


Abstract
Chronic pain is a public health problem because current treatments are unsatisfactory with small therapeutic index. Although pregabalin is effective for treating chronic pain, the clinical use is limited because of its side effects. Therefore, improving its therapeutic index is essential. In this study, HSK16149 was found to be a novel ligand of voltage-gated calcium channel (VGCC) α2δ subunit. HSK16149 inhibited [3H]gabapentin binding to the α2δ subunit and was 23 times more potent than pregabalin. In two rat models of neuropathic pain, the minimum effective dose (MED) of HSK16149 was 10 mg/kg, and the efficacy was similar to that of 30 mg/kg pregabalin. Moreover, the efficacy of HSK16149 could persist up to 24 hours postadministration at 30 mg/kg, whereas the efficacy of pregabalin lasted only for 12 hours at 30 mg/kg in streptozotocin-induced diabetic neuropathy model, indicating that HSK16149 might be a longer-acting drug candidate. HSK16149 could also inhibit mechanical allodynia in intermittent cold stress model and decrease phase II pain behaviors in formalin-induced nociception model. In addition, the locomotor activity test showed that the MED of HSK16149 was similar to that of pregabalin, whereas in the Rotarod test, the MEDs of HSK16149 and pregabalin were 100 and 30 mg/kg, respectively. These findings indicated that HSK16149 might have a better safety profile on the central nervous system. In summary, HSK16149 is a potent ligand of VGCC α2δ subunit with a better therapeutic index than pregabalin. Hence, it could be an effective and safe drug candidate for treating chronic pain.
SIGNIFICANCE STATEMENT As a novel potent ligand of voltage-gated calcium channel α2δ subunit, HSK16149 has the potential to be an effective and safe drug candidate for the treatment of chronic pain.
Introduction
Chronic pain is a long-term debilitating disease that affects normal work and daily life of patients (Tsuda et al., 2011; Vicuña et al., 2015; Tramullas et al., 2018) and is mainly categorized as inflammatory or neuropathic (Liu et al., 2008; Beggs, et al., 2012). However, the exact pathologic mechanisms underlying chronic pain remain to be unmasked, which impedes the development of new treatments for chronic pain (Li et al., 2011; Zhao et al., 2013). Presently, the common medication management for chronic pain consists of nonsteroidal anti-inflammatory drugs, tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, and opioids (Cohen et al., 2015). Unfortunately, a subset of patients is refractory to the currently available treatments with limited clinical applicability because of severe side effects (Wang et al., 2011). Moreover, opioids cannot be continually used because of tolerance and physical dependence. Therefore it is necessary to develop a new treatment of chronic pain.
Gabapentin and pregabalin belonging to gabapentinoids are selective ligands of voltage-gated calcium channel (VGCC) α2δ subunit (Boroujerdi et al., 2011) and were originally used for the treatment of epilepsy (Coderre et al., 2005; Kavoussi, 2006). At a later stage, the antinociceptive effect of these drugs was detected (Sills, 2006). Since approved, gabapentin and pregabalin have extremely improved the life quality of patients suffering from chronic pain, especially neuropathic pain. However, the use of these drugs is usually accompanied by some undesirable side effects, such as dizziness, somnolence, and peripheral edema. Hence, it is advisable to improve this class of drugs (i.e., retain or increase their efficacy and decrease the side effects).
In this study, HSK16149 bound to α2δ subunit with a high affinity in vitro. In addition, the in vivo assays also proved the potential analgesic effects of HSK16149 in neuropathic pain, fibromyalgia, and inflammatory pain. Furthermore, HSK16149 showed fewer effects on the central nervous system (CNS) in the Rotarod and the locomotor activity tests. Therefore, HSK16149 is a novel and potent ligand for the α2δ subunit of VGCCs with a better therapeutic index than pregabalin.
Materials and Methods
Animals.
Male Sprague-Dawley (SD) and Wistar rats weighing 160–180 g were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. Male C57/BL6 mice weighing 18–25 g and male ICR mice weighing 25–35 g were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. All animals were maintained on a standard 12-hour light/12-hour dark cycle with free access to food and water. Those who conducted pain assessment were blinded to the treatment conditions. All experimental procedures were performed in accordance with the guidelines of National Institutes of Health for the handling and use of laboratory animals and the Guidelines of the Institutional Animal Care and Use Committee of Haisco Pharmaceutical Group Co., Ltd. (China).
Drugs and Reagents.
