


Abstract
Toll-like receptor (TLR) 7 and TLR8 are transmembrane receptors that recognize single-stranded RNA. Activation of these receptors results in immune cell stimulation and inflammatory cytokine production, which is normally a protective host response. However, aberrant activation of TLR7/8 is potentially pathogenic and linked to progression of certain autoimmune diseases such as lupus. Thus, we hypothesize that an inhibitor that blocks TLR7/8 would be an effective therapeutic treatment. Prior efforts to develop inhibitors of TLR7/8 have been largely unsuccessful as a result of the challenge of producing a small-molecule inhibitor for these difficult targets. Here, we report the characterization of M5049 and compound 2, molecules which were discovered in a medicinal chemistry campaign to produce dual TLR7/8 inhibitors with drug-like properties. Both compounds showed potent and selective activity in a range of cellular assays for inhibition of TLR7/8 and block synthetic ligands and natural endogenous RNA ligands such as microRNA and Alu RNA. M5049 was found to be potent in vivo as TLR7/8 inhibition efficaciously treated disease in several murine lupus models and, interestingly, was efficacious in a disease context in which TLR7/8 activity has not previously been considered a primary disease driver. Furthermore, M5049 had greater potency in disease models than expected based on its in vitro potency and pharmacokinetic/pharmacodynamic properties. Because of its preferential accumulation in tissues, and ability to block multiple TLR7/8 RNA ligands, M5049 may be efficacious in treating autoimmunity and has the potential to provide benefit to a variety of patients with varying disease pathogenesis.
SIGNIFICANCE STATEMENT This study reports discovery of a novel toll-like receptor (TLR) 7 and TLR8 inhibitor (M5049); characterizes its binding mode, potency/selectivity, and pharmacokinetic and pharmacodynamic properties; and demonstrates its potential for treating autoimmune diseases in two mouse lupus models. TLR7/8 inhibition is unique in that it may block both innate and adaptive autoimmunity; thus, this study suggests that M5049 has the potential to benefit patients with autoimmune diseases.
Introduction
Toll-like receptor (TLR) 7 and TLR8 are receptors for single-stranded RNA that reside in the endosome. TLR7 is expressed primarily in B cells, monocytes, and plasmacytoid dendritic cells (pDCs), whereas TLR8 is found more in neutrophils, monocytes, and myeloid dendritic cells. The two TLRs share some common downstream signaling, but TLR7 activation is more biased to signaling in the interferon regulatory factor (IRF) pathway, resulting in production of type I interferon (IFN), whereas TLR8 activation results in greater nuclear factor-κB (NF-κB) activation and production of other inflammatory cytokines (Gorden et al., 2005; Bender et al., 2020). Because of these differences in expression and downstream signaling, TLR7 and TLR8 activation can have very different outcomes, and the two receptors likely play nonredundant roles in immunity.
Although TLR7 and TLR8 are involved in antiviral defense, their aberrant activation can contribute to autoimmunity. Single nucleotide polymorphisms in both TLR7 and TLR8 have been associated with lupus (Lee et al., 2016). TLR7 has been implicated as a driver of lupus, and data from mouse studies with TLR7 overexpression and TLR7 knockout (KO) have shown that TLR7 may promote disease (Christensen et al., 2006; Fairhurst et al., 2008; Lee et al., 2008; Savarese et al., 2008). Activation of TLR7 can trigger pDCs to produce large amounts of IFN, which is recognized as a significant contributor to lupus development. TLR7 can also activate B cells; the hallmark of TLR7-induced B-cell activation is the presence of antibodies against RNA-containing proteins such as anti-SmRNP and anti-RiboP that contain TLR7 ligands.
The activity of TLR8 in mice is controversial, and there are significantly fewer preclinical disease model data regarding its role in autoimmunity compared with TLR7. However, reports have suggested that TLR8 expression correlates with disease in BXSB-Yaa mice (Kimura et al., 2014). Also, in normally healthy C57BL/6 mice, TLR8 overexpression can trigger disease development (Guiducci et al., 2013). Although the data implicating TLR8 are less extensive than for TLR7, it is a reasonable hypothesis that TLR8 may also play a role in human autoimmunity, as some of the same RNA ligands that activate TLR7 can also activate TLR8 (Gorden et al., 2005). Given that TLR7 and TLR8 are implicated as drivers of autoimmunity, the two receptors may be considered valuable drug targets, and dual inhibition may provide greater efficacy than inhibiting either receptor alone.
TLR7 and TLR8 are both activated by single-stranded RNA molecules, but they have some differences in their ligand binding specificities. Studies have been conducted to identify the ligand features that drive TLR7/8 binding and activation. Interestingly, both small-molecule agonists and larger RNA molecules have been shown to activate TLR7/8. It is accepted that RNA molecules that exist in immune complexes with RNA protein-binding autoantibodies (Hung et al., 2015) gain access to the endosome when they are taken up via B-cell receptors or Fc receptors in autoimmunity (Avalos et al., 2010; Green and Marshak-Rothstein, 2011). RNA species that activate in this manner include U1 RNAs, Y1 RNAs, and others. More recently, emerging data suggest that RNA molecules from retroelements such as Alu RNA (Mavragani et al., 2016) and microRNA (miRNA) molecules (Lehmann et al., 2012; Feng et al., 2017; Salvi et al., 2018) may also activate TLR7 and TLR8. Inhibiting TLR7/8 to effectively treat disease likely requires compounds that block ligands with varying structures from binding to the receptors.
Developing TLR7 and TLR8 selective inhibitors has been challenging for several reasons. TLR7/8 are membrane-bound receptors that are difficult to recombinantly express, and producing purified protein has been difficult, hindering binding studies and crystallographic structural determination. Despite these difficulties, efforts have been made to produce TLR7/8 single and dual inhibitors, and a few molecules have been developed. However, a molecule targeting these TLRs has never been tested in patients with lupus, advanced to phase III trials, or gained FDA approval. Because of the scarcity of drugs inhibiting these promising targets, we have endeavored to develop a dual TLR7/8 inhibitor for the treatment of autoimmunity. Herein, we report the discovery of M5049 and compound 2 (CMPD 2), which are potent and selective dual TLR7/8 inhibitors. Characterization of M5049 has found it to be a molecule with drug-like properties and a long duration of action that effectively reduces disease in lupus preclinical models.
Materials and Methods
Compounds
M5049 and CMPD 2, as well as selective agonists for TLR7 (CL-087) and TLR8 (see Supplemental Fig. 1 for structures), were synthesized by EMD Serono (a business of Merck KGaA, Darmstadt, Germany).
