


Abstract
Multiple clinical and preclinical studies have demonstrated that plasma levels of asymmetric dimethylarginine (ADMA) are strongly associated with hypertension, diabetes, and cardiovascular and renal disease. Genetic studies in rodents have provided evidence that ADMA metabolizing dimethylarginine dimethylaminohydrolase (DDAH)-1 plays a role in hypertension and cardiovascular disease. However, it remains to be established whether ADMA is a bystander, biomarker, or sufficient contributor to the pathogenesis of hypertension and cardiovascular and renal disease. The goal of the present investigation was to develop a pharmacological molecule to specifically lower ADMA and determine the physiologic consequences of ADMA lowering in animal models. Further, we sought to determine whether ADMA lowering will produce therapeutic benefits in vascular disease in which high ADMA levels are produced. A novel long-acting recombinant DDAH (M-DDAH) was produced by post-translational modification, which effectively lowered ADMA in vitro and in vivo. Treatment with M-DDAH improved endothelial function as measured by increase in cGMP and in vitro angiogenesis. In a rat model of hypertension, M-DDAH significantly reduced blood pressure (vehicle: 187 ± 19 mm Hg vs. M-DDAH: 157 ± 23 mm Hg; P < 0.05). Similarly, in a rat model of ischemia-reperfusion injury, M-DDAH significantly improved renal function as measured by reduction in serum creatinine (vehicle: 3.14 ± 0.74 mg/dl vs. M-DDAH: 1.1 ± 0.75 mg/dl; P < 0.01), inflammation, and injured tubules (vehicle: 73.1 ± 11.1% vs. M-DDAH: 22.1 ± 18.4%; P < 0.001). These pharmacological studies have provided direct evidence for a pathologic role of ADMA and the therapeutic benefits of ADMA lowering in preclinical models of endothelial dysfunction, hypertension, and ischemia-reperfusion injury.
SIGNIFICANCE STATEMENT High levels of ADMA occur in patients with cardiovascular and renal disease. A novel modified dimethylarginine dimethylaminohydrolase by PEGylation effectively lowers ADMA, improves endothelial function, reduces blood pressure and protects from ischemia-reperfusion renal injury.
Introduction
Asymmetric dimethylarginine (ADMA), also known as a cardiotoxin, exhibits a strong association with cardiovascular and renal disease. ADMA levels correlate with increased cardiovascular morbidity and mortality (Böger et al., 2005). High plasma concentrations of ADMA are found in a number of disease states involving vascular dysfunction, including coronary artery disease (Krempl et al., 2005), hypertension (Goonasekera et al., 1997; Perticone et al., 2005), heart failure (Zoccali et al., 2001), peripheral arterial disease (Böger et al., 1997), chronic kidney disease (Ravani et al., 2005), and pregnancy-related hypertension (Pettersson et al., 1998). In diabetic hypertensive patients, high ADMA was associated with chronic kidney disease progression (Triches et al., 2018). In large prospective studies, plasma ADMA was independently associated with cardiovascular mortality in patients with ischemic heart disease (Meinitzer et al., 2007) and increased incidence of myocardial infarction and stroke in women (Leong et al., 2008). These studies have suggested that ADMA is a biomarker and perhaps a risk factor contributing to microvascular dysfunction and cardiovascular disease.