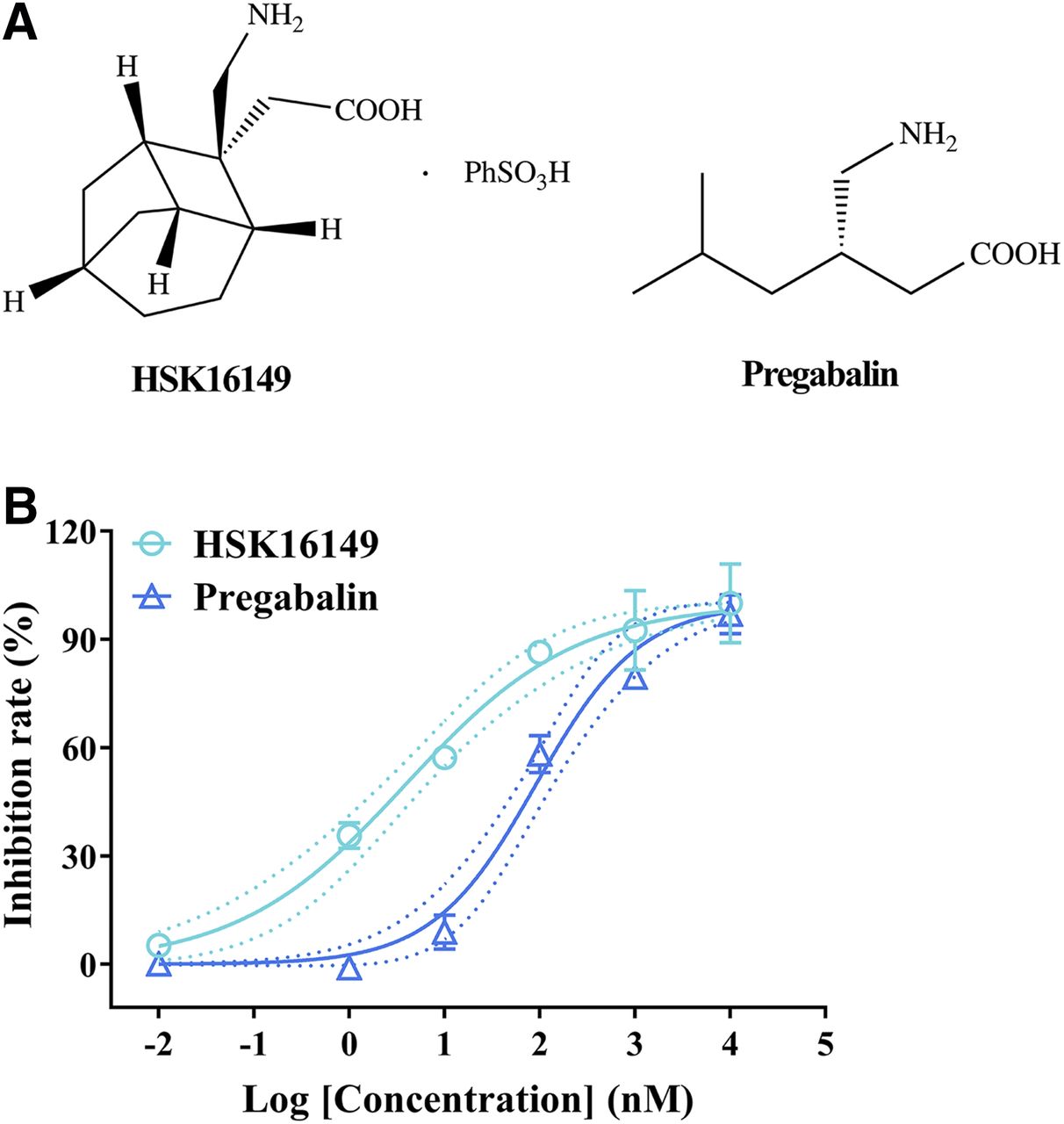
HSK16149 [2-((1S,2S,3R,6S,8S)-2-(aminomethyl)tricyclo[4.2.1.03,8]nonan-2-yl)acetic acid ben-zenesulfonic acid (1:1)] was synthesized in Haisco Pharmaceutical Group Co., Ltd., and the chemical structure of HSK16149 is shown in Fig. 1A. Pregabalin was obtained from Hunan Boheng Pharmaceutical Co., Ltd. HSK16149 and pregabalin were solubilized in DMSO for in vitro assays. In animal experiments, the two compounds were suspended in 0.5% carboxymethylcellulose sodium or methylcellulose and orally administered at a volume of 10 μl/g. Streptozotocin (STZ) was purchased from Chengdu Dingdang Pharmaceutical Co., Ltd. All reagents were of analytical grade unless otherwise stated.