Crystallography
The TLR8 protein used in this study was purchased from the laboratory of Professor Toshiyuki Shimizu at the University of Tokyo. The protein was produced as previously described (Tanji et al., 2013). Recombinant human TLR8 (amino acids 27–827) was crystallized by hanging-drop vapor diffusion by mixing 1 μl of a reservoir solution (100 mM Tris, 11.5% PEG 8000, 200 mM NaCl, pH 8.0) with 1 μl of concentrated protein solution (10.6 mg/ml protein in 10 mM Tris, 150 mM NaCl, 10% glycerol, 128 μM poly-dT, 820 μM CL075, pH 7.5). The resulting crystals of TLR8 were soaked with a solution containing 2 mM M5049 with 2% DMSO for 24 hours. After soaking, the crystals were mounted on a cryoloop for X-ray data collection and flash cryocooled by holding it in liquid nitrogen. Crystallographic data collection was done on the synchrotron radiation source at Swiss Light Source in Villigen, Switzerland. The wavelength of radiation used was 1.00000 Å, and the data were collected using a Pilatus 6M area detector. Data processing and structure refinement were carried out according to standard crystallographic methods. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID: 6ZJZ).
Cell-Based Assays
Peripheral Blood Mononuclear Cells.
M5049 potency was tested using freshly isolated human PBMCs seeded in complete RPMI 1640 medium (Gibco, Waltham, MA) supplemented with 10% fetal bovine serum (Corning, Corning, NY). M5049 dilutions were added to cells and incubated at 37°C for 30 minutes prior to stimulation. The following final concentrations of ligands were added to the PBMCs: 5 µM TLR7 ligand CL-087, 2 µM TLR8 ligand (see Supplemental Fig. 1 for structures), 4 µg/ml TLR3 ligand polyinosinic:polycytidylic acid (Poly I:C) (InvivoGen, San Diego, CA), 50 ng/ml TLR4 ligand lipopolysaccharide (Escherichia coli 0111:B4, List Biologics, Campbell, CA), and 75 µg/ml TLR9 ligand ODN2006 (InvivoGen). RNA molecules (miRNA or Alu RNA) used for stimulation were synthesized with phosphodiester backbones (Sigma, St. Louis, MO) and were preincubated with dioleoyl-3-trimethylammonium propane (DOTAP; Roche, Basel, Switzerland) at room temperature for 15 minutes prior to adding to the cells. The final concentration of the RNA molecules was 50 nM, and the final dilution of DOTAP was 1:200. After addition of M5049 and ligands, the cells were incubated at 37°C and 5% CO2 overnight, and cytokines were measured in culture supernatants by AlphaLISA (PerkinElmer, Waltham, MA). All treatments were performed in duplicate or, in some instances, in triplicate.
Human Embryonic Kidney 293 Cells.
HEK293 cells were stably transfected with either TLR7 or TLR8 and an NF-κB-luciferase reporter gene. For compound testing, cells were seeded in 384-well, black, clear-bottom plates (Corning), and after an overnight incubation at 37°C and 5% CO2, M5049 dilutions were added in duplicate. The cells were stimulated with 10 µM R848 (InvivoGen) or 30 μM R848 for testing in HEK TLR7 or HEK TLR8 cells, respectively. After incubation for 5 hours at 37°C and 5% CO2, SteadyGlo substrate reagent (Promega, Madison, WI) was added to each well, and luminescence was measured using the Perkin Elmer Envision Multilabel Reader.
Whole Blood.
Blood was drawn from healthy donors into vacutainer tubes with EDTA as the anticoagulant (BD Biosciences, San Jose, CA) and used within 2 hours of collection. Four volumes of blood were diluted with one volume of PBS, and 150 µl per well of the diluted blood was dispensed per well of 96-well plates. M5049 dilutions were added in triplicate, and plates were incubated at 37°C and 5% CO2 for 30 minutes. DMSO, the TLR7 ligand CL-087 (3 µM), or the TLR8 selective agonist described above (0.5 µM) were added to the cells for stimulation. The treated blood was incubated overnight at 37°C and 5% CO2, and cytokines were measured in the plasma by AlphaLISA.
Sequences of RNA ligands used for stimulation are as follows: Let-7a, UGAGGUAGUAGGUUGUAUAGUU; Let-7b, UGAGGUAGUAGGUUGUGUGGUU; Let-7c, UGAGGUAGUAGGUUGUAUGGUU; Let-7e, UGAGGUAGGAGGUUGUAUAGUU; Let-7f, UGAGGUAGUAGAUUGUAUAGUU; miR-122, UGGAGUGUGACAAUGGUGUUUG; miR-223, CGUGUAUUUGACAAGCUGAGUU; miR-21, UAGCUUAUCAGACUGAUGUUGA; miR-574, UGAGUGUGUGUGUGUGAGUGUGU; and Alu motif B, UUUUUUUUUUUUUUUUUUUUUUGAGACGGAGUCUCGCUCUGUCGCC.
Absorption, Distribution, Metabolism, and Excretion
Serum Protein Binding.
Compound serum protein binding was measured using rapid equilibrium dialysis (10 kDa) following the method of Banker et al. (2003).
Caco-2 Permeability/Efflux.
Caco-2 permeability and efflux ratio were determined at a concentration of 1 µM following the method of Feng et al. (2008).
Microsomal Clearance.
Microsomal clearance was determined at a concentration of 1 µM in pooled liver microsomes following the method of Obach (1999).
Hepatocyte Clearance.
Hepatocyte clearance was determined at a concentration of 1 µM in pooled hepatocytes following the method of McGinnity et al. (2004).
Human Cytosol Clearance (Aldehyde Oxidase).
Human cytosol clearance was determined at a concentration of 1 µM in pooled (N = 200) human cytosol in the presence/absence of hydralazine following the method of Zientek et al. (2010).
Cytochrome P450 Inhibition.
Cytochrome P450 inhibition was determined in pooled human microsomes at concentrations ranging from 0 to 50 µM in a cocktail format following the methods of Kozakai et al., 2012) for cytochrome P450s 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A.
Cytochrome P450 Induction.
Cytochrome P450 induction was determined at concentrations ranging from 0 to 50 µM in a HepaRG cell line following the method of McGinnity et al. (2009).
Animal Studies
All procedures using animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals and all local and national laws and regulations regarding animal care under protocols approved by the local Institutional Animal Care and Use Committee.
Pharmacokinetic Studies.
Animals were dosed intravenously or by oral gavage with M5049 or CMPD 2 formulated in 0.1 M Na-citrate buffer pH 4.5 (i.v.)or pH 3.0 (oral). Blood was collected via different routes for each species and processed to obtain plasma that was subsequently analyzed for compound concentrations by liquid chromatography-mass spectrometry. Mouse (CD1; Charles River Laboratories, Margate, UK) blood was collected via submandibular bleeds, and rat (Wister; Charles River Laboratories, Wilmington, MA) blood was collected via femoral catheter. Blood was collected from dogs (beagle; Nuvisan Pharma Services, Neu Ulm, Germany) and monkeys (cynomolgus; Aptuit, an Evotec Co., Verona, Italy) by femoral bleeds. For these studies, female mice, rats, and dogs and male monkeys were used.