Initially identified as an endogenous inhibitor of nitric oxide synthases (Leiper and Nandi, 2011; Schwedhelm and Boger, 2011), ADMA also inhibits arginine transport and increases oxygen free radicals, mitochondrial dysfunction, and transforming growth factor-β expression (Druhan et al., 2008; Fabrice et al., 2011; Sun et al., 2013). Further, nitric oxide (NO)-independent cellular actions of ADMA have been identified (Jarzebska et al., 2019). The major pathway for ADMA metabolism is by dimethylarginine dimethylaminohydrolase (DDAH) enzymes (Leiper and Nandi, 2011; Schwedhelm and Boger, 2011). Two isoforms of DDAH (DDAH-1 and DDAH-2), encoded by distinct genes, exist (Leiper and Nandi, 2011). In preclinical models of cardiac or renal ischemia-reperfusion, DDAH-1 is reduced with a concomitant increase in tissue ADMA (Stühlinger et al., 2007; Nakayama et al., 2014). DDAH gene knockout and DDAH overexpression, either in transgenic mice or by adenoviral gene delivery in rodents, have provided evidence of a pathologic role of ADMA (Leiper et al., 2007; Xu et al., 2017). Although, these DDAH gene knockout and overexpression studies have produced strong evidence for the pathologic role of ADMA in cardiovascular and renal disease, the results of other genetic studies have shown contradictory phenotypes (Nakayama et al., 2014; Tomlinson et al., 2015; Rodionov et al., 2019; Wetzel et al., 2020). Thus, modification of pathologic state by pharmacological lowering of ADMA would provide direct evidence for its role in cardiovascular and renal disease. To date, specific lowering of ADMA by pharmacological treatment has not been achieved. Previous attempts to discover small-molecule modulators of DDAH activity have not been successful (Ghebremariam et al., 2012; Linsky and Fast, 2012). Thus, at the present time, pharmacological agents that can specifically lower pathologic ADMA are not available. The goal of the present study was to develop a pharmacological molecule to specifically lower ADMA and test the hypothesis that ADMA lowering would reduce pathologic state. Here, we show for the first time that a novel M-DDAH with drug-like pharmacological properties effectively reduced ADMA in preclinical models. ADMA lowering improved endothelial function ex vivo and significantly reduced blood pressure in hypertensive rats. In a rat model of ischemia-reperfusion injury, M-DDAH protected from renal injury. These data show that pharmacological lowering of ADMA may provide a new mechanism for targeting disease states associated with high ADMA, including cardiovascular disease, heart failure, vascular complications of diabetes, and kidney disease.
Methods
Cloning, Expression, and Purification of DDAH and Mutants.
Pseudomonas aeruginosa (PA)-DDAH, PA-DDAH mutants, and human DDAH-1 were cloned and expressed in Escherichia coli. Coding sequence of DNA was synthesized and ligated into a pE-SUMO vector. PA-DDAH mutants were prepared by individually replacing lysine residues 3, 25, 103, and 159 with cysteine. Plasmid containing DDAH or mutant genes was transformed into BL21 (DE3) E. coli cells (EMD Millipore), plated on LB agar plate with 50 μg/ml of kanamycin, and grown overnight. The colonies expressing highest levels of DDAH were then used for large-scale production. For preparation of purified DDAH, 50 ml culture was inoculated in 1 L of LB with 50 μg/ml kanamycin and grown to 0.8 OD at 600 nm and then induced with isopropyl β-D-1-thiogalactopyranoside (IPTG). After 16 hours, cells were collected by centrifugation, resuspended, and lysed by sonication. DDAH in the supernatant was then purified using a Ni-Sepharose column. DDAH activity was determined by generation of l-citrulline from ADMA using a colorimetric assay (Knipp and Vasak, 2000).
Site-Specific PEGylation of DDAH Mutant and Generation of M-DDAH.
The 159Lys-Cys mutant cloned and expressed as described above was selected for site-specific PEGylation. The 159Lys-Cys DDAH protein was dialyzed against 0.1 M phosphate buffer (pH 6.3). mPEG40k-maleimide (Nanocs) (10× molar excess) was added to the dialyzed protein and incubated for 4 hours at room temperature. The PEGylated protein was purified using HiTrap Q FF anion exchange chromatography (Sigma-Aldrich). The column was equilibrated with 20 mM Tris (pH 8). PEGylation reaction mixture was applied to the column. The PEGylated protein was eluted in 175 mM NaCl. The PEGylated protein was evaluated for purity, molecular weight, and DDAH activity.
For the generation of M-DDAH, PA-DDAH was PEGylated at the lysine residues using mPEG10K-NHS (Nanocs). PEGylation was initiated after extensive dialysis of DDAH against 0.1 M phosphate buffer (pH 8) to remove amine-containing substances. mPEG10K-NHS (Nanocs) (20× molar excess) was then added and incubated for 4 hours at room temperature. The reaction mixture was purified using 50-kDa Amicon Ultra centrifugal filter unit (EMD Millipore) and centrifugation at 4500 rpm for 15 minutes at 4°C. Excess and unreacted mPEG10K-NHS was removed by three cycles of purification using the 50-kDa Amicon filter. SDS gel electrophoresis showed multiple bands of PEGylated DDAH after one cycle of PEGylation. To achieve maximum PEGylation and homogenous preparation, the partially PEGylated protein was subjected to additional cycles of PEGylation as described above until the protein was fully PEGylated. The optimized PEGylation reaction protocol achieved PEGylation of all accessible lysine residues, yielding a protein band at 250 kDa in SDS gels. The purified M-DDAH was then evaluated for molecular weight and DDAH activity.