Chemical structures (A) and in vitro potency (B) of HSK16149 and pregabalin. The in vitro potency was measured using [3H]gabapentin binding assay in cerebral cortical membranes of male Wistar rats. Data are expressed as mean ± S.D. of duplicate determinations. Dotted lines indicate 95% CI.
[3H]gabapentin Binding Assay.
Male Wistar rats were killed by decapitation, and craniotomy was conducted for each animal. The whole brain was collected, the meninges were peeled off, and the cortex was removed with forceps and tweezers. Fresh cerebral cortical membranes were homogenized in modified 10 mM HEPES buffer (pH 7.4) with Bertin Precellys Evolution. The pellet was collected by centrifugation of the homogenate at 12,000g for 20 minutes at 4°C. An equivalent of 0.02 mg of the membrane was incubated with 20 nM [3H]gabapentin in the presence of varying concentrations of test compounds for 30 minutes at 25°C. Bound and free fractions were separated by vacuum filtration through a GF/B filter pretreated with 0.3% polyetherimide. Then the filters were washed with ice-cold buffer. Bound radioactivity was determined using liquid scintillation counting (Suman-Chauhan et al., 1993; Gee et al., 1996). Nonspecific binding was defined in the presence of 100 μM gabapentin (Vincent et al., 2016). The percentage inhibition of [3H]gabapentin binding was calculated as follows: inhibition rate (%) = (CPMtotal-CPMcompound)/(CPMtotal-CPMnonspecific), in which CPMtotal = total [3H]gabapentin bound (membrane + 20 nM [3H]gabapentin) and CPMnonspecific = nonspecific [3H]gabapentin bound (membrane + 20 nM [3H]gabapentin + 100 μM gabapentin). The IC50 was determined by nonlinear, least-squares-regression analysis using GraphPad Prism 8.3.0 (San Diego, CA).
In Vitro Off-Target Pharmacological Profile.
The in vitro pharmacological activities of HSK16149 on 105 receptors, ion channels, transporters, and enzymes were evaluated based on radioligand binding and enzyme assays at Eurofins Panlabs Discovery Services Taiwan, Ltd. The assay services included SafetyScreen87 Panel (Item PP223) and 18 additional radioligand binding and enzyme assays (Domon et al., 2018).
Chronic Constriction Injury Model.
The chronic constriction injury (CCI) model of neuropathic pain was established on the left side. Briefly, under inhaled isoflurane anesthesia, the femoral skin was incised, and the sciatic nerve was exposed with a pair of forceps (Ghoreishi-Haack et al., 2018). A 2-mm-long polyethylene cuff was successively implanted around the nerve (Bailey and Ribeiro-da-Silva, 2006; Balasubramanyan et al., 2006). Then the incision was closed with a skin stapler, and the rats were returned to their cages after recovering from anesthesia. The pharmacological effects of mirogabalin were evaluated on day 17 post-CCI. On the test day, the 50% paw withdrawal threshold (PWT) was determined using Dixon’s up-down method before dosing (baseline value). The animals were grouped according to the baseline value and orally administered 0.5% carboxymethylcellulose sodium solution or mirogabalin at a volume of 10 μl/g. Subsequently, PWTs were measured at 2, 4, and 6 hours postdosing. The area under the curve (AUC) (50% PWT vs. time) was calculated using the trapezoid rule. The satellite groups were set (N = 4), and plasma was collected at the following time points: 0, 0.083, 0.25, 0.5, 1, 2, 6, 12, and 24 hours postdosing. The plasma concentrations of HSK16149 and pregabalin were detected using a validated liquid chromatography–tandem mass spectrometry method.
STZ-Induced Diabetic Neuropathy.
Male SD rats were acclimated to the laboratory for 5–7 days before study initiation, and the mechanical pain thresholds of rats were detected three times. Rats whose 50% PWT was not smaller than 15 g were intraperitoneally administered 70 mg/kg of STZ in 0.1 mol/l citrate buffer (pH 4.4) for three successive days. A total of 22 days after the first STZ injection, the levels of blood glucose were measured using a glucose meter, and the animals with fasted glucose levels >11.1 mmol/l were defined as diabetic rats. Subsequently, the pain thresholds of diabetic rats were measured and defined as baseline PWT values (predose). The animals were randomized based on the baseline and orally administered 0.5% carboxymethylcellulose sodium solution (vehicle control group) or test compounds (treatment groups) at a volume of 10 μl/g. Then the PWTs were measured at different time points. The AUC (50% PWT vs. time) was calculated by a trapezoid rule. The satellite groups were set (N = 4), and the plasma was collected at the following time points: 0, 0.083, 0.25, 0.5, 1, 2, 6, 12, and 24 hours postdosing. The plasma concentrations of HSK16149 and pregabalin were detected with a validated liquid chromatography–tandem mass spectrometry method.
