


Visual Overview

Abstract
Blebbistatin, para-nitroblebbistatin (NBleb), and para-aminoblebbistatin (AmBleb) are highly useful tool compounds as they selectively inhibit the ATPase activity of myosin-2 family proteins. Despite the medical importance of the myosin-2 family as drug targets, chemical optimization has not yet provided a promising lead for drug development because previous structure-activity-relationship studies were limited to a single myosin-2 isoform. Here we evaluated the potential of blebbistatin scaffold for drug development and found that D-ring substitutions can fine-tune isoform specificity, absorption-distribution-metabolism-excretion, and toxicological properties. We defined the inhibitory properties of NBleb and AmBleb on seven different myosin-2 isoforms, which revealed an unexpected potential for isoform specific inhibition. We also found that NBleb metabolizes six times slower than blebbistatin and AmBleb in rats, whereas AmBleb metabolizes two times slower than blebbistatin and NBleb in human, and that AmBleb accumulates in muscle tissues. Moreover, mutagenicity was also greatly reduced in case of AmBleb. These results demonstrate that small substitutions have beneficial functional and pharmacological consequences, which highlight the potential of the blebbistatin scaffold for drug development targeting myosin-2 family proteins and delineate a route for defining the chemical properties of further derivatives to be developed.
SIGNIFICANCE STATEMENT Small substitutions on the blebbistatin scaffold have beneficial functional and pharmacological consequences, highlighting their potential in drug development targeting myosin-2 family proteins.
Introduction
Myosin-2 family proteins are the final effectors of cellular and intracellular contractile processes and tension generation ranging from cell division (Ma et al., 2012) to cell motility (Vicente-Manzanares et al., 2009) and neuronal plasticity (Kubo et al., 2008; Seabrooke and Stewart, 2011; Briggs et al., 2017) to skeletal, cardiac, and smooth muscle contraction (Szent-Gyorgyi, 1951, 1952; Johnson et al., 1994; Geeves and Holmes, 1999; Sellers, 2000; Eddinger and Meer, 2007; Brozovich et al., 2016; Tang et al., 2017). Therefore, myosin-2 isoforms are promising drug targets in treatment of various myosin- and cytoskeleton-related diseases, such as cancer, neuropathies, and myopathies (Straight et al., 2003; Newell-Litwa et al., 2015; Rauscher et al., 2018; Roman et al., 2018). As a myosin-2–specific inhibitor (Straight et al., 2003; Limouze et al., 2004), blebbistatin (Straight et al., 2003) can serve as a starting point for the development of clinical drug candidate (Rauscher et al., 2018; Roman et al., 2018). It has already been an excellent tool compound since its discovery in unveiling the role of the myosin-2 family in various biologic processes. Although the chemical optimization of blebbistatin was apparently fruitful in creating more photostable, less fluorescent, noncytotoxic, and more water-soluble tool compounds for research (Rauscher et al., 2018; Roman et al., 2018) through numerous chemical optimization efforts (Lucas-Lopez et al., 2005, 2008; Lawson et al., 2011; Képiró et al., 2012, 2014; Várkuti et al., 2016; Verhasselt et al., 2017a,b,c; Roman et al., 2018), no lead compound suitable for drug development have yet been published.
However, structure-activity-relationship studies of blebbistatin derivatives (Lucas-Lopez et al., 2005; Rauscher et al., 2018; Roman et al., 2018) suggest that a substantial chemical space available on the D-ring of compounds can be exploited to fine-tune the biologic and physicochemical properties of compounds. The structure-activity-relationship studies are in line with the crystal structure where A-B-C tricyclic core of blebbistatin fits tightly into the binding pocket and the D-ring protrudes out of the binding pocket (Allingham et al., 2005), providing substantial space for chemical alterations on this part of the molecule.
Moreover, blebbistatin and our recently developed derivatives azidoblebbistatin (Képiró et al., 2012), para-nitroblebbistatin (NBleb) (Képiró et al., 2014), and para-aminoblebbistatin (AmBleb) (Várkuti et al., 2016) have been applied as myosin-2 inhibitors in a series of in vivo disease models (blebbistatin: Zhang et al., 2009, 2011a,b; Gavin et al., 2011; Young et al., 2014, 2016, 2017; Briggs et al., 2017, 2018; Chen et al., 2018a,b; ABleb: Zenker et al., 2018; NBleb: Letelier et al., 2018; AmBleb: van Opbergen et al., 2018; Pénzes et al., 2020).
After our publications on the original synthesis, physicochemical properties, and toxicity assessment of NBleb and AmBleb (Képiró et al., 2014; Várkuti et al., 2016), NBleb has recently also been used in a drug development study focusing on the applicability of myosin-2 inhibitors in substance use relapse (patent WO2019/241469A1). In this patent NBleb was identified as a useful compound “for practice of an embodiment of the methods of the invention.”
Moreover, we have recently shown that AmBleb can be successfully used in ischemic stroke interventions due to its direct relaxing effect on precapillary smooth muscle cells (Pénzes et al., 2020), which otherwise remain permanently closed after stroke, thereby hindering the restart of healthy blood circulation at the capillary level even after recanalization of large vessels (Hall et al., 2014; Hill et al., 2015). Despite these promising effects in living systems, very little information is available about the pharmacological properties of blebbistatin, NBleb, and AmBleb.
The above observations motivated us to perform in-depth characterization of these tool compounds, blebbistatin, NBleb, and AmBleb, including their pharmacokinetic and pharmacodynamic properties, in vivo distribution, genotoxicity, and myosin-2 isoform specificity, which are essential to judge the feasibility of developing a drug candidate and to delineate routes for development.
Materials and Methods
Materials
High-pressure liquid chromatography (HPLC)–grade acetonitrile, chloroform and water were purchased from VWR (PA). Other chemicals were purchased from Sigma-Aldrich (Germany) if not otherwise stated. Blebbistatin was purchased from Sellekchem (TX), and isoflurane was purchased from Rotacher-Medical GmbH (Germany). Williams Medium E for freshly isolated hepatocytes was purchased from ThermoFischer (MA). Ames Microplate Format Mutagenicity Assay kit was purchased from Xenometrix.
Inhibitory Properties of Blebbistatin, NBleb, and AmBleb on Seven Myosin-2 Isoforms
Expression of Nonmuscle Myosin-2 Isoforms (Dictyostelium Myosin-2, NM2A, NM2B, and NM2C).
A single-tryptophan mutant Dictyostelium motor domain construct (W501+) (Málnási-Csizmadia et al., 2000) was expressed in Dictyostelium AX2-ORF+ cells and purified with His-Tag affinity chromatography as described previously (Gyimesi et al., 2008). The Sf9 codon optimized sequences for nonmuscle myosin-2 isoform (NM2) motor domains [M1-R775 for NM2A, M1-R782 for NM2B (B0), and M1-R799 for NM2C (C0)] fused to an Ala-Ser linker and an artificial α-actinin lever arm [as described earlier for NM2C (Heissler and Manstein, 2011)] with C-terminal FLAG-tag sequences were synthesized and cloned into pFastBac1 expression vector by Biomatik Co. The pFastBac1/NM2 constructs were overexpressed in Sf9 cells after bacmid preparation form DH10Bac cells. Sf9 cells were infected with P3 virus stocks, and myosin constructs were expressed for 48 hours, 27°C, 135 rpm. To reduce proteolytic digestion during expression, Pepstatin-A (0.1 mg/l) was added to Sf9 cells (Gotoh et al., 2001).
Purification of Actin and Sarcomeric Myosin-2 Isoforms (Skeletal, Cardiac, and Smooth Muscle Myosin-2s).
Rabbit actin was prepared according to an earlier published protocol (Spudich and Watt, 1971). Skeletal muscle myosin-2 was isolated from rabbit fast muscles (m. psoas, m. latissimus dorsi, m. biceps femoris) according to an earlier published protocol (Margossian and Lowey, 1982), and subfragment-1 (SkS1) was prepared by applying α-chymotryptic digestion (0.05 mg/ml α-chymotrypsin, 10 minutes, 25°C; reaction was stopped with 3 mM PMSF). Cardiac muscle myosin (CM) was isolated from left ventricle of porcine heart after cardiac myofibril preparation protocol (Tong et al., 2008) with the modification that 20 mM ATP (in Krebs-Hensleit buffer) was added to the final washed myofibril pellet, and the clear, myosin-containing supernatant was dialyzed against Storage buffer (20 mM Tris pH 7.5, 200 mM NaCl, 5 mM MgCl2, 2 mM β-mercaptoethanol) before freezing in liquid nitrogen. Smooth muscle myosin was prepared from freshly slaughtered chicken gizzard according to an earlier protocol (Trybus, 2000), and subfragment-1 (SmS1) was prepared by activated papain digestion according to a previously published protocol (Seidel, 1980) (0.2 mg/ml papain, 12 minutes, 25°C; reaction was stopped with 5 mM sodium iodoacetate).
ATPase Activity Measurements.
Steady-state ATPase measurements were carried out in 50 μl volume in a flat-bottom 384-well plate (Nunc-Thermo Fischer) using an NADH-PK/LDH coupled assay described earlier (Gyimesi et al., 2008) at 25°C in the presence of 0.5 mM ATP and F-actin (25 μM for W501+, NM2s; 11.5 μM for CM; 20 μM for SkS1; and 33 μM for SmS1) in a low ionic strength buffer (10 mM MOPS pH 7.0, 4 mM MgCl2, 2 mM β-mercaptoethanol) for 15 minutes. Blebbistatin derivatives were added to the reaction in 0.5 μl DMSO (1% of total volume), and three parallels were measured for each point. Controls containing DMSO with myosin but no inhibitor and actin-control containing actin and DMSO but no myosin were measured in all measurement sets. Applied myosin concentrations (100 nM SkS1, 200 nM W501+, 500 nM CM and SmS1, 1 μM NM2s) were used to fit quadratic function. ATPase activity was calculated from the slope of the linear fit to the time-dependent absorbance data collected at 340 nm.