Mouse Pharmacodynamic Studies.
Female C57BL/6 mice (Charles River Laboratories) were dosed via oral gavage with M5049 formulated in 0.1 M Na-citrate buffer (pH 3). At different time points after dosing, the mice were challenged with an intraperitoneal injection of 25 µg R848 dissolved in water. At 2 hours after R848 challenge, the mice were euthanized, and serum was collected by cardiac puncture. Mice were also challenged with miR-122 formulated in Invivofectamine 3.0 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The miRNA was injected at 2 mg/kg i.v. via the tail vein, and at 4 hours after injection, blood was collected by cardiac puncture. Serum interleukin-6 (IL-6) and IFN-α (PBL, Piscataway, NJ) concentrations were measured by ELISA (BD, San Jose, CA), and the concentration of M5049 was measured by liquid chromatography-mass spectrometry.
BXSB-Yaa Model.
BXSB-Yaa studies were conducted as previously described (Bender et al., 2016). Male BXSB-Yaa mice were purchased from Jackson Laboratories (Bar Harbor, ME), and at 7 to 8 weeks of age, treatment was started. Mice were dosed once per day via oral gavage with M5049 formulated in 0.1 M Na-citrate buffer (pH 3). After 14 weeks of treatment, mice were euthanized via CO2 asphyxiation, and blood was collected via the vena cava. Plasma was collected from a portion of the blood, and 100 µl of blood was preserved in animal blood protect tubes (Qiagen, Hilden, Germany) for gene expression analysis. For monitoring of proteinuria in BXSB-Yaa mice, urine was collected by bladder massage in the morning on two consecutive days, and the samples were pooled. The levels of albumin and creatinine in the urine were determined using the ADVIA 1800 clinical chemistry analyzer (Siemens, Berlin, Germany). The urinary albumin-to-creatinine ratio was calculated as the ratio of milligrams of albumin per gram of creatinine per deciliter of urine.
IFN-α NZB/W Model.
IFN-α NZB/W model studies were conducted as previously described (Haselmayer et al., 2019). Briefly, 10-week-old female NZB/W F1 mice (Jackson Laboratories) were given two intravenous injections on day 0 and day 1 of an adenovirus encoding for mouse IFN-α (BioFocus, Saffron Walden, UK) in saline or were left untreated (sham). Treatments were initiated at 2 weeks after adenovirus injection, and mice were treated for 10 weeks. Mice were dosed once per day orally with M5049 formulated in 0.1 M Na-citrate buffer (pH 3) or mycophenolate mofetil (MMF, CellCept; Roche, Basel, Switzerland) at 300 mg/kg. After 10 weeks of treatment, mice were euthanized via CO2 asphyxiation, and blood was drawn for serum collection; 100 µl of the blood was preserved in animal blood protect tubes (Qiagen) for gene expression analysis. Spleen cells were analyzed for B- and T-cell subsets on the final day using flow cytometry. Over the course of the study, urine samples were collected, and proteinuria was determined by measuring the ratio of urinary protein to creatinine using the ADVIA 1800 clinical chemistry analyzer (Siemens).
Autoantibody Measurements.
Autoantibodies were measured in mouse lupus plasma samples as previously described (Bender et al., 2016). Briefly, a custom-manufactured 5-plex Meso Scale Discovery (Kenilworth, NJ) plate for assessment of five different autoantibodies (anti-dsDNA, anti-histone, anti-Ro/SSA, anti-SmRNP, and anti-RiboP) was used to test plasma samples collected at the start of treatment or at the end of the study. Plasma samples were diluted 1:100 or 1:200 with PBS containing 0.5% bovine serum albumin. Plasma samples from MRL/lpr mice with high autoantibody titers were pooled to create a standard curve that was used for quantitative comparison of the lupus study plasmas, and values were expressed as autoantibody units per milliliter of plasma (units per milliliter).
Gene Expression Analysis.
Gene expression analysis of lupus mouse blood samples was performed using the NanoString platform (NanoString, Seattle, WA). The RNeasy Protect Animal Blood System (Qiagen) was used for RNA isolation. A custom-designed gene panel for monitoring TLR7 and TLR8 activation along with general inflammation was used as previously described (Bender et al., 2020). For each study, all samples were normalized to the included housekeeping genes, and a Log2-fold change was calculated for the treatment groups relative to the vehicle group. IFN gene signature scores were calculated for all lupus mice relative to a healthy control group of DBA/1 mice.
Data Analysis
For in vitro assays testing M5049 against TLR7 and TLR8 in whole blood, or M5049 against RNA ligands in PBMCs, experiments were performed in duplicate or triplicate, and IC50 values were calculated using a four-parameter fit of data in Prism (GraphPad, San Diego, CA). The percent inhibition was determined for stimulation in the presence of compound compared with stimulation in the absence of compound. Potency of compounds in HEK293 cells and PBMCs against TLR7- and TLR8-specific agonists was calculated with curve fitting in Genedata Screener and the Smart Fit function with the Neutral Controls normalization method. For mouse PD studies measuring inhibition of cytokine production, the percent inhibition was calculated for M5049-treated mice relative to mice dosed with R848 alone, and the group means are reported with S.D.. In lupus model studies, proteinuria and autoantibody data were analyzed with group medians, and statistical significance was determined using a Kruskal-Wallis test or ANOVA for autoantibodies. Gene expression analysis for lupus studies was performed by calculating the Log2-fold change for each mouse relative to the vehicle group, and statistical significance for M5049-dosed mice relative to the vehicle group was determined by t test.
Results
Discovery of TLR7/8 Selective Inhibitors.
A screening campaign was performed to identify compounds that function as dual TLR7/8 inhibitors without activity against other TLRs. The campaign and additional structure-activity relationship optimization led to the discovery of two promising candidate drugs: M5049 and CMPD 2 (see Fig. 1A for structures). Both compounds were found to be competitive reversible inhibitors, and the Schild plot for M5049 is shown in Supplemental Fig. 1 Supplemental Figs.2

TLR7/8 inhibitor structures and crystallography. (A) The structures of M5049 and CMPD 2 are shown. (B) The crystal structure of M5049 in complex with human TLR8 shows the bound inhibitor at the interface between the two dimers presented as a ribbon diagram. (C) The TLR8 residues contacting M5049 at the binding site are shown with the two different monomers colored as pink or magenta (PDB ID: 6ZJZ).