Intermittent Cold Stress Model.
C57/BL6 mice were exposed to intermittent cold stress (ICS) as previously reported (Nishiyori and Ueda, 2008; Mukae et al., 2016). Firstly, the mice were kept in stainless cages at an overnight temperature of 4°C from 16:30 on day 0 to 10:00 on day 1, after which the animals were placed in an environment with room temperature (24 ± 2°C) and returned to the cold environment (4°C) after 30 minutes. The mice were then transferred between the room temperature and the cold environment every 30 minutes until 16:30 on day 1. The above procedures were repeated once from day 1 to day 2. Subsequently, the mice were kept again in the cold environment from 16:30 on day 2 to 10:00 on day 3 (Saeki et al., 2019). Finally, the mice were returned and adapted to the room temperature. The pain test (baseline value) was implemented on day 4, and mice with 50% PWT over 0.5 g were excluded. Finally, the animals were divided into different groups according to the baseline value and orally administered 0.5% methylcellulose solution or test compounds. The mechanical pain thresholds were detected at 2 hours post–drug administration.
Mechanical Pain Threshold Test.
Paw withdrawal responses to mechanical stimuli were measured using a set of von Frey filaments (Stoelting). Before each testing cycle, each animal was habituated to a Plexiglas chamber on a metallic mesh floor for a 30-minute to 1-hour period (Kawasaki et al., 2008; Sakai et al., 2013; Gazzo et al., 2019). For mice, a series of calibrated von Frey filaments in log increments of force (0.02–1.4 g) was applied to the plantar surface of the affected paws below the mesh floor. The paw withdrawal responses of rats were determined in the same manner but using von Frey filaments over a range of 1–15 g (Lai et al., 2006; Xie et al., 2017; Zhao et al., 2017). The 50% PWT was determined using Dixon’s up-down method (Chaplan et al., 1994; Weir et al., 2017).
Formalin-Induced Nociception.
After administration of the test compounds or vehicle (0.5% methylcellulose solution), ICR mice were subcutaneously injected with 15 μl of 2.5% formalin solution into the back of the right hind paw and placed into an automatic detector (Ponsati et al., 2012). Licking or biting of the injected paw (motion counts) was recorded as a nociceptive response, and the mice were observed from 0 to 9 minutes (neurogenic phase, phase I) and 10–45 minutes (inflammatory phase, phase II) post–formalin injection (Brittain et al., 2011; Shin et al., 2012).
Rotarod Test.
An accelerating Rotarod apparatus was used to measure the impact of the test compounds on motor coordination (Shiotsuki et al., 2010; Sakai et al., 2017; Slivicki et al., 2018). Rats were placed on a 6-cm-diameter rod that was accelerated to 15 rpm. The duration of staying on the rod was recorded for all the rats. Each rat was evaluated three times, and the average value was defined as the fall-off latency. Rats were trained for three successive days, and those with fall-off latency of >90 seconds (baseline value) on day 3 were moved on to the test session. On day 4, the rats were grouped according to the baseline value and orally administered 0.5% methylcellulose solution or test compounds. At 2 hours after dosing, the fall-off latency for each rat was recorded again. The maximum fall-off latency was set at 120 seconds.
Locomotor Activity Test.
The locomotor activity was tracked and analyzed using an ANY-maze video tracking system. After acclimatizing to the chamber for 2 days, the rats were administered either vehicle or test compounds and placed into the detection system at 2 hours postdosing. Each rat was then allowed to explore the field for 1 hour, and the total distance traveled was analyzed (Kraeuter et al., 2019).
Statistical Analysis.
All data were expressed as mean ± S.D., and no data were excluded. Shapiro-Wilk test was employed to assess whether the data followed a normal distribution. All data were analyzed by two-tailed, unpaired t test; unpaired Mann-Whitney U test; one-way ANOVA with Dunnett’s comparisons; Kruskal-Wallis test with Dunn’s comparisons; or two-way ANOVA with Dunnett’s comparisons. All statistical tests were performed using GraphPad Prism 8.3.0 (San Diego, CA). P < 0.05 indicated statistical significance.
Results
Binding Affinity to VGCC α2δ Subunit.