Inhibition of Muscle Force and In Vitro Motility by Blebbistatin, NBleb, and AmBleb
Force Measurements on Muscle Fibers.
Synaptically evoked isometric force in the semi-intact larval bodywall preparation (Paterson et al., 2010) of Drosophila was measured as described previously (Ormerod et al., 2013). Briefly, Canton S strain Drosophila early wandering stage third instar larvae were dissected in Hemolymph-like (HL3.1) saline (10 mM NaHCO3, 115 mM sucrose, 5 mM trehalose, 70 mM NaCl, 5 mM KCl, 4 mM MgCl2, 5 mM HEPES, 1.5 mM CaCl2 × 2H2O, ≤0.1% DMSO). Synaptic potentials were elicited by stimulating a group of several segmental motoneurons via a glass suction electrode, Grass S88 stimulator, and stimulus isolation unit (Grass Technologies, West Warwick, RI). Single impulses were generated at 0.2 Hz, 0.3ms pulse duration, and ∼125% of the voltage needed to attain maximal compound excitatory junction potential amplitude. A custom designed force transducer (Micron Instruments, Simi Valley, CA) was used in all experiments to record evoked contractions. Treatment with blebbistatin, NBleb, and AmBleb followed the following routine: 1) baseline force generating ability was determined after 10–15 minutes in HL3.1 saline; 2) response to blebbistatin, NBleb, and AmBleb was determined during treatment (1–1000 μM inhibitor in HL3.1 saline, ≤0.1% DMSO) for 10–20 minutes; and 3) reversibility of inhibition and baseline force generating ability was evaluated after 10–15-minute washout in HL3.1 saline.
In Vitro Motility.
Fluorescent actin filaments were made by combining 1 μM tetramethyl-rhodamine with 1 μM actin in in vitro motility assay buffer (25 mM imidazole, pH 7.4, 25 mM KCl, 4 mM MgCl2, 1 mM EGTA, 1 mM DTT) containing 1 mM ATP. Movement of polymerized F-actin filaments over full-length myosin-coated surfaces was achieved using a modification to the method of Uyeda et al. (1990). Movies were collected on an ImageXpress XL high content imaging system at 25°C with a frame rate of 3 Hz and a 40× air objective. Compound dose responses were collected at a final concentration of 2% DMSO, 40 μM 2× serial dilution. Custom analysis software was created by VigeneTech, in which images were thresholded based on pixel intensity, filaments were identified, trajectories were determined for each filament, and filament velocities for each movie were calculated. Only filaments >2 μm and velocity of >25 nm/s were analyzed. Three to four movies, each with 500–2000 filament trajectories, from different surfaces were analyzed, and the median velocities of these were averaged for a single n. Each data point shown is a combination of three to four individual experiments (n = 3 to 4, each containing 4000–20,000 filament tracks per data point).
Molecular Dynamics Simulations
Molecular Dynamics Simulations for Blebbistatin Derivatives.
Molecular dynamics simulations and evaluations were carried out using AMBER16. For the derivatives, partial charges were calculated with the AM1-BCC charge model using the antechamber program. Force field parameters for GAFF force field were appended using parmchk2 based on atomic type similarity. Molecules were explicitly solvated with three-site model (TIP3P) waters and energy-minimized in 2000 steps of steepest descent followed by 4000 steps of conjugate gradient minimization. After minimization, a 10-nanosecond NPT simulation was carried out for each molecule at 1 bar pressure and 300 K temperature using Langevin dynamics for temperature coupling with 5 picosecond−1 collision frequencies. Samples were collected for the charge distribution calculation every 10 picosecond.
Calculation of Time-Averaged Charge Distributions.
The structures of the trajectory were RMS-fitted to three atoms of the tricyclic ring: the N atoms in the tricyclic ring and C6 atom of the A-ring. Since these atoms are part of a conjugate system, they remain fixed relative to one another during the simulations. We defined a 1-Å resolution rectangular with the tricyclic rings plane as the x, z plane of the ordinate system and the N linking the C and D rings as its origin. We then derived the time-averaged charge distribution by integrating the average density of each atom multiplied by its respective partial charge.
Simulations of the Myosin-Blebbistatin Complexes.
The ff14SB force field was used in all subsequent simulations to model protein interactions. The initial structure of the Dictyostelium discoideum myosin-2 head – blebbistatin complex was based on crystal structure 3mjx.pdb. ADP.VO4 was replaced with ADP.PO4. The water molecules resolved by crystallography were retained, and the model was expanded in an 8-Å clearance dodecahedron box with three-site model (TIP3P) water molecules for explicit solvation. The complex was minimized and heated in three 100-K 20-picosecond steps to 300 K under NVT conditions, then subsequently equilibrated to 1 bar in 20 picoseconds under NPT conditions. The system was relaxed for 500 picoseconds under NVT conditions at 300 K, then further equilibrated for 60 nanoseconds under NPT conditions, by which time the structure’s root-mean-square deviation (RMSD) compared with the initial structure had converged. Blebbistatin was replaced with the nitro- and amino derivatives at this point to obtain the initial structure for their relaxation. All three variants were re-equilibrated using the same equilibration protocol as described above up to the 500-picosecond 300-K NVT step. The complexes were then further equilibrated for 6 nanoseconds under NPT conditions to obtain the final structures. The trajectories were sampled every 10 picoseconds during the last 1 nanosecond of the simulation. The time-averaged charge distribution within the protein was calculated using the coordinates of blebbistatin atoms in these 100 frames with the method described for water solvent–only simulations. Side chain binding enthalpy contributions were calculating with the molecular mechanics energies combined with the generalized Born and surface area continuum solvation (MM-GBSA) method using the mmpbsa programs of AMBER.
In Vitro Metabolic Stability of Blebbistatin, NBleb, and AmBleb
Pharmacokinetic Studies.
After 2 days of accommodation the rodents were randomly divided into three groups. Compounds were administered intraperitoneally to each group 2.5, 5, 10, 15, 25, and 45 minutes before tissue sampling. Control animals were treated with 0.9% NaCl solution. Blood samples were collected from the heart under isoflurane inhalation anesthesia into test tubes containing 200 μl (500 IU/ml) heparin. Tissue samples from brain, heart, liver, kidney, spleen, muscle, and urine were collected from different animals after a 2-minute perfusion with Krebbs-Henseleit solution via the aorta. Tissue samples were stored in 1 ml chloroform at −80°C.
Sample Preparation for HPLC–Mass Spectrometry Analysis.
Tissue samples mixed with chloroform were minced, vortexed, and sonicated for 30 minutes. Samples were centrifuged (60,000g, 20 minutes, 4°C). The organic phase was collected and dried under a fume-hood. To the dried tissue samples, appropriate amount of acetonitrile-water mixture (50:50, v/v%) was added to the dried material; samples were sonicated for 30 minutes and ultracentrifuged (84,000g, 45 minutes, 10°C). After ultracentrifugation, supernatant was collected into already weighed eppendorfs, and net weight of the samples prepared for HPLC–mass spectrometry (MS) was determined. Ten microliters of supernatant was injected for HPLC-MS analysis.
HPLC-MS Conditions.
Chromatographic separation of the compounds and metabolites was carried out using an HP Agilent 1100 series HPLC system consisting of G1312A binary pump, G1365B multiwavelength detector, G1322A Degasser, G1313A auto sampler, and Waters SQ Mass Spectral Detector (Waters Corporation, Milford, MA). Chromatographic separation was achieved on an analytical C18 Merck Purospher STAR RP-18 endcapped (250 mm × 4.6 mm, 5 μm) column maintained at room temperature. Isocratic separation was carried out with acetonitrile:water (50:50 v/v%) as the mobile phase with the flow rate of 0.5 ml/min. The injection volume was 10 μl. Mass detection of samples were conducted utilizing an electrospray source in positive ion mode. Blebbistatin, NBleb, and AmBleb and their metabolites were quantified based on peak areas of their respective m/z values by extracted ion chromatograms from the single quadrupole scan measurements. For blebbistatin, NBleb, and AmBleb, reference materials were readily available, and their limit of quantification was at least 300 pg [30 ng/ml sample concentration (equal to 0.1 μM) with the 10 μl injection volume applied]. The MassLynx 4.1 software was used for instrument control, data acquisition, and evaluation.
High-Resolution Mass Spectrometry.
High-resolution mass spectrometric measurements were run on a SciexTripleTOF 5600+ hybrid quadrupole time-of-flight mass spectrometer (Sciex, MS) equipped with TurboV ion source. Samples were measured under electrospray condition in positive ion detection mode. The resolution of the instrument was 35,000. Source conditions were curtain gas: 45 arbitrary unit (AU), spray voltage: 5500 V, nebulizer gas: 40 AU, drying gas: 45 AU, source temperature: 450°C, collision energy in MS/MS experiment: 35 eV, scan time: 1 second. A Perkin Elmer Series 200 micro HPLC system with binary pumps and an autosampler was used for online HPLC-HRMS measurements. A Merck Purospher Star C18 (55 × 2 mm, 3 µm) was used for the separation. The mobile phases were water containing 0.1 v/v% formic acid (eluent A) and acetonitrile containing 0.1 v/v% formic acid (eluent B). The flow rate was 0.5 ml/min linear gradient was used starting with 20% B and increasing to 90% B by 8 minutes. This was followed by a 1-minute washing period with 90% B and returning to the initial conditions for 5 minutes for equilibrating the system. The HPLC-MS system was controlled by Analyst TF (Sciex, MA) software. Data were processed by PeakView and MasterView software (Sciex).
Isolation of Primary Hepatocytes.