To characterize the molecular interactions of M5049 with its target TLRs, the three-dimensional structure of M5049 bound to recombinant human TLR8 was determined by single-crystal X-ray diffraction from a TLR8 crystal soaked in a solution containing M5049 (Fig. 1, B and C). It was found that two molecules of M5049 bound in the mirrored hydrophobic pockets of the TLR8 dimer interface (herein referred to as TLR8 and TLR8* along with their corresponding amino acids). Similar to previous TLR8 inhibitors, M5049 has van der Waal interactions with Phe346, Val378, Ile403, Phe494*, Ala518*, and Tyr567* (Zhang et al., 2018). Further, H-bond interactions are seen between the M5049 N of the quinoline ring and backbone of Gly351, and a charged interaction between the aminopiperidine and the backbone carbonyl of Ser516*. Finally, offset π-π stacking can be seen between the M5049 quinoline ring and Tyr348 and Phe495*. As has been seen with previous TLR8 antagonists, inhibition by M5049 is likely the result of the compound binding the open form of the dimer and thus stabilizing the receptor’s resting (inactive) state, which prevents activation of the receptor. Supporting this idea, it was observed that the C termini of the two monomers in our structure were 51 Å apart. This is in accordance with the observations of Zhang et al. (2018), who noted that the C termini of TLR8 bound to inhibitor are 51 Å apart compared with a distance of 34 Å in the agonist-bound activated dimer. Although a structure could not be generated for either inhibitor complexed with TLR7, it is likely that the binding mode for M5049 to TLR7 is similar to that of TLR8.
M5049 and CMPD 2 were characterized for their inhibitory activity against different TLRs in a variety of cellular systems (Table 1). Both compounds were found to potently inhibit TLR7 and TLR8 in HEK NF-κB-luciferase reporter cells with IC50 values in the low nanomolar range. Additionally, both compounds were found to be potent against TLR7 and TLR8 in PBMCs as well as whole blood. There appears to be an increased selectivity for TLR7 inhibition relative to TLR8 for both M5049 and CMPD 2 in whole blood compared with PBMCs and transfected HEK cells. Although different ligand concentrations were used for stimulation in the various systems, we think it is unlikely that this alone explains the increased TLR7 selectivity in whole blood. One possible reason for the selectivity shift is higher protein binding of the TLR7 agonist compared with the TLR8 agonist in whole blood, which could lead to less competition for M5049 or CMPD 2 at TLR7. Another reason could be that neutrophils, which primarily express TLR8, are present in whole blood but not in PBMCs, so the activation of these additional cells may require more inhibitor activity in whole blood.
TLR7/8 inhibitor potency and selectivity
Values are means ± S.D.
The compounds showed good selectivity and were found to be inactive against TLR3, TLR4, and TLR9 in vitro (Table 1) and in vivo (Supplemental Fig. 2). Selectivity was also established by testing for activity against a panel of 389 kinases as off-target profiling (Reaction Biology, Malvern, PA), and at 1 μM, M5049 inhibited the activity of only one kinase more than 50% (CAMKK2), and CMPD 2 inhibited none. M5049 was also tested for off-target effects in the CEREP Extended Receptor Profile assay (Eurofins, Saint Etienne, Luxembourg), and 5-hydroxytryptamine (5-HT) uptake was the only activity affected out of the many tested. Lack of off-target activity was confirmed by profiling using the BioMAP Diversity PLUS panel (Berg et al., 2010) of primary human cell coculture systems using a wide concentration range (10 nM to 10 μM). No significant activity of M5049 was observed in the panel, and no cytotoxic activity was detected either (Supplemental Fig. 3).
Evaluation of M5049 Drug-Like Properties and Duration of Action In Vivo.
Studies were conducted to characterize the absorption, distribution, metabolism, and excretion properties of M5049 and CMPD 2, and the compounds showed drug-like features. They both showed favorable in vitro characteristics, such as low microsomal clearance, good Caco-2 permeability, and no relevant P450 inhibition or induction liabilities. However, M5049 showed much lower clearance in the human cytosol assay, indicating greatly reduced AOX-mediated clearance compared with CMPD 2 (Table 2). Preclinical PK assessments were conducted in CD1 mice and Wistar rats for M5049 and CMPD 2, and additional studies were conducted for M5049 in beagle dogs and cynomolgus monkeys (Fig. 2A; Supplemental Table 1). Both compounds showed high bioavailability with moderate half-life and clearance (Table 2). Neither compound was positive in human ether-à-go-go–related gene (hERG) or Ames tests, which are considered important safety features. The compounds both have high bioavailability but a relatively short half-life. As M5049 was found to have superior PK properties in mice because of its much greater exposure [6-fold higher area under the curve (AUC)], and CMPD 2 was found to be a human AOX substrate, it was determined that M5049 is a more promising drug candidate and was advanced for further in vivo testing.
TLR7/8 inhibitor physicochemical, absorption, distribution, metabolism, and excretion, and safety properties

M5049 pharmacokinetic/pharmacodynamic activity in mice. (A) To determine the PK properties of M5049, mice were dosed with M5049 at 1 mg/kg via oral gavage or i.v., and blood samples were collected over time for the measurement of plasma concentrations. (B) To test the PD activity of M5049, mice were given a single oral injection of M5049 and 1 hour later were dosed i.p. with 25 μg per mouse of R848. After 2 hours, blood samples were collected, and plasma concentrations of M5049, IFN-α, and IL-6 were measured. The percent inhibition of cytokine release was calculated relative to mice dosed with R848 alone. (C) The duration of inhibition of M5049 was determined by dosing mice with M5049 at 1 mg/kg oral, and at 24, 12, 8, 4, and 1 hour after dosing, an i.p. injection of 25 μg R848 was given. After 2 hours, blood samples were collected, and plasma concentrations of M5049, IFN-α, and IL-6 were measured. (D) A plot was constructed overlaying the IC50 curve for M5049 inhibition of R848 stimulation in mouse blood in vitro and M5049 inhibition of R848 stimulation in vivo [from (B)]. Groups of five mice were used, and each study is representative of two separate experiments. Group means ± S.D. are shown.
M5049 was tested in mice to characterize its PD properties and duration of action. The TLR7/8 agonist R848 can effectively stimulate both interferon-α (IFN-α) and IL-6 production in mice in vivo, and the ability of M5049 to block an R848 challenge was used as a PD assessment. When M5049 was administered 1 hour prior to R848 challenge, it dose-dependently decreased IFN-α and IL-6 production (Fig. 2B). Inhibition of cytokine release was significant even at dosages of M5049, at which the resulting plasma levels of M5049 were very low (i.e., 0.1 and 0.3 mg/kg).