The binding affinity of the compound to VGCC α2δ subunit was assayed by a competitive [3H]gabapentin binding assay. As shown in Fig. 1B, both HSK16149 and pregabalin could prevent [3H]gabapentin from binding in a dose-dependent manner. The IC50 of HSK16149 and pregabalin were 3.96 nM (95% CI: 2.28–6.63 nM) and 92.12 nM (95% CI: 61.40–141.2 nM), respectively. Therefore, HSK16149 is a potent ligand of VGCC α2δ subunit, which exhibited stronger pharmacological activity than pregabalin.
In Vitro Off-Target Pharmacological Profile.
To assess the in vitro target selectivity of HSK16149, its pharmacological activities on 105 targets were evaluated, and an inhibition or stimulation rate of >50% was considered as a significant response. Surprisingly, HSK16149 had no significant effects on any of the targets at the concentration of 10 μM (Supplemental Table 1). Therefore, it can be concluded that HSK16149 is selective ligand for the α2δ subunit of VGCCs.
Analgesic Effects in CCI-Induced Neuropathic Pain Model.
As one of the most commonly used neuropathic pain models, the CCI model was employed to evaluate the analgesic effects of HSK16149 and pregabalin. Seventeen days after the surgery, HSK16149 and pregabalin were orally administered, and the effects on the mechanical pain threshold were determined after dosing. The data showed that both HSK16149 and pregabalin could increase the PWT in a dose-dependent manner (Fig. 2). In the 30 mg/kg HSK16149 group, the 50% PWT value was 3.24-fold larger than that in the vehicle-treated group (P < 0.001) at 2 hours and peaked at 6 hours after dosing (Fig. 2A). The effects of 10 mg/kg HSK16149 on mechanical pain thresholds were similar to the 30 mg/kg HSK16149–treated group at 2 hours (12.93 vs. 11.93 g) or 4 hours (13.66 vs. 13.55 g). Although the 50% PWT value in 10 mg/kg HSK16149–treated group was lower than that in the 30 mg/kg HSK16149–treated group (P = 0.004, t test) at 6 hours, the effect of 10 mg/kg HSK16149 was similar to that of the 30 mg/kg pregabalin–treated group (P = 0.29, Mann-Whitney test). In addition, compared with the vehicle-treated group, 3 mg/kg of HSK16149 induced a significant increase of 50% PWT value at 4 hours (7.33 vs. 3.11 g, P = 0.008). Typically, the pharmacological activities were comparable with each other in 10 or 30 mg/kg HSK16149– or 30 mg/kg pregabalin–treated groups (Fig. 2D). On the other hand, the effect of 10 mg/kg pregabalin was lower than that of 30 mg/kg pregabalin (43.90 vs. 64.82, P = 0.0012; Fig. 2E). When administered at the maximal dose (30 mg/kg), HSK16149 and pregabalin exhibited significant effects on the 50% PWT values with a 2-fold increase at 8 hours postdosing. To calculate the therapeutic index, the minimum effective dose (MED) was obtained by defining the lowest dose that markedly increased the AUC (50% PWT vs. time). The MED was 10 mg/kg for both HSK16149 and pregbalin. Considering the weaker activity of 10 mg/kg pregabalin (vs. 30 mg/kg pregabalin) and the comparability of the AUC value between 10 mg/kg HSK16149 and 30 mg/kg pregabalin, we speculated that HSK16149 might provide better improvement for patients suffering from neuropathic pain.

Analgesic effects of HSK16149 and pregabalin in CCI model. (A–C) Time course for the effects of HSK16149 and pregabalin on neuropathic pain in rats. HSK16149 and pregabalin were orally administered, and the mechanical thresholds were evaluated at different time points after dosing. The AUCs (50% PWT vs. time) were calculated with a trapezoid rule and are shown in (D–F). Data are expressed as mean ± S.D. (n = 10/group). *P < 0.05; **P < 0.01; ***P < 0.001 vs. vehicle, two-way ANOVA with Dunnett’s multiple comparisons (A–C); *P < 0.05; ***P < 0.001 vs. vehicle, Kruskal-Wallis test with Dunn’s multiple comparisons (D and E); *P < 0.05 vs. vehicle, one-way ANOVA with Dunnett’s multiple comparisons (F); #P < 0.05; ##P < 0.01 vs. 10 mg/kg pregabalin, two-tailed, unpaired t test (E); $P < 0.05 vs. 30 mg/kg HSK16149, two-tailed, unpaired t test (F).
The pharmacokinetic parameters of HSK16149 and pregabalin are shown in Table 1. The exposure levels (Cmax and AUC0–24 h) in the plasma increased in a dose-proportional manner, and the AUC0–24 h of HSK16149 was 2.25–4.73 times lower than that of pregabalin.