Primary hepatocytes were isolated from male Wistar rats (Toxi-Coop Toxicological Research Center, Budapest, Hungary) and human tissue donors (Department of Transplantation and Surgery, Semmelweis University, Budapest, Hungary) using the collagenase perfusion method of Bayliss and Skett (1996). Briefly, the liver tissues were perfused through the portal vein with Ca2+-free medium (Earle’s balanced salt solution) containing EGTA (0.5 mM) and then with the same medium without EGTA, finally with the perfusate containing collagenase (Type IV, 0.25 mg/ml) and Ca2+ at physiologic concentration (2 mM). The perfusion was carried out at pH 7.4 and at 37°C. Softened liver tissue was gently minced and suspended in ice-cold hepatocyte dispersal buffer. Hepatocytes were filtered and isolated by low-speed centrifugation (50g) and washed three times. The yield and percent of cell viability according to the trypan blue exclusion test were determined (Berry et al., 1997). For pharmacokinetic studies, the hepatocytes were suspended at 2 × 106 cells/ml concentration in culture medium (Ferrini et al., 1998). Incubations with rat hepatocytes isolated from four animals were performed individually, whereas human hepatocytes pooled from three tissue donors were applied.
In Vitro Pharmacokinetics of Blebbistatin, NBleb, and AmBleb.
Time courses of the unchanged pharmacons (blebbistatin, NBleb, and AmBleb) in primary hepatocytes were obtained. Each compound was incubated with cell suspension (2 × 106 cells/ml) at 37°C in a humid atmosphere containing 5% CO2. The parent compounds dissolved in DMSO were added directly to the cell culture medium at the final concentration of 30 µM. The final concentration of DMSO was 0.1%. At various time points (at 0, 5, 10, 20, 30, 45, 60, 90, 120, 180, 240 minutes), the incubation mixtures were sampled (aliquots: 0.25 ml) and terminated by the addition of 0.25 ml ice-cold dichloromethane containing the internal standard, carbamazepine (0.13 µM). Blebbistatin and its derivatives were also incubated in cell-free medium and sampled at 0 and 240 minutes. The liquid-liquid extraction step was repeated two times, and the organic phases were collected and evaporated. The extract was dissolved in 100 μl of acetonitrile-water (50:50, v/v) and was analyzed by liquid chromatography–tandem MS for quantitation of the parent compound.
Estimation of Pharmacokinetic Parameters.
The intrinsic clearance (Clint) for hepatocytes [ml/(min×2 × 106 cells)] was calculated from the decrease in the concentration of the parent compound as follows (Obach, 1999): where the dose (D) was the target 30 nmol (in 1 ml) and
where the dose (D) was the target 30 nmol (in 1 ml) and The concentration at 0 minute (B) was 30 µM (30 nmol/ml), and β was determined by fitting exponentials to the measured drug candidate disappearance. As in our case D was numerically equal to B, and Clint was equal to β per hepatocyte concentration (2 × 106 cells/ml):
The concentration at 0 minute (B) was 30 µM (30 nmol/ml), and β was determined by fitting exponentials to the measured drug candidate disappearance. As in our case D was numerically equal to B, and Clint was equal to β per hepatocyte concentration (2 × 106 cells/ml): For scaling up the Clint value to obtain Clint per whole liver (g)/bw (kg), the cell concentration in the liver (cell number in rat liver: 1.17 × 108cells/g liver, in human liver: 1.39 × 108cells/g liver), the ratio of the average liver weight and average body weight parameters (for rat: 40 g/kg, for human: 23.7 g/kg) were used. The value for predicted hepatic clearance (ClH) was calculated as follows (Houston, 1994; Sohlenius-Sternbeck, 2006):
For scaling up the Clint value to obtain Clint per whole liver (g)/bw (kg), the cell concentration in the liver (cell number in rat liver: 1.17 × 108cells/g liver, in human liver: 1.39 × 108cells/g liver), the ratio of the average liver weight and average body weight parameters (for rat: 40 g/kg, for human: 23.7 g/kg) were used. The value for predicted hepatic clearance (ClH) was calculated as follows (Houston, 1994; Sohlenius-Sternbeck, 2006): where the hepatic plasma flow rate (HPF) is
where the hepatic plasma flow rate (HPF) is To calculate ClH, the hepatic flow rate (QH for rat: 55.2 ml/min per kilogram; for human: 20.7 ml/min per kilogram), plasma/blood ratio (for rat: 0.63; for human: 0.57) and the unbound fraction of the compound (fu) values were used (Davies and Morris, 1993; Szakács et al., 2001). For hepatocyte binding, the unbound fraction was calculated:
To calculate ClH, the hepatic flow rate (QH for rat: 55.2 ml/min per kilogram; for human: 20.7 ml/min per kilogram), plasma/blood ratio (for rat: 0.63; for human: 0.57) and the unbound fraction of the compound (fu) values were used (Davies and Morris, 1993; Szakács et al., 2001). For hepatocyte binding, the unbound fraction was calculated: The fu values for blebbistatin, AmBleb, and NBleb were 0.805, 0.916, and 1.0, respectively. The bioavailability (%) was determined by using the equation
The fu values for blebbistatin, AmBleb, and NBleb were 0.805, 0.916, and 1.0, respectively. The bioavailability (%) was determined by using the equation where E is the hepatic extraction ratio:
where E is the hepatic extraction ratio:
In Vivo Pharmacokinetics of Blebbistatin, NBleb, and AmBleb
Animals.
Male Wistar rats (220–250 g) were obtained from Toxi-Coop (Hungary). Animals were maintained under standard conditions (air-conditioned animal house at 25–28°C, relative humidity of 50%, and a 12:12 hour light/dark cycles). The animals were provided with water and diet pellets ad libitum. All experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health publication no. 85-23, revised 1996) and conformed to Directive 2010/63/EU of the European Parliament. The protocols were approved by the review board of the Department of Animal Health and Food Control of the Ministry of Agriculture and Rural Development, Hungary (XIII./1211/2012). Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines were adhered to during the study (NC3Rs Reporting Guidelines Working Group, 2010).
Compound Stability Tests.
Thirty micromolars (diluted from 1 mM DMSO stock) compounds in 0.5 ml blood sample were incubated for 0, 20, 40, and 60, 120, 240 minutes at room temperature in an Eppendorf tube supplemented with 30 IU/ml heparin. At each time point 50 μl chloroform was added to 50 μl sample aliquots. Samples were vortexed, sonicated, and centrifuged (60,000g, 20 minutes), and organic layer containing compound was collected. Organic layer of the respective samples was dried under fume-hood, and then dried samples were redissolved in acetonitrile-water mixture (50:50, v/v). Samples were then sonicated and ultracentrifuged (84,000g, 45 minutes, 10°C), and supernatant was collected. Net weight of the supernatant of each sample was determined. Stability of compounds was determined based on their peak area in HPLC chromatograms using HPLC-MS protocol described under pharmacokinetic studies in the following text.
In Vivo Pharmacokinetics: Time-Dependent Distribution of Blebbistatin, NBleb, and AmBleb in Rats.
All applied animals were littermates and weighed between 220 and 230 g upon arrival to the test facility. After 1 week of acclimatization, rodents were randomly divided into three groups of seven intraperitoneally receiving 1 mg of blebbistatin, NBleb, or AmBleb dissolved in 100 μl DMSO (34 mM for blebbistatin, 30 mM for NBleb, and 33 mM for AmBleb), which DMSO volume is in the safe range for single intraperitoneal dose (Bartsch et al., 1976; Gad et al., 2006). Due to homogenous weights upon arrival and identical housing conditions, all animals were 280 ± 5 g at the day of experiments. Note that 3.6 mg/kg is below the adverse cardiovascular and respiratory effect level determined for AmBleb in a separate study (Gyimesi et al., 2020). Control animals were treated with DMSO. Blood samples were collected from the heart under isoflurane inhalation anesthesia into test tubes containing 200 μl (500 IU/ml) heparin. Tissue samples from brain, heart, liver, kidney, spleen, muscle, and urine were collected from different animals after a 2-minute perfusion with Krebbs-Henseleit solution via the aorta. Tissue samples were stored in 1 ml chloroform at −80°C. We note that adverse effects were not observed at the site of DMSO injection in any organs, and no signs of inhibitor precipitation was observed in the intraperitoneum after necropsy.
Sample Processing for HPLC-MS.
Tissue samples were minced, vortexed, and sonicated in chloroform for 30 minutes. Samples were centrifuged (3000g, 20 minutes, 4°C). The organic phase was collected and dried under a laminar box. Two hundred microliters acetonitrile-water mixture (50:50, v/v) was added to the dried material and ultracentrifuged (45,000g, 45 minutes, 10°C). Ten microliters supernatant was injected for HPLC-MS analysis.
Mutagenicity Test of Blebbistatin, NBleb, and AmBleb
Ames Reverse Mutagenicity Test.
Ames microplate format (Xenometrix) reverse mutagenicity assay was performed according to the manufacturer’s guide on TA98 and TA100 bacterial strains in the absence and presence of phenobarbital/β-naphtoflavone-induced rat liver S9 fraction. We used phenobarbital/β-naphtoflavone–induced liver S9 fractions due to the higher structural similarity of these two componds to blebbistatin than that of Aroclor 1254, another possibly applicable substance. Solubility-dictated maximal applied concentrations of the inhibitors were 100 μM for blebbistatin and NBleb (29 and 34 μg/ml, respectively) and 400 μM (123 μg/ml) for AmBleb in 4% DMSO solutions. Note that slight precipitation occurred at the 100 μM NBleb–containing wells in the case of TA100 strain and the 400 μM AmBleb–containing wells in both TA98 and TA100 strains; therefore, those data points were not included in the analysis.
Results
ATPase Inhibition of Different Myosin-2 Isoforms by Blebbistatin Derivatives.
