investigation. Moreover, heart rate in the conscious condition was not different between Ang II and Ang II + BRL mice in our study. A progressive increase in BP in sheep during chronic Ang II infusion was not followed by bradycardia, which indicated reset of the baroreflex control of heart rate during infusion of Ang II (Hood et al., 2007).
To the best of our knowledge, the present study is the first to show cardioprotective effects of a β3-AR agonist by evaluation of cardiac performance using a conductance catheter system in a chronic HFpEF model. HF in Ang II–induced cardiomyopathy in mice resulted in increased LVEDP and wet lung weight, and systolic function parameters were not significantly different between Ang II and Ang II + BRL mice, as shown by echocardiography and an LV catheter. These results suggested that more severe HFpEF occurred in Ang II mice compared with Ang II + BRL mice. In C57BL/6J mice, cardiac systolic function is relatively preserved in response to Ang II infusion (Xu et al., 2008; Yagi et al., 2008; Peng et al., 2011; Valero-Munoz et al., 2017; Brenes-Castro et al., 2018). The present study, which used direct methodology with pressure-volume loops acquired during a loading intervention, showed an increased end-diastolic pressure-volume relation in Ang II mice. This finding indicated LV stiffness as the main abnormality in HFpEF. LVEDP and min LV dP/dt are diastolic load-dependent parameters, but diastolic stiffness is independent of load (Chang et al., 2009). In our study, min LV dP/dt and τ were not different among the groups, but increased diastolic stiffness might have contributed to increased LVEDP and pulmonary congestion in Ang II mice. Pulmonary congestion, alveolar edema, and lung water content are increased in the dilated cardiomyopathy mouse model (Gladysheva et al., 2013). Furthermore, in TAC-induced HF in mice with increased lung weight and LVEDP, profound pulmonary remodeling with muscularized lung vessels, marked lung fibrosis, and leukocyte infiltration occurred as a result of long-term pulmonary congestion (Chen et al., 2012). Increased diastolic stiffness significantly modulates clinical symptoms in patients with HFpEF. A reduction in diastolic stiffness is one of the targets for treating HFpEF (Westermann et al., 2008). In patients with amyloidosis, a progressive increase in myocardial stiffness as measured by intrinsic cardiac elastography was correlated with worsening of N-terminal probrain natriuretic peptide levels (Pislaru et al., 2019). Diastolic stiffness was correlated with pulmonary congestion and lower in Ang II + BRL mice compared with Ang II mice in the present study. An increase in passive stiffness can be induced by abnormalities in cardiac myocytes, the extracellular matrix, or both of these factors (Zile and Brutsaert, 2002; Burkhoff et al., 2005; Mehta and Griendling, 2007). Cardiac fibrosis was lower in Ang II + BRL mice compared with Ang II mice, which was probably associated with decreased diastolic stiffness.
Among patients with HFpEF, the use of renin-angiotensin system antagonists is associated with lower all-cause mortality (Lund et al., 2012). However, a previous study showed that a β-blocker (carvedilol) did not improve the prognosis in the clinical setting in patients with HFpEF (Yamamoto et al., 2013). β3-AR is an attractive target for novel pharmacological approaches in several clinical areas, including metabolic, ocular, cardiovascular, and urinary diseases (Schena and Caplan, 2019). The selective β3-AR agonist mirabegron has already been approved for overactive bladder syndrome in some countries (Imran et al., 2013). Recently, clinical studies examining potential beneficial effects of mirabegron for HF were performed. The first-in-human randomized trial of mirabegron in chronic HF (BEAT-HF trial: BEta 3 Agonists Treatment in HF trial) was conducted in 70 patients with New York Heart Association class II–III HF (Bundgaard et al., 2017). However, the primary endpoint of an increase in LVEF after 6 months was not reached. Exploratory data analysis indicated that β3-AR stimulation by mirabegron increased LVEF in patients with a baseline ejection fraction lower than 40%. Moreover, mirabegron therapy did not change BP or heart rate compared with placebo, as found in our study. Another clinical trial on mirabegron that is currently ongoing is Beta3-LVH (Beta3-left ventricular hypertrophy) trial, which is a randomized, placebo-controlled, double-blind, multicenter trial on 296 patients (Pouleur et al., 2018). Patients with LVH were treated with 50 mg mirabegron for 12 months. This trial aimed to investigate the effects of mirabegron on the LV mass index and diastolic function in patients with LVH. This trial is expected to be completed in 2020. The role of β3-AR agonists has not been determined in a human HFpEF study. The clinical value of attenuated myocardial stiffness by β3-AR agonists requires investigation.
In summary, the current study shows that BRL, which is a β3-AR agonist, attenuates cardiac fibrosis and improves diastolic dysfunction independently of BP in an Ang II–induced hypertensive mouse model. β-Blockers have become the standard treatment of chronic HF since 1990, but they have not shown an improvement in prognosis of HFpEF in the clinical setting. Our findings suggest a clinical benefit of β3-AR agonists in the treatment of HFpEF.
Acknowledgments
The authors thank the staff of the Maruyama Memorial laboratory at Nippon Medical School for their help.
Authorship Contributions
Participated in research design: Kamiya, Asai, Shimizu.
Conducted experiments: Kamiya, Asai, Maejima, Shirakabe, Murai, Noma, Komiyama.
Performed data analysis: Kamiya, Asai, Maejima, Shirakabe, Murai, Noma, Komiyama, Sato, Shimizu.
Wrote or contributed to the writing of the manuscript: Kamiya, Asai, Maejima, Shirakabe, Murai, Noma, Komiyama, Sato, Mizuno, Shimizu.
Footnotes
- Received May 31, 2020.
- Accepted November 11, 2020.
No author has an actual or perceived conflict of interest with the contents of this article.
This work was previously presented as an abstract at the European Society of Cardiology Congress in 2013 and published as follows: Masataka Kamiya, Kuniya Asai, Satsuki Noma, Hidenori Komiyama, Akihiro Shirakabe, Naoki Sato, and Kyoichi Mizuno (2013) Beta-3 adrenergic receptor agonist prevents diastolic dysfunction in angiotensin II-induced cardiomyopathy mouse model. European Heart Journal 34: P3332.
Abbreviations
- Ang II
- angiotensin II
- AR
- adrenergic receptor
- BP
- blood pressure
- BRL
- RR + SS-(±)-4-(2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl)phenoxyacetic acid
- dP/dt rate of pressure development, E/A
- peak velocities of early and late diastolic filling ratio
- HF
- heart failure
- HFpEF
- heart failure with preserved ejection fraction
- HFrEF
- heart failure with reduced ejection fraction
- IVST
- interventricular septal thickness
- LV
- left ventricular, LVD LV diameter.
- LVEDP
- left ventricular end-diastolic pressure
- LVEF
- left ventricular ejection fraction
- LVH
- left ventricular hypertrophy
- min LV dP/dt
- peak negative left ventricular dP/dt
- PWT
- posterior wall thickness
- TAC
- transverse aortic constriction
- τ
- time constant relaxation
- TGF
- transforming growth factor
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}