Pharmacokinetic parameters of HSK16149 and pregabalin in two rat models of neuropathic pain
Analgesic Effects in STZ-Induced Diabetic Neuropathy Model.
The analgesic effects produced by HSK16149 and pregabalin were tested in STZ-induced diabetic neuropathy model. On the day of testing, different doses of HSK16149 and pregabalin were orally administered to SD rats. As shown in Fig. 3, both HSK16149 and pregabalin increased the PWT in a dose-dependent manner. Compared with the vehicle group, HSK16149 at 30 mg/kg obviously increased the 50% PWT value at 2 hours postdosing (10.35 vs. 3.91 g, P = 0.021), and the efficacy could persist up to 6 hours postdosing with a slight increase. Although no statistical significance was detected, the 50% PWT value in the 10 mg/kg HSK16149 group was 2.4 times larger than that of the vehicle group at 2 hours postdosing (P = 0.051), and HSK16149 at 10 mg/kg could significantly increase the 50% PWT value at 4 hours (10.55 vs. 4.35 g, P = 0.016) or 6 hours (11.74 vs. 4.24 g, P = 0.0026) after dosing. In the 3 mg/kg HSK16149 group, the PWT value was approximately doubled at 6 hours postdose (vs. vehicle group; Fig. 3A). The MED was defined as the lowest dose level that statistically increased the AUC (50% PWT vs. time). The MED of HSK16149 was 10 mg/kg (P = 0.016 vs. vehicle), and no statistically significant difference was detected in the efficacy between 10 and 30 mg/kg, which was comparable to that of pregabalin at 30 mg/kg (Fig. 3D). On the other hand, the MED of pregabalin was also determined to be 10 mg/kg (P = 0.0035 vs. vehicle). However, the efficacy of 10 mg/kg pregabalin was remarkably inferior to 30 mg/kg pregabalin (P = 0.0008, Fig. 3, B and E). In addition, the efficacy of a single dose of HSK16149 could persist up to 8 hours at 10 mg/kg and 24 hours at 30 mg/kg. In comparison, the analgesic effect of pregabalin was lost at 8 hours for 10 mg/kg and at 24 hours for 30 mg/kg after drug administration (Fig. 3C). The data showed that HSK16149 might have a strong potency that was longer-acting than pregabalin.

Analgesic effects of HSK16149 and pregabalin in STZ-induced diabetic neuropathy. (A–C) Time course for the effects of HSK16149 and pregabalin on neuropathic pain in rats. HSK16149 and pregabalin were orally administered, and the mechanical thresholds were evaluated at different time points after dosing. The AUCs (50% PWT vs. time) were calculated with trapezoid rule and are shown in (D–F). Data are expressed as mean ± S.D. (n = 10/group). *P < 0.05; **P < 0.01; ***P < 0.001 vs. vehicle, two-way ANOVA with Dunnett’s multiple comparisons (A–C); **P < 0.01; ***P < 0.001 vs. vehicle, Kruskal-Wallis test with Dunn’s multiple comparisons (D–F); #P < 0.05; ###P < 0.001 vs. 10 mg/kg pregabalin, two-tailed, unpaired t test (E); $$$P < 0.001 vs. 30 mg/kg HSK16149, &&P < 0.01 vs. 30 mg/kg pregabalin, two-tailed, unpaired t test (F).
The pharmacokinetic parameters of HSK16149 and pregabalin are shown in Table 1. The data are similar to those in the CCI-induced neuropathic pain model. The exposure levels (Cmax and AUC0–24 h) in plasma increased in a dose-dependent manner, and the AUC0–24 h of HSK16149 was 2.67–4.33 times lower than that of pregabalin.
Antinociceptive Effects in a Mouse Model of Fibromyalgia.
The antinociceptive effects of HSK16149 and pregabalin in fibromyalgia were evaluated using an ICS model, which is one of the well established mouse models of fibromyalgia. On the day of the test, different doses of HSK16149 and 30 mg/kg pregabalin were orally administered to the mice at 2 hours pretesting. As shown in Fig. 4A, intermittent cold stress exposure dramatically induced hypersensitivity to mechanical stimulation in mice with an obvious decrease in 50% PWT (from 1.22 to 0.32 g). Furthermore, HSK16149 inhibited mechanical allodynia in a dose-dependent manner. The MED of HSK16149 was 30 mg/kg in this assay (P = 0.0005 vs. vehicle). Compared with the vehicle group, HSK16149 at 30 mg/kg induced a 2.6-fold increase in 50% PWT, and the efficacy was comparable with 30 mg/kg pregabalin (Fig. 4B).