Actin-activated ATPase activities of seven different myosin-2 isoforms in the presence of different concentrations of blebbistatin, NBleb, and AmBleb were measured (Fig. 1; Table 1), and their solubility under assay conditions was confirmed (Supplemental Methods; Supplemental Table 1). As expected based on earlier results (Képiró et al., 2014; Várkuti et al., 2016), both derivatives inhibited myosin-2 isoforms similarly to blebbistatin (Straight et al., 2003; Limouze et al., 2004; Eddinger et al., 2007; Wang et al., 2008b; Heissler and Manstein, 2011; Zhang et al., 2017). However, on skeletal muscle myosin-2, NBleb showed reduced IC50 value compared with those of blebbistatin and AmBleb. These results indicate that the electron-withdrawing group in the para position of the D-ring positively influences the inhibitory properties of the molecule. The electron-donating amino group in AmBleb resulted in significantly lower maximal ATPase inhibition with similar IC50 values for the NM2A and NM2B isoforms (Fig. 1; Table 1), further confirming that electron distribution in blebbistatin’s D-ring is an important determinant of the inhibitory mechanism. We also analyzed the inhibitory efficiency, defined as the maximal extent of inhibition divided by the inhibitory constant (kinh = Imax/IC50) (Table 1), which corresponds to the initial slope of the fitted hyperbola. The ratio of the inhibitory efficiencies on skeletal and cardiac muscle myosin-2s showed drastic differences among the three inhibitors. The ratio is 56 for NBleb, whereas it is only 9.7 and 5.8 for blebbistatin and AmBleb, respectively. This finding suggests that skeletal muscle myosin specificity may be achieved by substituting the D-ring of blebbistatin with electron-withdrawing groups in the para position. Moreover, these results suggest that D-ring substitutions in the para position provide opportunities to fine tune inhibition and enhance isoform specificity among myosin-2s.

Inhibition of the actin-activated ATPase activity of seven myosin-2 isoforms. We measured the inhibitory effect of blebbistatin (A), NBleb (B), and AmBleb (C) on the F-actin–activated ATPase activities of seven myosin-2 isoforms, as indicated. Hyperbolic functions were fitted to the relative ATPase activity data points to determine IC50 and maximal inhibition (Imax) values for each myosin-2 isoform (Table 1). IC50 values for skeletal muscle myosin-2 were lower than the applied protein concentration; thus, a quadratic function was used to fit ATPase data to determine the IC50 and Imax parameters. Data points represent averages ± S.D. (n = 3–12) on (A–C).
Inhibitory parameters (IC50, maximal inhibition, and inhibitory efficiency) of blebbistatin, NBleb, and AmBleb for seven different myosin-2 isoforms
AmBleb Inhibits Force Generation.
We characterized how AmBleb inhibits force generation in in vivo muscle preparations using motoneuronal stimulation of muscle fibers. Briefly, neuromuscular preparations from Drosophila larval bodywall muscle were tested for force generating ability during incubation with AmBleb. As control, force generating ability was tested prior to incubation and after washout (Fig. 2A) and compound excitatory junction potentials were recorded before (pre-), during, and after (post-) application (Fig. 2B). AmBleb inhibited force generation, whereas it did not exhibit statistically significant change in amplitude of the synaptic voltages recorded at the neuromuscular junction (pre- 29.7 ± 1.8 mV and post- 30.1 ± 0.8 mV; ANOVA P = 0.55). Blebbistatin and NBleb effects were not characterized due to possible solubility-related precipitation of these inhibitors to the surface of fibers hindering appropriate measurements. This effect was not observed with AmBleb due to the sixfold higher solubility of Ambleb in HL-3.1 saline buffer than blebbistatin and NBleb (Supplemental Table 1). Concentration dependence of relative isometric force gives 9 ± 2 µM IC50 for AmBleb (Fig. 2C).

Blebbistatin, NBleb, and AmBleb inhibit force generation and motility. (A) Isometric force generation in larval Drosophila bodywall neuromuscular preparation is reversibly inhibited by AmBleb (red). Pretreatment (gray) and post treatment (black) traces are shown for comparison. (B) The intracellular voltage recordings remain unaffected by AmBleb treatment. (C) Concentration dependence of the relative isometric force in the presence of AmBleb (average ± S.E.M.). (D) Concentration dependence of the sliding velocity of actin filaments in motility assay with cardiac myosin in the presence of blebbistatin (blue), NBleb (green), and AmBleb (red) (mean ± S.D.).
Blebbistatin Derivatives Inhibit In Vitro Motility.
We next characterized the motion-generating capability of myosin in an in vitro motility assay by measuring the movement of individual rhodamine-phalloidin–labeled actin filaments over a myosin-coated surface (Uyeda et al., 1990). Actin filament velocity movies were automatically and objectively analyzed by a custom commercial software created by VigeneTech to measure two different velocity parameters. Briefly, these parameters are MVEL and TOP5%; MVEL is the mean velocity of all moving filaments and TOP5% is the mean of the top 5% of the velocity distribution across different actin filament lengths. The mean values from many actin velocity measurements using bovine β-cardiac full-length myosin at 25°C were as follows: MVEL: 350 ± 50 nm/s; TOP5%: 800 ± 70 nm/s (n = 9 exp, three preps). We next measured the motility of myosin in the presence of different concentrations of blebbistatin, NBleb, and AmBleb up to the solubility limit of the inhibitors (Fig. 2D; Supplemental Table 1). Both derivatives inhibited the velocity of actin gliding similarly to blebbistatin in a dose-dependent fashion. The IC50 of inhibition for blebbistatin, NBleb, and AmBleb was measured to be 0.7 ± 0.1, 4 ± 1, and 3 ± 1 µM, respectively.
Charge Distribution of Blebbistatin Derivatives in the Blebbistatin Binding Pocket Influences the Conformation of Key Functional Residues in Myosin-2.
Previously it was shown that the blebbistatin binding pocket is large enough to easily accommodate D-ring substitutions (Képiró et al., 2014; Verhasselt et al., 2017a). Here we calculated how D-ring substitutions with different electron profiles influence the interaction of the inhibitor with the protein. We calculated the time-averaged charge distribution around the three inhibitors in an explicit water box and in simulated myosin-bound structures. In water box, AmBleb showed a drastically different charge density profile around the para position of the D-ring, as compared with the other two inhibitors, whereas it was very similar for blebbistatin and NBleb (Fig. 3B). However, time-averaged charge densitiy of NBleb within the blebbistatin binding pocket of Dictyostelium myosin-2 became more similar to that of AmBleb (Fig. 3B). More importantly, the different charge densities around the D-ring of the inhibitors resulted in significant differences in the conformation of key functional residues of myosin-2 (Fig. 3C).

Time averaged charge distribution of blebbistatin, NBleb, and AmBleb in water and in myosin-2–bound forms. (A) Chemical structure of blebbistatin with A-D rings indicated. NBleb and AmBleb harbor a nitro and an amino group in para position of the D-ring, respectively. (B) Time-averaged charge distribution of blebbistatin, NBleb, and AmBleb in water (upper three structures) and docked and equilibrated in Dictyostelium myosin-2 structure in the ADP.Pi-bound pre-powerstroke state (lower three structures; transparent myosin structure is shown as cartoon with the relay helix colored in salmon). The three inhibitors are shown as green sticks and the time-averaged charge distributions are shown as spheres colored red for negative and blue for positive average charge values in a voxel (arbitrary units). Dotted surfaces represent the simulated charge envelope in both conditions. (C) Structure of the blebbistatin-binding pocket of Dictyostelium myosin-2 in the ADP.Pi-bound pre-powerstroke state, relaxed using molecular dynamics. Interacting side chains are shown as transparent sticks. The two side chains with significantly different binding energy contributions, Phe466 in the tip of the relay helix (salmon) and Lys587, are highlighted as solid orange sticks.
The major difference was observed in the inhibitor binding energy contribution of Lys587, which was practically negligible in blebbistatin and AmBleb (ΔGbleb = 0.006 ± 0.009 kcal/mol and ΔGAmBleb = 0.03 ± 0.03 kcal/mol, calculated from two independent runs on each inhibitor), whereas it was significant in NBleb (ΔGNBleb = −0.5 ± 0.3 kcal/mol). This residue plays important role in phosphate (Pi) release during the chemomechanical cycle of the myosin ATPase by blocking the Pi release route during the power-stroke of myosin (Gyimesi et al., 2008; Cecchini et al., 2010).
The other significant difference appeared in the orientation of Phe466, which has been described as a residue interacting with the D-ring of blebbistatin (Allingham et al., 2005). In the NBleb structure the phenyl ring of Phe466 develops more contact with the inhibitor, whereas it folds outwards in the blebbistatin and AmBleb structures (Fig. 3C). These differences may be of importance because Phe466 is in a key position between the switch-2 loop and the relay helix of myosin, structural elements that are responsible for the initiation of the powerstroke during myosin’s ATPase cycle (Málnási-Csizmadia and Kovács, 2010).
Metabolism of Blebbistatin, NBleb, and AmBleb in Primary Hepatocytes.
We characterized the pharmacokinetic properties in primary rat and human hepatocytes (Fig. 4, A–C) to determine the metabolic stability and to identify the major metabolites (Fig. 4, D–I) for all three tool compounds.