The duration of action of M5049 was determined by dosing M5049 at 1 mg/kg at different time points before R848 challenge (−1, −4, −8, −12, −24 hours). Despite the relatively short half-life of M5049 in mice (<3 hours, Fig. 2A), the compound was found to have a long duration of action, as IL-6 production was inhibited more than 50% when the compound was dosed 24 hours prior to challenge and more than 80% when dosed 12 hours prior to challenge. In both cases, the plasma concentrations of M5049 were low as 6 and 33 nM plasma M5049 were found at the time of blood collection for the 24-hour and 12-hour groups, respectively (Fig. 2C). Although these plasma concentrations are low, the compound will still likely provide TLR7/8 inhibitory activity, as the IC50 for M5049 in mouse blood in vitro has been found to be 1.9 nM against R848. If the values for the plasma concentrations of M5049 versus the percentage inhibition of IL-6 from the PK/PD time course study (Fig. 2C, results) are overlaid on the in vitro dose-response curve, there is a nearly perfect overlap (Fig. 2D), suggesting that the in vitro and in vivo potencies are highly similar and that a very good PK/PD correlation exists for M5049. These results suggest that M5049 has a long duration of action and is capable of substantial target inhibition with once-per-day dosing in mice.
Effects of M5049 on Disease in Mouse Lupus Models.
The ability of M5049 to reduce disease was tested in two different mouse lupus models: the BXSB-Yaa model and the IFN-α accelerated NZB/W model. In the BXSB-Yaa model, male mice have a Y-linked autoimmune accelerator locus (Yaa) on their Y chromosome that normally resides on the X chromosome, which results in a duplication of genes such as TLR7 that drive autoimmunity, and the mice spontaneously develop a lupus-like disease (Pisitkun et al., 2006; Deane et al., 2007). Treatment of BXSB-Yaa mice with M5049 led to an improvement in survival (Fig. 3A) and reduction in kidney disease as evidenced by lower proteinuria (Fig. 3, B and C). M5049 was also tested in the IFN-α accelerated NZB/W F1 model and again showed significant efficacy. The pathogenesis of the IFN-α accelerated NZB/W model is different from that of the BXSB-Yaa model. The model uses treatment with an adenovirus expressing IFN-α at the beginning of the study to induce a transient elevation of IFN-α, accelerating disease development in NZB/W F1 mice, which normally require many months to spontaneously manifest disease (Dubois et al., 1966). TLR7 has not been demonstrated to drive disease in this model (or in the standard NZB/W model without disease acceleration), in contrast to the BXSB-Yaa model. In the IFN-α accelerated NZB/W model, M5049 treatment also improved survival (Fig. 3D) and reduced kidney damage as evidenced by reduced proteinuria (Fig. 3, E and F) and lower histopathological glomerulonephritis scores.

The effect of M5049 on disease in mouse lupus models. BXSB-Yaa mice were dosed starting at 7 to 8 weeks of age with M5049 or vehicle (Veh) via oral gavage. NZB/W mice were treated with an injection of an IFN-α adenovirus to trigger disease development, and 14 days later, treatment was started with M5049 or with MMF at 300 mg/kg. Survival was tracked over time (A and D), and kidney disease was assayed by measuring proteinuria as defined as the albumin/creatinine (Albu/Creat) or protein/creatinine (Prot/Creat) ratio, and the data are plotted as a time course (B and E) or the AUC for individual mice (C and F). For each study, 10–12 mice per group were used, and results are representative of two separate studies for each model. The proteinuria time course graph presents means ± S.E.M., and AUC data are shown as medians. Statistical significance was determined by Kruskal-Wallis (*P < 0.05; **P < 0.01). Survival statistical significance was determined using a Mantel-Cox test (**P < 0.01).
Thedosages that were efficacious in the BXSB-Yaa and IFN-α NZB/W models resulted in low 24-hour plasma concentrations of M5049 in lupus mice (Supplemental Fig. 4) and are at the low end of the dose-response curve where inhibition of R848 in vitro or in vivo is seen (Fig. 2D). This suggests that 24-hour target inhibition is not needed for efficacy or that the disease-driving TLR7/8 ligands naturally existing in lupus mice are different from R848 in their levels or nature. It is also possible that the primary site of action of M5049 is localized in tissues instead of in the circulation. Supporting this idea are results from a whole-body radiography study with [14C]M5049 (Supplemental Fig. 5). When rats were dosed with [14C]M5049, it was found that there was preferential accumulation of the compound in tissues as evidenced by the high AUC and Cmax ratios for different tissues compared with blood. Tissue accumulation of M5049 could be the reason for the disconnect between plasma concentrations and efficacy in lupus mice.
In the BXSB-Yaa model, there is a robust increase in multiple autoantibodies, and M5049 significantly reduced both DNA-containing reactivities such as anti-dsDNA and anti-histones (Fig. 4, A and B), as well as RNA-binding protein reactivities such as anti-RiboP and anti-SmRNP (Fig. 4, C and D). At high dosages of M5049, the effect on autoantibodies is so profound that titers are reduced below the baseline levels. In contrast, there is much less development of autoantibodies in the IFN-α NZB/W model, and titers are not largely increased over the course of the study from baseline (Fig. 4, E–H). The lack of an increase in autoantibody titers in the IFN-α NZB/W mice may indicate that the disease pathogenesis is more localized in the tissues and less systemic, and autoreactive antibody-producing B cells play a smaller role in disease pathogenesis. M5049 had a negligible effect on autoantibody titers in this model, which is consistent with the hypothesis that the compound may be efficacious in these mice because of effects in tissues.

The effect of M5049 on autoantibodies in mouse lupus models. At the beginning of treatment (baseline) with M5049 or vehicle (Veh) and at the end of the BXSB-Yaa and IFN-α NZB/W studies, plasma samples were collected, and autoantibodies were measured using a custom Meso Scale Discovery multiplex assay for various reactivities. Titers of anti-dsDNA (A and E), anti-histones (B and F), anti-SmRNP (C and G), and anti-RiboP (D and H) were measured. Bars show group medians, and statistical significance was determined by one-way ANOVA (*P < 0.05; **P < 0.01). For each study, 10–12 mice were used per group, and results are representative of two separate studies for each model.
Effects of M5049 on Gene Expression in Mouse Lupus Models.