Antinociceptive effects of HSK16149 and pregabalin in ICS-induced fibromyalgia. HSK16149 and pregabalin were intragastrically administered at 2 hours before the pain test was performed. Data are expressed as mean ± S.D. (n = 10/group). ***P < 0.001 vs. naïve, Kruskal-Wallis test with Dunn’s multiple comparisons (A); ***P < 0.001 vs. vehicle, ###P < 0.001 vs. naïve, Kruskal-Wallis test with Dunn’s multiple comparisons (B).
Effects on Pain Behaviors in an Inflammatory Pain Model.
A formalin-induced pain model was used to assess the pharmacological effects of HSK16149 and pregabalin. In this model, different doses of HSK16149 and 30 mg/kg pregabalin were administered at 2 hours pretesting after formalin injection into the mice’s hind paws, causing a two-phase pattern of behavior responses (phase I and phase II). Consequently, we observed that HSK16149 decreased formalin-induced pain behaviors in phase II in a dose-dependent manner. Both 10 and 30 mg/kg HSK16149 significantly decreased formalin-induced pain behaviors in phase II with a 1.6- and 2.2-fold decrease in motion counts, respectively (P < 0.0001 vs. vehicle). The effect of 30 mg/kg HSK16149 was similar to that of pregabalin at the same dose (P = 0.56, Fig. 5B). Both HSK16149 and pregabalin had slight effects on formalin-induced pain behaviors in phase I at a dose of 30 mg/kg, although not significantly (Fig. 5A).

Effects on pain behavior of HSK16149 and pregabalin in formalin-induced inflammatory pain, Phase I (A) and Phase II (B).HSK16149 and pregabalin were intragastrically administered at 2 hours before formalin was injected. Data are expressed as mean ± S.D. (n = 10/group). ***P < 0.001 vs. vehicle, one-way ANOVA with Dunnett’s multiple comparisons.
Effects on Central Nervous System.
Reportedly, VGCC inhibition is related to the side effects on CNS, such as ataxia and sedation. A Rotarod test was used to test the ataxic effects of HSK16149 and pregabalin. In this assay, different doses of HSK16149 and pregabalin were orally administered to SD rats at 2 hours before the test. At a dose of 100 mg/kg, both HSK16149 and pregabalin produced ataxic side effects with a 1.8- and 3.6-fold decrease in the fall-off latency, respectively, and HSK16149 had a milder effect on ataxia than pregabalin (P = 0.012). Notably, pregabalin at 30 mg/kg attenuated the ability of rats to maintain their position on an accelerating Rotarod (P = 0.043 vs. vehicle, a 1.4-fold decrease). However, HSK16149 at 30 mg/kg did not show any ataxic effect (P = 0.28 vs. vehicle, Fig. 6A).

Ataxic (A) and sedative (B) side effects of HSK16149 and pregabalin. The tests were performed at 2 hours postadministration. Data are expressed as mean ± S.D. (n = 10/group). *P < 0.05; **P < 0.01; ***P < 0.001 vs. vehicle, Kruskal-Wallis test with Dunn’s multiple comparisons, #P < 0.05 vs. 100 mg/kg HSK16149, two-tailed, unpaired Mann-Whitney U test (A); ***P < 0.001 vs. vehicle, one-way ANOVA with Dunnett’s multiple comparisons (B).
The sedative effects of HSK16149 and pregabalin at 2 hours postdosing were evaluated by a locomotor activity test. As shown in Fig. 6B, both HSK16149 and pregabalin reduced the total distance traveled by the rats within 1 hour in the chambers in a dose-dependent manner. The MED was 100 mg/kg for both HSK16149 and pregabalin (P < 0.0001 vs. vehicle, a 3-fold decrease), and the effect of HSK16149 on sedation was the same as that of pregabalin at the same dose.
Discussion
Pregabalin was approved for treating neuropathic pain in 2004 and has significantly improved the life quality of patients who suffered from chronic pain. However, undesirable side effects, such as dizziness, somnolence, and peripheral edema, are exposed in treatment (Calandre et al., 2016). Therefore, developing new treatments for chronic pain with improved efficacy and fewer side effects is a great challenge. This study aimed to find a novel potent VGCC α2δ subunit ligand with better efficacy and safety profiles.
The in vitro binding studies showed that both HSK16149 and pregabalin showed a high affinity to the α2δ subunit of VGCCs. HSK16149 was more potent (23-fold) than pregabalin. In addition, HSK16149 showed no activities on 105 other targets, indicating a good safety profile.