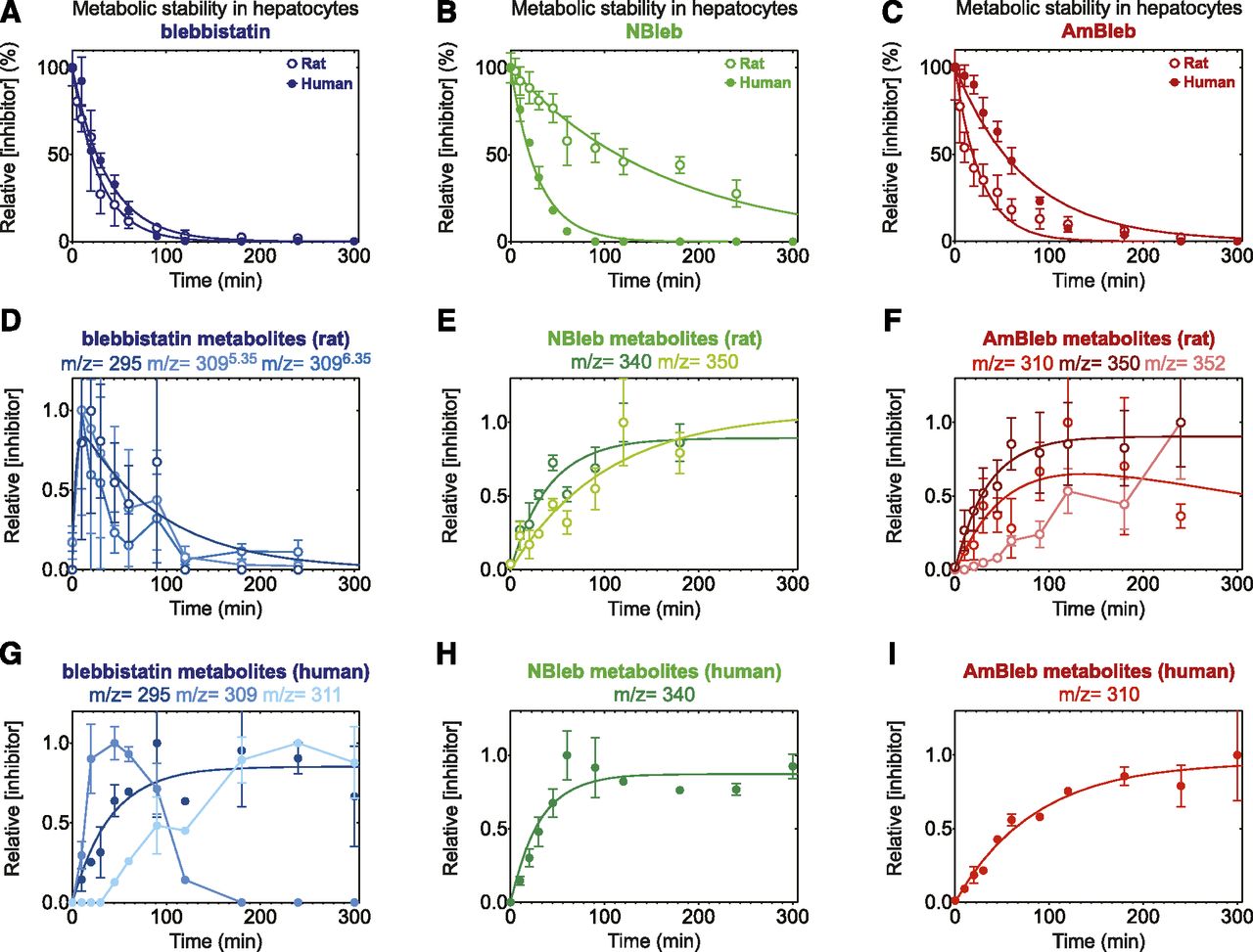
Biologic stability of blebbistatin, NBleb, and AmBleb in rat and human hepatocytes. Time-dependent changes in the concentration of blebbistatin (A), NBleb (B), and AmBleb (C) in the presence of rat (open circles) and human (solid circles) hepatocytes. Data points were fitted with single exponential functions (Table 2). Relative concentrations of the major metabolites detected in rat (D–F) or human (G–I) hepatocytes are shown. OH-bleb (m/z = 309) is a mixture of two identified isomers (cf. Fig. 5; Supplemental Fig. 2), in case of rat hepatocytes, whereas only one isomer could be detected with human hepatocytes. (D) Double exponential function was fit to blebmz295, which showed quick formation (t1/2 = 3.7 minutes) and elimination (t1/2 = 87 minutes) of this metabolite. Both metabolites with metabolites with m/z = 309 showed similar kinetics indicating that all major metabolites could be fully eliminated by rat hepatocytes in 300 minutes. (E) Single exponential functions were fit to data points showing that NBlebmz350 formed more slowly (t1/2 = 70 minutes) than NBlebmz340 (t1/2 = 28 minutes). (F) Single exponential function fit to AmBlebmz350 (t1/2 = 24 minutes) indicates that this metabolite could not be completely eliminated by rat metabolic enzymes, and formation of AmBlebmz352 is much slower than the formation of the other two metabolites. (G) Single exponential function fit to blebmz295 indicate that—contrary to rat hepatocytes—human enzymes could not eliminate this metabolite in 300 minutes. Formation of blebmz309 followed similar kinetics as with rat hepatocytes, and the rate of formation of blebmz311 followed inverse kinetics as that of blebmz309, indicating that blebmz311 is formed from blebmz309 and that blebmz311 could not be further metabolized by human hepatocytes. (H and I) The major difference between rat and human hepatocytes was the absence of the metabolite with m/z = 350 from both NBleb and AmBleb samples. Both NBlebmz340 and AmBlebmz310 formed with similar kinetics as with rat hepatocytes, but the elimination phase of AmBlebmz310 was not present until 300 minutes. Data points represent averages ± S.D. (n = 4–12).
In case of rat hepatocytes, pharmacokinetics of blebbistatin could be fitted with a single exponential function with half-life (t1/2) of t1/2,bleb = 20.2 minutes (Table 2). Three detectable metabolites appeared in the experiments with m/z = 295 [M + H]+ and m/z = 309 [M + H]+ (Fig. 4D). Importantly, we detected the metabolite m/z = 309 [M + H]+ at two different retention times (5.35 and 6.35 minutes), which suggests that two different species with the same molecular mass were formed in these experiments. All three detectable metabolites of blebbistatin were formed quickly (t1/2 between 3 and 10 minutes) and decomposed with similar rates than that of the original blebbistatin. We only determined the relative concentrations of these metabolites due to possible difference in molar extinction coefficients between blebbistatin and the metabolite.
Pharmacokinetic parameters of blebbistatin, NBleb, and AmBleb from experiments using rat and human hepatocytes
Clint = (ln2/t1/2) × ((cell number/tissue weight)/(cell number/incubation volume)) × (liver weight/body weight), where t1/2 is the time required to achieve 50% of the initial concentration at t = 0 min. ClH = (Clint × fu × HPF)/(Clint × fu + HPF), where HPF = QH × plasma/blood ratio, where QH = 55.2 ml/min per kilogram for rat, 20.7 ml/min per kilogram for human, plasma/blood ratio = 0.63 for rat, 0.57 for human. EH = ClH/QH. F (bioavailability) = 100*(1−EH).
Compared with blebbistatin, NBleb showed markedly slower elimination kinetics by rat hepatocytes with t1/2,NBleb = 114 minutes (Fig. 4B; Table 2), which clearly suggests that the D-ring substitution affects not only the inhibitory functions but also the pharmacokinetic properties of blebbistatin. Similarly to blebbistatin, metabolites could be detected with m/z = 340 [M + H]+ and m/z = 350 [M + H]+ (Fig. 4E). In agreement with slower elimination kinetics of NBleb compared with blebbistatin, the half-lives of formation of these metabolites were also significantly slower. Although only relative concentrations were determined for these metabolites, we could suspect that the metabolite with m/z = 350 [M + H]+ may be the major metabolite in case of rat hepatocytes, as the rate of formation of this molecule was similar to the elimination kinetics of NBleb (t1/2,NBleb = 114 minutes, t1/2,mz350 = 70 minutes). In contrast to those of blebbistatin, neither metabolites of NBleb decomposed during the 240-minute experiment, which suggests that these metabolites are not substrates for the enzymes transforming blebbistatin metabolites.
Interestingly, AmBleb decomposition by rat hepatocytes showed a markedly higher rate than that of NBleb but very similar kinetics to that of blebbistatin (t1/2,AmBleb = 19.0 minutes) (Fig. 4C; Table 2), confirming that D-ring substitution at least in the para-position has functional and pharmacological consequences by modulating pharmacokinetic properties of blebbistatin derivatives. Three detectable metabolites with m/z = 310 [M + H]+, m/z = 350 [M + H]+, and m/z = 352 [M + H]+ could be detected during AmBleb biotransformation (Fig. 4F). Moreover, similarly to that of NBleb, the metabolite with m/z = 350 [M + H]+ could be the major metabolite of AmBleb because the half-life of its formation (t1/2,mz350 = 24 minutes) was almost identical to that of AmBleb elimination.
To investigate differences between the metabolic properties of the three inhibitors in the presence of rat and human hepatocytes, we performed the same experiments with primary human hepatocyte samples (Fig. 4, A–C). Similar decomposition kinetics was observed for blebbistatin and AmBleb as with the rat hepatocytes, although AmBleb elimination kinetics was slightly slower by the human hepatocytes (Table 2). However, the more than sixfold slower elimination of NBleb could not be detected in case of human hepatocytes; rather, a very similar rate was observed to that of blebbistatin. Moreover, characteristic differences between the two species could be observed in the metabolite profiles of the three inhibitors (Fig. 4, G–I).
Although the metabolite of blebbistatin with m/z = 295 [M + H]+ could be detected, the rate of formation was 10-fold slower (t1/2,mz295,rat = 3 minutes, t1/2,mz295,human = 28 minutes) and it did not disappear until 300 minutes. Moreover, only one species of metabolite with m/z = 309 [M + H]+ could be detected, which elimination was concomitant with the formation of a new metabolite (not detected with rat hepatocytes) with m/z = 311 [M + H]+, which metabolite did not disappear until 300 minutes either.