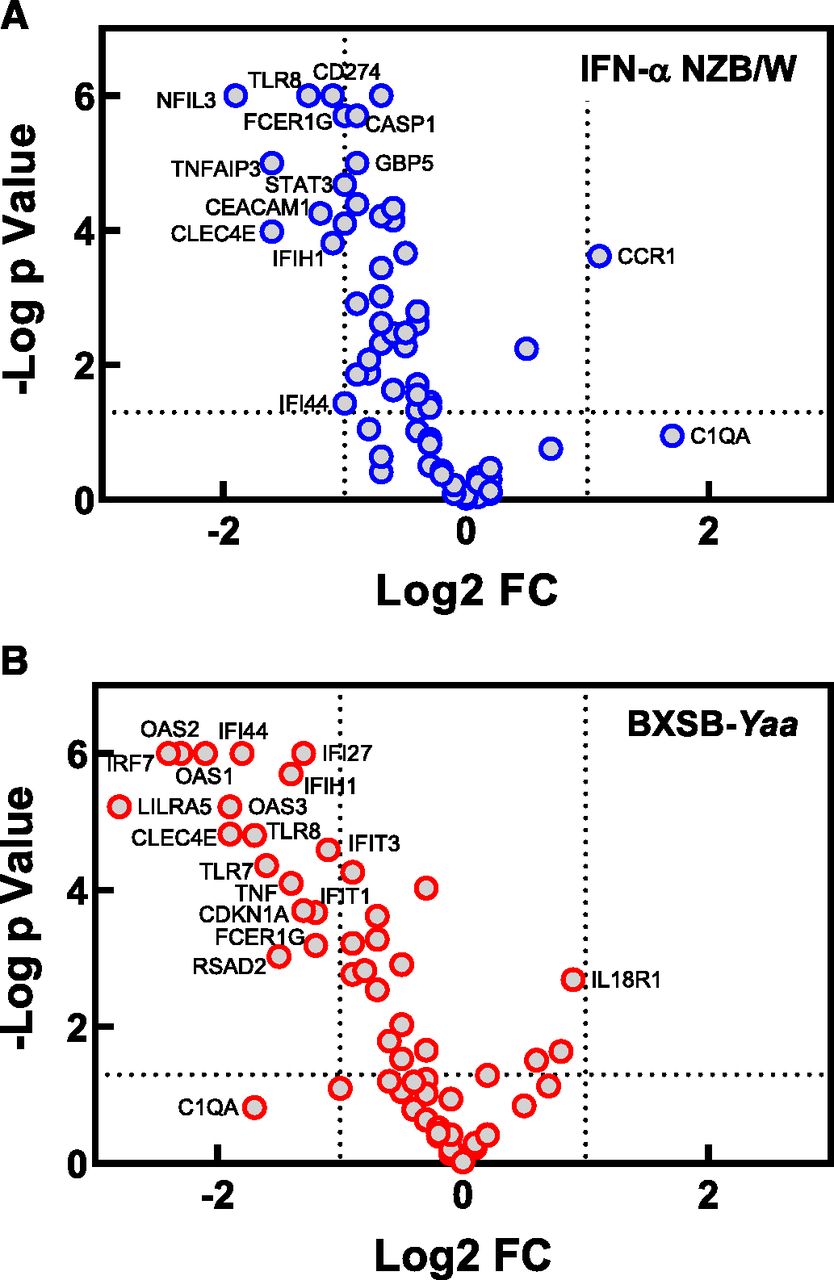
Gene expression analysis was performed using NanoString on blood samples collected from the BXSB-Yaa and IFN-α NZB/W lupus mice treated with M5049 at the end of the studies to try and better characterize the effects of compound treatment on disease. A custom NanoString panel of 97 genes was used for the analysis, and the genes selected were previously identified to be regulated downstream of TLR7/8 signaling and also disease-relevant (Bender et al., 2020). It was found that there were 66 genes expressed above the lower limit of detection in both models, and these genes were analyzed in detail. A Log2-fold change in gene expression was calculated for the compound-treated groups in each model compared with the vehicle control group to determine the effect of M5049 treatment. In both models, M5049 impacted many of the same genes, although there were some differences in the magnitude of the effect (Fig. 5). Volcano plots were constructed for the group receiving 10 mg/kg M5049 for each study to best identify the most significantly affected genes (Fig. 6, A and B). In the IFN-α NZB/W model, the most highly affected genes were markers for myeloid and granulocyte populations, such as CEACAM1, CLEC4E, FCER1G, and TLR8. In the BXSB-Yaa model, some of the same markers for those subsets were also decreased, such as CLEC4E, TLR8, and FCER1G, along with other myeloid markers, such as LILRA5 as well as TLR7. However, in the BXSB-Yaa mice, there was a much more significant effect of treatment on IFN-regulated genes such as IFI44, OAS1, OAS2, OAS3, IFI27, IFIH1, and IRF7. Given the high degree of efficacy at the dose of 10 mg/kg, it is a little surprising that larger effects on gene expression were not observed and that more genes were not seen to be affected significantly and to a larger degree in the volcano plot. It is possible that the mechanism of action of M5049 leading to efficacy was more at work in tissues and less in the circulation, and this could be the reason for the modest effect on gene expression in the blood.

The effect of M5049 on gene expression in mouse lupus models. Blood samples were collected from IFN-α accelerated NZB/W or BXSB-Yaa model mice at the conclusion of each study. RNA was extracted and analyzed by NanoString using a custom-designed panel to measure gene expression. The Log2-fold change in expression was calculated relative to the vehicle group (V) for each study, and the results are presented in a heat map. For each study, 10–12 mice were used per group, and results are representative of two separate studies for each model.

Genes significantly affected by M5049 in mouse lupus models. Blood samples were collected from IFN-α accelerated NZB/W (A) or BXSB-Yaa (B) model mice at the conclusion of each study, and gene expression was determined by NanoString. A standard t test was performed to determine statistical significance for difference relative to the vehicle group, and a volcano plot showing the resulting P values vs. the Log2-fold change (Log2 FC) was constructed for the 10 mg/kg M5049 group of the studies. For each study, 10–12 mice were used per group, and results are representative of two separate studies for each model. The data presented are the same as those presented in the heat map in Fig. 5.
To more fully compare the strength of IFN signaling in these models and the effect of M5049 on IFN activity, a panel of 10 IFN-regulated genes (OAS1, OAS2, OAS3, CMPK2, GBP5, IFI44, IFIH1, ISG15, IFI27, IRF7) that were significantly increased with disease in both models were used to calculate an IFN gene signature score for each mouse (Supplemental Fig. 6). The IFN-α NZB/W model mice had higher IFN gene signature scores (vehicle group median of 4.6) compared with the BXSB-Yaa mice (vehicle group median of 1.2). M5049 had no effect on IFN scores in the IFN-α NZB/W model but did significantly reduce scores in the BXSB-Yaa model. The source of IFN in the IFN-α NZB/W model leading to the robust upregulation of IFN-regulated genes at the end of the study is unclear. We have observed that adenovirus injection of these mice results in a very high, but transient, peak in IFN-α protein that is greatly reduced within days. It is possible that the early disease-triggering adenovirus injection is the reason for the strong IFN gene signature in these mice at the end of the study and the reason why M5049 failed to suppress it, since it would not be TLR7-dependent. Taken together, these results indicate that M5049 efficacy may be achieved in the IFN-α NZB/W model by a mechanism other than inhibition of IFN production.
Activation of TLR7/8 by Natural RNA Molecules and Inhibition by M5049.