Notably, the in vitro pharmacological activities are not completely in accordance with the in vivo pharmacological effects in many cases, and binding to the α2δ subunit of VGCC might not correlate with in vivo analgesic efficacy (Lynch et al., 2006). Therefore, the pharmacological effects of HSK16149 were evaluated in four animal models, including CCI-induced neuropathic pain, STZ-induced diabetic neuropathy, ICS-induced FM-like pain, and formalin-induced nociception. In all these models, HSK16149 exhibited analgesic efficacy in a dose-dependent manner, and the pharmacological activity of HSK16149 was similar to or slightly more potent than that of pregabalin at the same dose. On the other hand, the exposure level of HSK16149 in plasma was markedly lower than that of pregabalin at the same dose. When administered at 30 mg/kg, the exposure level of HSK16149 (AUC0–6 h) in the brain tissue was 18-fold lower than that of pregabalin (Supplemental Table 2). These findings indicated that HSK16149 might have fewer side effects at a dose at which the pharmacological activities between HSK16149 and pregabalin are equipotent.
As previously reported, the major side effects of pregabalin were its effects on CNS. Therefore, the Rotarod test and locomotor activity test were performed. The results showed that the ataxic side effects of HSK16149 were weaker than those of pregabalin, and the sedative side effects of HSK16149 were similar to those of pregabalin. The in vivo pharmacological and side-effect profiles of HSK16149 and pregabalin are summarized in Table 2. The therapeutic indexes were obtained by calculating the ratio of the MEDs producing side effects in the Rotarod test or locomotor activity test and the MED in the neuropathic pain model. Strikingly, the safety profiles of HSK16149 were better than those of pregabalin.
Therapeutic index of HSK16149 and pregabalin
Recently, mirogabalin was approved in Japan as another ligand of VGCC α2δ subunit. The efficacy and safety profile of mirogabalin was also tested in our laboratory. The IC50 of mirogabalin was 6.24 nM (95% CI: 4.47–8.64 nM) to inhibit [3H]gabapentin binding to VGCC α2δ subunit (Supplemental Fig. 1), which was comparable with that of HSK16149. The in vivo efficacy of mirogabalin was evaluated in CCI hyperalgesia model in rats. The MED of mirogabalin was 10 mg/kg in this model (Supplemental Fig. 2). The effects of mirogabalin on CNS were also tested in the Rotarod and locomotor activity tests. The MEDs of mirogabalin were 10 and 25 mg/kg, respectively (Supplemental Fig. 3). The therapeutic indexes were obtained by calculating the ratio of the MEDs producing side effects in the Rotarod test or the locomotor activity test and the MED in the CCI model, thereby indicating that the therapeutic index of HSK16149 is larger than that of mirogabalin (10-fold or 4-fold, Table 2).
Although it is usual that some candidates with good preclinical data show little or no benefits for patients in clinical trials, we await the results from phase II clinical trials of HSK16149. The subsequent data might prove that HSK16149 is a more potent and safer analgesic drug.
In conclusion, HSK16149 showed promising efficacy in different in vitro assays and in vivo pain models with fewer CNS side effects. Currently, HSK16149 is under phase II/III clinical trial in China (CTR20202015, https://www.wuxuwang.com/linchuang/49a9d19a-0c7c-11eb-a061-00,163e 0eafb3). The data strongly support continued clinical trials in various chronic pain conditions.
Acknowledgments
The authors thank Eurofins Panlabs Discovery Services Taiwan Ltd. and Wuxi Apptec Co., Ltd. for their assistance with the experiments.
Authorship Contributions
Participated in research design: Gou, Ye, Liang, Ni.
Conducted experiments: Gou, Yu, Bai, Tan, Cao, Qian, Zheng.
Contributed new reagents or analytic tools: Chen, Shi, Li.
Performed data analysis: Gou, Ye, Ni.
Wrote or contributed to the writing of the manuscript: Gou, Li, Ye, Ni.
Footnotes
- Received August 31, 2020.
- Accepted December 4, 2020.
This work received no external funding.
All authors were employees of Haisco Pharmaceutical Group when the study was conducted.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- CCI
- chronic constriction injury
- CI
- confidence interval
- CNS
- central nervous system
- ICS
- intermittent cold stress
- MED
- minimum effective dose
- PWT
- paw withdrawal threshold
- SD
- Sprague-Dawley
- STZ
- streptozotocin
- VGCC
- voltage-gated calcium channel
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}