In contrast to the experiments with the rat hepatocytes, only one metabolite was formed from NBleb with m/z = 340 [M + H]+ and from AmBleb with m/z = 310 [M + H]+. Interestingly, the metabolite with m/z = 350 [M + H]+ could not be detected in either NBleb or AmBleb, which indicates that the same metabolite is formed from NBleb and AmBleb with rat hepatocytes and further suggests the absence of that reaction pathway in the human hepatocytes.
Pharmacokinetic analyses were performed on results from both rat and human primary hepatocytes, and pharmacokinetic parameters have been calculated for all three inhibitors (Table 2). As expected from the similar kinetics of blebbistatin and AmBleb in rat samples, hepatic extraction ratio (EH) was almost identical for these two inhibitors, whereas NBleb had somewhat lower EH levels. Consequently, the bioavailability of blebbistatin and AmBleb was around 60%, whereas that of NBleb was more than 80%. In human hepatocytes, pharmacokinetic behavior of blebbistatin was similar to that in rat hepatocytes (t1/2 values in rat and human hepatocytes were similar), whereas substantial differences were observed in the rates of elimination of NBleb between rat and human. On the other hand, the lower hepatic clearance (ClH) of all three compounds in human hepatocytes than in rat cells was considered to be associated with lower hepatic flow rate in human (QH for rat: 55.2 ml/min per kilogram; for human: 20.7 ml/min per kilogram). Regarding the bioavailability (F), it should be noted that prediction from hepatic clearance may result in an overestimation. Bioavailability is related to the total clearance (the sum of all clearances in the organs that participate in the elimination of drugs, e.g., intestine and kidney); therefore, intestinal metabolism can be expected to decrease the bioavailability of orally administered drugs, and renal clearance can also modify the bioavailability in case of extensive renal elimination. Although the assessment of total clearance would be optimal, much information can be obtained from hepatic clearance as well. In conclusion, all three compounds should still be considered as drugs with high extraction ratios, where elimination is mainly determined rather by the hepatic blood flow extraction ratio than by the activities of the metabolizing enzymes. It also indicates that further modifications of blebbistatin should be performed to have more favorable pharmacokinetic properties and to achieve higher bioavailability values.
Identification of Major Metabolites.
Pharmacokinetic analysis of blebbistatin, NBleb, and AmBleb in freshly isolated hepatocytes showed that all three compounds were extensively metabolized forming several metabolites (Fig. 4). To identify these metabolites, the rat hepatocyte-incubated samples were analyzed by high-resolution MS and MS/MS (Fig. 5).

Identification of major metabolites of blebbistatin, NBleb, and AmBleb. Major metabolites of blebbistatin (1), NBleb (2), and AmBleb (3). The exact positions of the hydroxyl groups on blebbistatin, NBleb, and AmBleb could not be determined; only the rings (A, C or D) that contained hydroxyl substituent were identified. The keto-group in the B-ring was reduced in blebbistatin (1a), NBleb (2a), and AmBleb (3b). Based on kinetic analysis in Fig. 4, the hydroxylation of blebbistatin on the C-ring (1e) occurs before the reduction of the B-ring keto-group (1b). N-Ac-AmBleb (3a) could be formed from NBleb by transient formation of AmBleb and further acetylation of the amine-group as in the case of AmBleb (cf. Fig. 4). N-Ac-AmBleb (3a) could be further reduced on the keto-group of the B-ring (3e), which metabolite might also be formed by acetylation of 3b. This latter scenario is supported by the kinetics of elimination of 4-OH-AmBleb=AmBlebmz310 (3b) and the formation kinetics of 4-OH-N-AcAmBleb=AmBlebmz352 (3e) shown on Fig. 4F.
The keto-group on the B-ring could be reduced to hydroxyl in all three inhibitors resulting in two enantiomer forms with different chromatographic retention properties. This reduced metabolite of all three inhibitors could be detected with HPLC-MS analysis above [4-OH-bleb, m/z = 295 [M + H]+ (molecule “1a” on Fig. 5); 4-OH-NBleb, m/z = 340 [M + H]+ (2a); 4-OH-AmBleb, m/z = 310 [M + H]+ (3b)]. These results indicate that the metabolites detected from the hepatocytes with the same m/z values are equivalent to these metabolites. The reduced derivatives are further hydroxylated either on the C-ring as in the case of blebbistatin (1b) and NBleb (2b), or on the A-ring as in the case of AmBleb (3c). Moreover, the C-ring hydroxylated reduced blebbistatin (1b) is further hydroxylated on the A-ring too, thereby forming a reduced dihydroxy-blebbistatin metabolite (1c). Dihydroxyl derivatives could not be identified among the metabolites of NBleb and AmBleb.
Direct hydroxylation of blebbistatin, NBleb and AmBleb was also detected, but with characteristic differences in the hydroxylation patterns (Fig. 5). Blebbistatin is hydroxylated on the A-, C-, and D-rings (1d, 1e, 1f), from which two species could be detected with HPLC-MS analysis above with m/z = 309 [M + H]+. Direct hydroxylation of the C- and D-rings of NBleb and AmBleb did not occur (Fig. 5), indicating that nitro- or amino-substitution on the D-ring not only hinders D-ring hydroxylation but also has an effect on the electron distribution of the C-ring as well, thereby preventing it from hydroxylation. However, the reduced NBleb derivative, that is, 4-OH-NBleb (2a), is hydroxylated on the C-ring as well (2b), which does not occur with the reduced AmBleb form. Instead, reduced AmBleb is hydroxylated on the A-ring (3c) similarly to direct hydroxylation of AmBleb.
We could also identify a metabolite of blebbistatin with m/z = 311 [M + H]+, which is the C-OH-blebbistatin with reduced keto-group on its B-ring (1b). From the formation end elimination kinetics of these metabolites with human hepatocytes (cf. Fig. 4G) we can assume that hydroxylation occurs first, which is further reduced on the B-ring.
The major metabolite formed from AmBleb in pharmacokinetic assays with m/z = 350 [M + H]+ was identified as N-acetylated metabolite of AmBleb (N-Ac-AmBleb) (3a). Furthermore, this metabolite was also identified as the metabolite produced from NBleb with m/z = 350 [M + H]+ (c.f. Fig. 4, E and F). This suggests that NBleb underwent reduction, forming AmBleb that was further acetylated. The formation of AmBleb from NBleb could not be detected, indicating that N-acetylation is a much faster reaction than the reduction of the nitro-group to amine-group. This fact was confirmed in the pharmacokinetic assays with AmBleb demonstrating the extensive formation of N-Ac-AmBleb. We also identified the metabolite with m/z = 352 [M + H]+ as the N-Ac-AmBleb with reduced keto-group on its B-ring (3e). From the formation end elimination kinetics of these metabolites with rat hepatocytes (cf. Fig. 4F) we can assume that B-ring keto-group reduction occurs first, which is further acetylated to the amine-group. We note that neither acetylated forms could be detected in human hepatocytes, indicating that this enzymatic route is missing from the human samples.
Time-Dependent Distribution of Blebbistatin, NBleb, and AmBleb in Rats.
To follow the time-dependent distribution of the three compounds and their metabolites in living rats, we injected 1 mg each of blebbistatin, NBleb, and AmBleb intraperitoneally into 280 ± 5 g animals, resulting in 3.6 mg/kg dose level with 2% accuracy due to weight differences in rats. Assuming an even distribution, the 1 mg inhibitor dose applied should result in ∼15 μM per animal concentration. At different time points after drug administration, samples were collected from heart, blood, skeletal muscle (m. quadriceps femoris), brain, kidney, lung, spleen, and liver tissues (Fig. 6).

Distribution of blebbistatin, NBleb, and AmBleb and their metabolites in different tissues of living rat. (Labeling in parentheses indicate the nomenclature used in Fig. 5.) Time-dependent concentrations of blebbistatin, NBleb, and AmBleb were determined in different tissue types (as indicated on the graphs) after intraperitoneal administration to living rats. Significant accumulation of blebbistatin and AmBleb could be observed in muscle tissue samples, whereas NBleb did not appear in muscle samples in a detectable amount at any time points. After blebbistatin injection (blue bracket), 4-OH-bleb (1a) and the mixed populations of hydroxy-blebbistatin metabolites (1d–1f) were detected (Supplemental Fig. 2). After NBleb injection (green bracket), 4-OH-NBleb (2a) could be detected in all tissue samples, whereas N-Ac-AmBleb (3a) was present only in the liver. Shaded arrows indicate the assumed formation of AmBleb on the route of N-Ac-AmBleb formation, as suggested by MS/MS analysis. After AmBleb injection (red bracket), N-Ac-AmBleb (3a) was detected in all tissue types except for muscle samples. 4-OH-N-Ac-AmBleb (3e) could also be detected in the tissue samples, and the arrow indicates the possible route of its formation form 4-OH-AmBleb (3b) as suggested by the kinetics in Fig. 4F. Data points represent averages ± S.D. (n = 4).
Inhibitor concentrations and metabolites were analyzed by HPLC-MS. Stability of all three compounds in blood (Supplemental Fig. 1) was similar, and the metabolism of all three inhibitors was rapid. Their concentrations were well below 5 μM at all time points in all tissue types except for muscle. Interestingly, blebbistatin and AmBleb concentrations increased over time in muscle samples, suggesting an accumulation of these two inhibitors in skeletal muscle tissue (Fig. 6). This phenomenon was not observed for heart muscle tissue. The substantially lower tissue concentration of NBleb compared with that of blebbistatin and AmBleb may indicate insufficient NBleb solubility at the point of administration. Even though NBleb solubility in general is only slightly worse than that of blebbistatin (Supplemental Fig. 1), NBleb can precipitate into needle-like crystals (Várkuti et al., 2016), which may negatively affect its effective concentration in vivo.