To better understand how effectively M5049 may inhibit natural TLR7/8 ligands activating the receptors in vivo, the ability of the compound to block miRNAs and Alu RNA was determined. Human PBMCs were treated with miR-122, Let7c, Alu RNA, or R848 in a dose response, and after an overnight incubation, the production of IL-6 and IFN-α was measured, and the EC80 was determined. A concentration of roughly the EC80 was chosen for each ligand and then used to stimulate PBMCs after a pretreatment of the cells with M5049 to determine whether the compound could inhibit the different ligands with equal potency. All the ligands tested were found to induce IL-6 and IFN-α production (Fig. 7, A and B), but with slight differences for the two cytokines. The miRNAs and Alu RNA induced less IL-6 but more IFN-α compared with the small-molecule agonist R848. M5049 inhibited production of IL-6 stimulated by all the ligands nearly equally, with IC50 values ranging from 35 to 45 nM (Fig. 7C). The M5049 IC50 values for inhibition of IFN-α by the miRNAs were similar (210 nM for miR-122, 161 nM for Let7c), whereas the value for Alu RNA was slightly lower (68.5 nM) (Fig. 7D). The IC50 value for R848-stimulated IFN-α production was the lowest (8.9 nM), and this may be due to its much weaker activity compared with the other ligands for stimulation of IFN-α. Several other miRNAs have been reported to be disease-associated (Wang et al., 2012), and their ability to stimulate PBMCs and their sensitivity to inhibition by M5049 was also tested (Supplemental Fig. 7). It was found that a variety of miRNA molecules could induce IL-6 and IFN-α production, and all were susceptible to inhibition by M5049. The differences in M5049 IC50 values for natural ligands and synthetic small-molecule agonists highlights the importance of using endogenous stimuli whenever feasible to evaluate candidate inhibitors of these targets.

Inhibitory effect of M5049 on endogenous natural TLR7/8 stimulatory RNA molecules. PBMCs were stimulated with Let-7c (2 μM), miR-122 (1.8 μM), R848 (1 μM), or Alu RNA (0.4 μM) complexed with DOTAP or were left unstimulated (Unstim). After an overnight incubation, IL-6 (A) and IFN-α (B) concentrations in the supernatants were measured by AlphaLISA and the AlphaLISA counts (CPS) are reported. Stimulation was also carried out ±10 μM M5049 pretreatment for 30 min (C and D). Values shown are means ± S.D. of two donors from two separate experiments. The ability of M5049 to inhibit stimulatory activity of miRNA in vivo was determined by injection of C57BL/6 mice with miR-122 (2 mg/kg) 30 min after pretreatment with M5049. The production of IFN-α (F) and IL-6 (E) was measured in the plasma at 4 hours after miR-122 injection. The results shown are representative of two separate experiments using six mice per group, and bars show group medians. Statistical significance was determined by ANOVA (**P < 0.01).
We expanded our testing of miRNA to the in vivo setting and found that miR-122 can also stimulate IL-6 and IFN-α production in mice. When mice were dosed with miR-122, increased IL-6 and IFN-α were both detected in the plasma (Fig. 7, E and F). Pretreatment with M5049 before miR-122 injection dose-dependently inhibited the production of IL-6 and IFN-α. These results indicate that miRNA may be TLR7 or TLR8 activators in vivo and are inhibitable by M5049 in such a setting as they were in vitro.
Discussion
Despite the challenges associated with developing antagonists of TLR7 and TLR8, we have successfully produced two compounds that are potent and selective dual TLR7/8 inhibitors. The question of which endosomal TLRs to target for most effective autoimmune disease treatment is not easy to answer. Prior studies using KO of TLR7, TLR8, or TLR9 in various combinations have suggested that there is crosstalk between them, and KO of one may unexpectedly exacerbate disease by increasing the activity of TLR7, as this has been observed with TLR9 KO (Christensen et al., 2006; Jackson et al., 2014; Nundel et al., 2015) and TLR8 KO (Demaria et al., 2010). However, dual KO of TLR7 and TLR8 or TLR7 and TLR9 was found to be protective, suggesting that dual TLR KO is a safer approach to reduce disease. We believe that inhibition of TLR8 may be important for achieving efficacy in patients with lupus and other autoimmune diseases, as it is expressed in a number of abundant immune cell types and its activation can lead to robust inflammatory cytokine production (Gorden et al., 2005; Bender et al., 2020). It is unclear how much TLR8 contributes to disease in our mouse models and how much of the efficacy observed for M5049 is due to TLR7 versus TLR8 inhibition, and this assessment is out of the scope of this publication. The activity of TLR8 in mice is controversial, but we did observe that TLR8 mRNA is detected and elevated with disease in the mice and that potential TLR8 ligands such as miRNA are present in their plasma. Moving forward, we believe that the dual TLR7/8 inhibitory activity of M5049 may allow it to block multiple aspects of autoimmunity in a variety of cell types to provide efficacy.
M5049’s novel mechanism of inhibiting TLR7/8 may make the compound valuable for patients, as it provides differentiation from existing standard of care and other emerging novel treatments for lupus. Many currently used treatments have weaknesses; for example, steroids and mycophenolate mofetil are associated with untoward side effects, and antimalarials are weakly efficacious. Other available lupus treatments such as belimumab and rituximab target only the B-cell component of disease, whereas newer anti-IFN treatments such as anifrolumab will inhibit only one disease driver downstream of TLR activation and not NF-κB–stimulated cytokines or direct B-cell activation. TLR7/8 inhibition is unique in that it may block both innate and adaptive autoimmunity and inflammation. M5049 may inhibit TLR7/8 activation in pDCs, neutrophils, and monocytes, thus reducing IFN and inflammatory cytokine production. In addition, M5049 may also block adaptive inflammation by inhibiting autoreactive B cells. Because of this broad inhibitory profile but nondepleting nature, we believe that TLR7/8 inhibition by M5049 is a promising therapeutic approach.
Based on the characteristics reported here, M5049 may have advantages over most existing TLR7/8 inhibitors. Synthetic oligodeoxynucleotide antagonists specific for TLR7 (IRS661) or TLR7/9 (IRS954) were produced and tested preclinically (Pawar et al., 2007), and an oligonucleotide-based TLR7/8/9 antagonist, IMO-8400, has been developed and tested in early clinical trials (Balak et al., 2017). However, oligo-based inhibitors require dosing regimens that are not very practical for clinical usage, with frequent subcutaneous injections required in contrast to M5049, which can be administered orally. Small-molecule TLR8-specific (Zhang et al., 2018) and TLR7-specific (Bou Karroum et al., 2019) inhibitors have also been reported, but the in vivo properties of these molecules have not been characterized, and their potency may not be sufficient for clinical use. More recently, phase I trial results in healthy volunteers have been reported for a TLR7/8 inhibitor (E6742) (Nakai et al., 2018), but there is only a minimal amount of published information on its potential. Some other previously characterized small-molecule TLR inhibitors have been found to accumulate inside cells in a compartment such as the endosome because of protonation at low pH. This has previously been observed for hydroxychloroquine, which is used clinically, and other endosomal TLR7 and TLR9 inhibitors (Lamphier et al., 2014). Part of the mechanism of action of these compounds may be general interference with lysosomal pH and function and consequently inhibition of multiple endosomal TLRs (Ohkuma and Poole, 1978). Given that M5049 does not inhibit TLR3 and TLR9 and was demonstrated by crystallography to directly bind TLR8, it is unlikely that the compound is sequestered in lysosomes in the same way, and thus it has a different mechanism of action than hydroxychloroquine and similar lysosomotropic compounds and an advantageously more targeted mechanism of action.