From the MS spectra the three OH-bleb metabolites (1d–1f) could be identified with characteristically different retention times (RT1 = 4.0 minutes, RT1 = 5.3 minutes, RT1 = 6.3 minutes) (Supplemental Fig. 2). The kinetics and tissue distribution of the three metabolites showed characteristically different properties (Fig. 6). The OH-blebRT4.0 appeared in the kidney in the highest concentration, whereas OH-blebRT6.3 was observed in the liver in the highest concentration. The OH-blebRT5.3 metabolite was quickly formed but also quickly eliminated from all tissues; however, a slight accumulation of this metabolite was observed in the muscle tissue samples similar to the original blebbistatin.
4-OH-NBleb (2a) metabolite of NBleb predominantly appeared in the liver samples, remained high for 60 minutes, and showed a tendency to increase in the kidney 10 and 60 minutes after NBleb injection (Fig. 6). In agreement with the results from the hepatocyte experiments, AmBleb could not be detected in tissue samples after NBleb injections; however, the N-Ac-AmBleb metabolite (3a) was observed in high concentration in the liver. The presence of N-Ac-AmBleb in the liver samples and the lack of detectable amount of N-Ac-AmBleb in any other tissues suggest that AmBleb is quickly formed in the liver and further acetylates in place.
However, N-Ac-AmBleb (3a) could be detected in all tissue samples after AmBleb injection although with drastically higher concentrations in the liver. Similarly to that observed for N-Ac-AmBleb metabolite formed from NBleb, N-Ac-AmBleb formed from AmBleb did not accumulate in any tissue types, and its concentration was significantly higher in the liver. In both cases, N-Ac-AmBleb is almost completely eliminated at 60 minutes, suggesting quick elimination of this metabolite from the body.
Reverse Mutagenicity (Ames) Test of Blebbistatin, NBleb, and AmBleb.
We tested the mutagenicity of all three compounds in a reverse mutagenicity Ames test using two Salmonella strains sensitive for frameshift (TA98 strain) and base-pair substitution (TA100 strain) mutations in the Ames microplate format mutagenicity assay (Flückiger-Isler et al., 2004; Flückiger-Isler and Kamber, 2012) in the absence and presence of liver S9 fraction (Fig. 7). A molecule is indicated Ames positive (i.e., probably mutagenic) if it shows drug concentration-dependent mutagenicity or any measured data point falls above the mutagenicity threshold defined as twice of the average plus S.D. of the solvent control (Fig. 7).
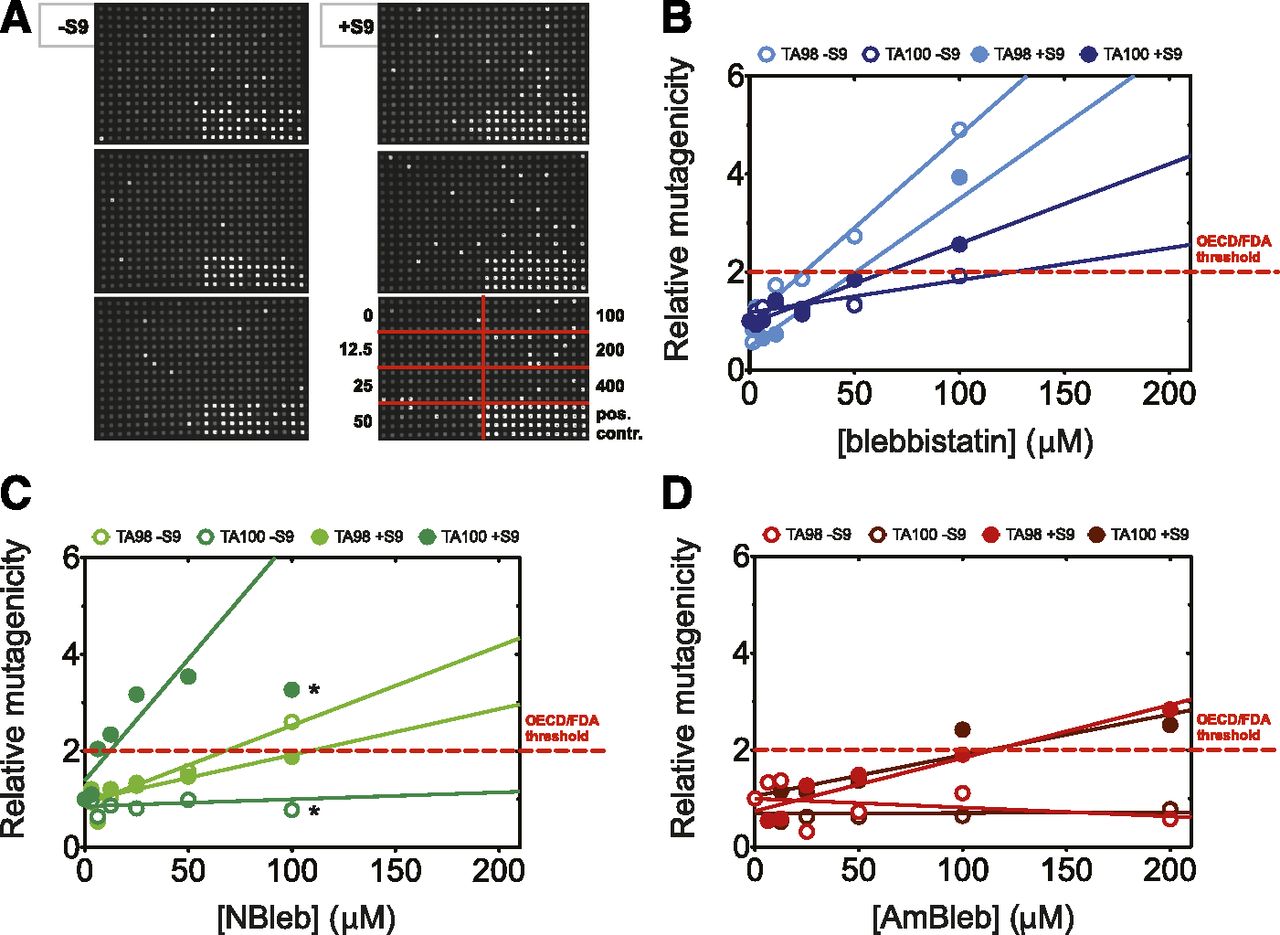
Reverse mutagenicity (Ames) test of blebbistatin, NBleb, and AmBleb in the absence and presence of induced rat liver S9 fraction. (A) Ames microplate format test plate triplicates with TA98 Salmonella strain in the presence of AmBleb (concentrations are indicated in micromolars). DMSO and 2-nitrofluorene were used as negative and positive controls, respectively, according to the manufacturer’s protocol. Dark wells represent conditions with no bacterial growth, whereas white wells contain growing bacteria assuming a reverse mutation in their genome. Relative mutagenicity of blebbistatin (B), NBleb (C), and AmBleb (D) tested on TA98 (lighter circles) and TA100 (darker circles) Salmonella strains in the absence (open circles) and presence (solid circles) of rat liver S9 fraction. The dashed red line indicates the mutagenicity threshold, defined as twice the average plus S.D. of the solvent control. All three inhibitors showed concentration-dependent increase in mutagenicity, and all three compounds produced values (individual data points) over the mutagenicity threshold. *Linear fits excluded measurements where precipitation was observed. Importantly, AmBleb is not mutagenic in the absence of rat liver S9 fraction; however, in the presence of rat liver S9 fraction even AmBleb shows concentration-dependent mutagenicity in both strains. FDA, US Food and Drug Administration; OECD, Organization for Economic Cooperation and Development.
Importantly, whereas blebbistatin and NBleb were mutagenic, AmBleb was not mutagenic in the absence of S9 fractions. Under these conditions, both for the TA98 and TA100 strains, the slope of the linear fit to concentration-dependent mutagenicity profiles of AmBleb was either negative or close to zero, respectively, indicating that the compound does not reach the mutagenicity threshold (Fig. 7D). However, in the presence of liver S9 fraction all three compounds showed concentration-dependent mutagenicity, and some data points fell above the mutagenicity threshold for all three compounds (Fig. 7, B–D). The same metabolites were produced during incubation with S9 liver fraction as in the experiments with the isolated hepatocytes (cf. Fig. 4) confirmed by high-resolution MS and MS/MS.
Among all investigated conditions, NBleb was the most mutagenic in the presence of S9 liver fraction in the TA100 strain. Whereas the nitro-group increased mutagenicity, especially base-pair substitution mutations, the amino group substitution significantly decreased blebbistatin’s mutagenicity, supporting our previous findings that the nitro-group has an inverse effect on key biologic and pharmacological processes (such as ATPase inhibition, cell permeability, and pharmacokinetic profile) as compared with the amino substitution (cf. Figs. 1⇑⇑⇑⇑–6). This finding implies that substitution of the D-ring of blebbistatin at the para position with electron-donating groups can be used to significantly decrease its mutagenic properties.
Discussion
Blebbistatin was discovered in a high-throughput assay screening for cytokinetic furrow ingression arrest, targeting nonmuscle myosin-2 functions (Straight et al., 2003). Blebbistatin has been further shown to influence additional NM2-related cellular functions (Melendez-Vasquez et al., 2004; Ponsaerts et al., 2008; Wang et al., 2008a; Walker et al., 2010). Moreover, NM2 isoforms—expressed in all human cell types—generate forces during cell division (Ma et al., 2012), regulate neuronal plasticity (Seabrooke and Stewart, 2011; Briggs et al., 2017), and contribute to cellular movements (e.g., in macrophages, fibroblasts, and cancer cell metastases) (Vicente-Manzanares et al., 2009).
Beside its effects on NM2 isoforms, blebbistatin has been in the focus of interest in developing myosin-2–specific drugs due to the involvement of myosin-2 motor proteins in several essential life processes. Myosin-2 isoforms are responsible for voluntary contraction of skeletal muscles (Szent-Gyorgyi, 1951; Geeves and Holmes, 1999) and that of the diaphragm (Johnson et al., 1994); cardiac myosin-2s drive the pumping of the heart (Szent-Gyorgyi, 1952; Tang et al., 2017), and smooth muscle myosin-2 is a key component of tension maintenance in blood vessels and the unconscious movements of our organs (Eddinger and Meer, 2007; Brozovich et al., 2016). Thus, blebbistatin is a highly potent nonselective myosin-2 inhibitor, indicating a general mechanism of inhibition on all myosin-2 isoforms (Kovács et al., 2004; Limouze et al., 2004; Wilson et al., 2014; Tang et al., 2017; Zhang et al., 2017). NBleb and AmBleb have also been shown as potent inhibitors of Dictyostelium and fast skeletal myosin-2s (Képiró et al., 2014; Várkuti et al., 2016; Verhasselt et al., 2017a), but their inhibitory properties have not been studied on other myosin-2 isoforms.
Thus, the development of drugs that could regulate myosin-2 functions—especially in a selective manner—is an important approach especially because myosins are the most downstream effectors of signaling pathways, and this way side effects related to upstream regulators can be avoided. For drug development purposes, isoform selectivity is an important feature; therefore, we investigated whether inhibitory efficiency and myosin-2 isoform specificity profile can be tuned by D-ring substitutions and how the D-ring substitutions affect the inhibition of in vivo force generation or in vitro motility.
In the present study, we show that essential inhibitory and absorption, distribution, metabolism, excretion, and toxicology (ADMET) properties can be improved by chemical optimization. Actin-activated ATPase measurements on seven myosin-2 isoforms, in vitro actin gliding measurements on cardiac myosin, and in vivo fiber studies on Drosophila skeletal muscle confirmed that D-ring–modified blebbistatin derivatives retain their inhibitory potential on the myosin-2 family, whereas it is possible to tune their selectivity toward the different myosin-2 isoforms. For skeletal muscle myosin NBleb showed a reduced IC50 value in the actin-activated ATPase assay and in vivo fiber studies (Fig. 1; Table 1), for the cardiac system, both NBleb and AmBleb have higher IC50 values as compared with blebbistain, as verified both by actin-activated ATPase and in vitro actin gliding measurements (Figs. 1 and 2A). Comparing the inhibitory efficiency of the three inhibitors on skeletal and cardiac muscle myosin-2s, we found that the ratio between skeletal and cardiac myosin-2s is 6 times and 10 times higher for NBleb than those for blebbistatin and AmBleb, respectively (Table 1). This feature highlights a very important indication for further drug development toward a skeletal muscle myosin-specific compound. This aspect is especially important as skeletal muscle myosin inhibition would be a novel approach for the treatment of spasticity in poststroke conditions or in patients with multiple sclerosis, cerebral palsy, or medication-induced spasms, where currently there is a huge unmet medical need for more efficient drugs without neurologic side effects (Chou et al., 2004; Chang et al., 2013).
The pharmacodynamics and metabolic stability studies indicate that although it is necessary to improve several properties of blebbistatin for drug development purposes, D-ring modifications can be very promising in terms of absorption-distribution-metabolism-excretion optimization. An important pharmacological parameter that needs to be improved is the ∼20-minute metabolic half-life of blebbistatin, as this rapid elimination could result in insufficient plasma concentration, hindering the medical application of the compound. Although rapid elimination of AmBleb similar to that os blebbistatin was demonstrated in rat hepatocytes, much lower elimination half-life and Clint was found in human cells. The rate of AmBleb metabolism would be close to sufficient for human uses with multiple doses per day prescription. In contrast, although NBleb was known to have increased chemical stability (Verhasselt et al., 2017a), and in rat hepatocytes, it showed increased biologic stability with elimination kinetics fivefold slower than that of blebbistatin (Fig. 4; Table 2), human liver cells were as active in metabolism of NBleb as in that of blebbistatin. Moreover, given that a small change such as amino-to-nitro substitution can have this remarkable effect on elimination time, we may expect to be able to find substituents with further improved pharmacokinetic properties. These findings, however, emphasized the importance of species differences in drug metabolism and careful extrapolation from laboratory animals to human.
Furthermore, parallel metabolic routes and variations in the activities of drug metabolizing enzymes can also contribute to interindividual differences in pharmacological efficacy or adverse effects. Similarly to NBleb metabolic pathways, nitro-group reduction to an amino-derivative by CYP3A4 that is further acetylated by N-acetyl transferase 2 (NAT2) are well described steps in clonazepam metabolism (Peng et al., 1984). However, hydroxylation pathway is considered to be negligible in case of clonazepam. Clonazepam plasma concentrations seem to be influenced by CYP3A4 activity, whereas 7-amino-clonazepam concentrations depend on the activities of both CYP3A4 and NAT2 (Tóth et al., 2016). Interindividual variations in the activities of CYP3A4 and NAT2 can results in clinical consequences. The metabolite 7-amino-clonazepam is pharmacologically inactive; however, it can competitively modify the effect of clonazepam, primarily when the concentration decreases after discontinuation of clonazepam therapy. Therefore, clinical consequences are anticipated for the patients with high levels of 7-amino-clonazepam during clonazepam withdrawal. In contrast, hydroxylation at various positions was the major route of blebbistatin metabolism, N-acetylation of the amino-group was the primary reaction of AmBleb metabolism, and in NBleb metabolism, nitro-reduction to amino-derivative (and further acetylation) and hydroxylation at various positions were observed. Various metabolic pathways and the activities of the phase I (mainly cytochrome P450 enzymes) and phase II N-acetyl transferase enzymes are assumed to be associated with different pharmacokinetic behavior of blebbistatin and two of its derivatives.
One of the most important tests at early stages of drug development is a mutagenicity assay. Due to the unambiguous correlation between mutagenicity and carcinogenicity (McCann et al., 1975), Organization for Economic Cooperation and Development, US Food and Drug Administration, and European Medicines Agency guidelines all require the clear demonstration that a lead compound is not mutagenic. The minimal requirement is to test mutagenicity in a reverse mutagenicity Ames test using two Salmonella strains sensitive for frameshift (TA98 strain) and base-pair substitution (TA100 strain) mutations. Guidelines also suggest repeating the test in the presence of induced S9 rat liver fraction to elucidate the mutagenicity of the potential metabolites produced in the liver during metabolism of the compound in the living organism. Although we found that blebbistatin and NBleb are clearly mutagenic, AmBleb is only mutagenic in the presence of S9 liver fraction, indicating that only AmBleb metabolites are mutagenic and AmBleb itself is not mutagenic. Although the genotoxicity of blebbistatin derivatives must be prudentially improved, given that the small difference between the amino- and nitro-substituted derivatives have drastic effect on mutagenicity, we may manage to design and synthesize future compounds with fully eliminated mutagenicity.
Conclusions
Blebbistatin derivatives are promising candidates for selective inhibition of myosin-2 isoforms. However, recently developed molecules do not meet the safety criteria to enter preclinical studies due to their harmful properties of cytotoxicity and genotoxicity described in this paper. Despite these current limitations, our results provide useful and promising indications for further development of drug candidates targeting myosin’s blebbistatin-binding site. Through different para substitutions on the D-ring, we could either increase the inhibitory efficiency, improve skeletal muscle selectivity, modify biologic stability, or drastically reduce mutagenicity—properties that are necessary for the development of a potentially useful lead compound to advance to clinical trials for severe medical indications. Based on these observations we believe that it is feasible to develop clinically applicable drug compounds.
Acknowledgments
We would like to thank Ádám István Horváth, Csilla Kapocsi-Kurdi, Zsuzsa Kállay, Wanda Imrich, and László Végner for their technical assistance on ATPase and Ames assays. We woul also like to acknowledge Caitlyn Sibiskie for assistance in collecting data from Drosophila neuromuscular preparation.
Authorship Contributions
Participated in research design: Gyimesi, Rauscher, Monostory, Nag, Krans, Kovács, Kornya, Málnási-Csizmadia.
Conducted experiments: Gyimesi, Suthar, Oravecz, Lőrincz, Borhegyi, Déri, Kiss, Szabó, Nag, Tomasic, Krans, Tierney, Málnási-Csizmadia.
Performed data analysis: Gyimesi, Rauscher, Suthar, Hamow, Oravecz, Lőrincz, Déri, Kiss, Monostory, Szabó, Nag, Tomasic, Krans, Tierney, Kovács, Kornya, Málnási-Csizmadia.
Wrote or contributed to the writing of the manuscript: Gyimesi, Rauscher, Monostory, Nag, Krans, Kovács, Kornya, Málnási-Csizmadia.
Note Added in Proof: One of the Hungarian National Research, Development and Innovation Office grants was accidentally not included in the Fast Forward version published January 19, 2021. The funding footnote has now been corrected.
Footnotes
- Received June 15, 2020.
- Accepted October 7, 2020.
This study has been supported by the Hungarian National Research, Development and Innovation Office [NVKP 16-1-2016-0051 to M.G., and A.M.-C.] and the Hungarian Ministry of Finance [GINOP-2.1.7-15-2016-02580 to M.G.].
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AmBleb
- para-aminoblebbistatin
- AU
- arbitrary unit
- ClH
- hepatic clearance
- Clint
- intrinsic clearance
- CM
- cardiac muscle myosin
- EH
- hepatic extraction ratio
- HL3.1
- Hemolymph-like
- HPF
- hepatic plasma flow
- HPLC
- high-pressure liquid chromatography
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- NAT2
- N-acetyl transferase 2
- NBleb
- para-nitroblebbistatin
- NM2
- nonmuscle myosin-2 isoform
- QH
- hepatic blood flow
- RT
- retention time
- Copyright © 2021 The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}