The detailed selectivity and potency characterization of M5049 along with its absorption, distribution, metabolism, and excretion and PK properties suggest that it is a strong drug candidate. In our studies, M5049 not only showed a long duration of action in PK/PD experiments after a single dose, but once-per-day dosing in lupus mice was sufficient to provide efficacy even at unexpectedly low dosages for which plasma levels are below the IC50 predicted by in vitro and in vivo PK/PD studies in healthy mice. We hypothesize that the long duration of action of M5049 in disease model studies may be due to its high potency and in part the result of the sustained presence of the compound in tissues, and this localization could be beneficial for treating organ inflammation in patients with lupus with disease manifestations in tissues such as skin, joints, or the kidney. Supporting this hypothesis, it has recently been reported that L1 mRNA transcripts are increased in lupus kidneys and in salivary glands of patients with Sjogren syndrome, and these RNAs can stimulate TLR7 (Mavragani et al., 2016). Our in vitro results demonstrate that M5049 effectively inhibits these natural endogenous TLR7/8 ligands, and in vivo activity against them may reduce disease.
The observed efficacy for M5049 was expected in the BXSB-Yaa model, as this model is recognized to be driven by TLR7, but the effectiveness of M5049 in the IFN-α NZB/W model was not anticipated. To date, TLR7 has not been reported to be a driver in the IFN-α NZB/W model, and these mice do not show high titers of canonical RNA-containing autoantibodies such as anti-SmRNP and anti-RiboP, which are hallmarks of TLR7-driven disease, such as that in the BXSB-Yaa and pristane models (Reeves et al., 2009; Perry et al., 2011). Previously it has been noted that NZB/W mice show an elevated IFN gene signature, although the mechanism for the increase is unclear (Zhuang et al., 2015). It is possible that TLR7 is activated in the IFN-α NZB/W model by a previously not recognized mechanism such as circulating miRNA or Alu RNA molecules or more localized TLR7 ligands. However, the presence of the IFN gene signature in these mice could also be due to persistent effects of the adenovirus injection.
The unexpected observed efficacy in the IFN-α NZB/W model may indicate that TLR7/8 inhibition could be effective in a large variety of patients with lupus and not only those with elevated RNA autoantibody reactivities. It is also notable that IFN activity is not reduced in the IFN-α NZB/W model by M5049, and we have also observed efficacy for CMPD 2 in the pristane model without a reduction in IFN activity (data not shown). It has previously been demonstrated that TLR7 activation by R848 in NZM2328 lupus-prone mice can drive disease in an IFN-independent manner (Wolf et al., 2018). These observations along with the observation that the elevation of autoantibody titers in our NZB/W studies is modest may further support the idea that M5049's activity is a consequence of inhibiting inflammation in tissues and less of a systemic TLR7-IFN–driven effect. Although we have extensively tested M5049 selectivity and have shown it to have very targeted activity against TLR7/8, we cannot completely rule out the possibility that some off-target effects contribute to its efficacy in vivo.
Overall, these results indicate that M5049 is a compound with characteristics that make it a promising drug candidate. Additionally, our studies provide new insight into the consequences of TLR7/8 activation by a variety of ligands and how the TLRs may contribute to disease.
Acknowledgments
The authors would like to acknowledge the contribution of Eurofins Discovery for compound testing using the BioMAP Diversity PLUS panel. Bioscript Science, Macclesfield, UK, provided medical editing support.
Authorship Contributions
Participated in research design: Vlach, Bender, Haselmayer, Sherer.
Conducted experiments: Bender, Przetak, Pereira, Deshpande, Johnson, Reissig, Tzvetkov, Musil, Morse, Haselmayer, Favre-Zimmerli, Okitsu, Walsky.
Performed data analysis: Bender, Przetak, Pereira, Deshpande, Johnson, Reissig, Tzvetkov, Musil, Morse, Haselmayer, Favre-Zimmerli, Okitsu, Walsky.
Wrote or contributed to writing of the manuscript: Bender, Przetak, Tzvetkov, Musil, Morse, Favre-Zimmerli, Okitsu, Walsky, Sherer.
Footnotes
- Received August 7, 2020.
- Accepted December 14, 2020.
↵1 J.V. and A.T.B. are co-first authors.
All work was funded by EMD Serono, Billerica (a business of Merck KGaA, Darmstadt, Germany) and Merck KGaA, Darmstadt, Germany. Bioscript Science, Macclesfield, UK, provided medical editing support funded by Merck KGaA, Darmstadt, Germany.
J.V., A.T.B., M.P., A.P., A.D., T.J., E.T., N.T.M., S.F.-Z., S.L.O., R.W., and B.S. are employees of EMD Serono, Billerica (a business of Merck KGaA, Darmstadt, Germany). S.R., D.M., and P.H are employees of Merck KGaA, Darmstadt, Germany.
Primary laboratory of origin: [EMD Serono, Billerica, MA (a business of Merck KGaA, Darmstadt, Germany)].
This work was due to be presented at SLEuro 2020, but the congress was cancelled because of the COVID-19 outbreak: Sherer B, Bender AT, Pereira A, et al. (2020) O37 M5049, a novel potent and selective inhibitor of toll-like receptors 7 and 8 (TLR 7/8). Lupus Science and Medicine; 7:doi: 10.1136/lupus-2020-eurolupus.48.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- Alu
- Alu DNA element
- AOX
- aldehyde oxidase
- AUC
- area under the curve
- CMPD 2
- compound 2
- DOTAP
- dioleoyl-3-trimethylammonium propane
- HEK
- human embryonic kidney
- IFN
- type I interferon
- IFN-α
- interferon-α
- IL-6
- interleukin-6
- IRF
- interferon regulatory factor
- KO
- knockout
- miRNA
- microRNA
- MMF
- mycophenolate mofetil
- NF-κB
- nuclear factor-κB
- P450
- cytochrome P450
- PBMC
- peripheral blood mononuclear cell
- PD
- pharmacodynamic
- pDC
- plasmacytoid dendritic cell
- PK
- pharmacokinetic RiboP Ribosomal P
- SmRNP Smith Ribonucleoprotein TLR
- toll-like receptor
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}