ADMA Measurement.
ADMA in plasma was analyzed by modification of a previously published HPLC-based method (de Jong and Teerlink, 2006; Teerlink, 2007). Test samples were prepared by solid-phase extraction using an Oasis MCX Cartridge (Waters). Briefly, the column was conditioned with 1 ml of elution buffer consisting of 30% ammonium hydroxide:water:methanol (10:40:50 by volume), followed by 1 ml of ultrapure water. Plasma (50 μl) was mixed, vortexed, and spun with cold methanol (100 μl) for extraction, and 100 μl was recovered, mixed with 200 μl of PBS, and loaded onto the column. The column was lightly centrifuged and rinsed with 1 ml of 0.1 M HCl and 1 ml of methanol two times. Analytes were eluted with 400 μl elution buffer and dried under nitrogen. Samples were reconstituted in 50 μl of ultrapure water and derivatized with ortho-phthaldialdehyde reagent (Sigma-Aldrich) (4:1 by volume). After mixing, the analytes were heated at 30°C for 1 minute, and 10 μl was injected into the HPLC system, equipped with a fluorescent detector RF-10AXL (Excitation wavelength: 340 nm, Emission wavelength: 455 nm) and Chromolith Performance RP-18e column (100 × 4.6 mm) (EMD Millipore). Mobile phase A consisted of 25 mM potassium phosphate buffer (pH 6.5), and mobile phase B was methanol/tetrahydrofuran (97:3 by volume). Chromatographic separation was performed at room temperature at a flow rate of 2 ml/min (10%–20% solvent A).
cGMP Measurement.
The effect of ADMA and ADMA + M-DDAH on NO generation in endothelial cells was determined using cGMP, the downstream marker of NO responses. HUVECs (1,000,000 per well) in a six-well plate were incubated in medium containing 0.5 mM IBMX (control) or IBMX + 100 μM ADMA for 5 minutes at 37°C. The medium was aspirated, and cells were incubated in fresh medium containing 0.5 mM IBMX or 0.5 mM IBMX + 10 μg/ml M-DDAH for 60 minutes. Cells were collected using trypsinization, and cGMP was analyzed using enzyme immunoassay kit (Arbor Assay).
In Vitro Angiogenesis.
Matrigel-coated plates were prepared by placing 200 μl Matrigel (Corning) into the 24-well plate using a method previously described (DeCicco-Skinner et al., 2014). The plates were incubated at 37°C for 45 minutes. HUVECs (60,000 cells per well) in 0.5% FBS medium or experimental groups were added and incubated at 37°C for 6 hours. The cells were imaged using 4× objective and were analyzed using ImageJ (Carpentier et al., 2012).
Animals.
All animal studies were approved by local institutional animal care and use committee (IUCAC) at Indiana University and the University of Mississippi. Sprague-Dawley (SD) rats (Envigo, Indianapolis) were maintained on normal chow (TD7034, 0.3% NaCl; Harlan Teklad) and water ad libitum on a 12-hour light/dark cycle. Dahl salt-sensitive hypertensive female rats were obtained from Dr. Michael Garrett at the University of Mississippi Medical Center (Rapp and Dene, 1985). Rats were euthanized in accordance with guidelines of the American Veterinary Medical Association.
Pharmacodynamic Studies.
Male SD rats (∼250–300 g) were catheterized in the jugular vein to collect blood. DDAH or M-DDAH (2 mg/kg) was administered by a single intravenous injection. Blood was drawn at various times, and plasma was prepared by centrifugation. DDAH activity was assayed by modification of a previously published method (Tain and Baylis, 2007). Plasma (70 μl) was incubated with 20 μl urease for 15 minutes before adding ADMA (10 μM). The mixture was incubated for 3 hours at 37°C. Samples were deproteinated by addition of 50 μl of 8% sulfosalicylic acid and centrifuged. Supernatant (80 μl) was used for determination of citrulline.
Blood Pressure Measurement.
Dahl salt-sensitive female rats were anesthetized using isoflurane (5% induction, 2% maintenance) and carprofen. Carotid artery and jugular vein catheters were inserted 1 day before the measurements (Younes et al., 2020). M-DDAH (1 mg/kg) or PBS vehicle was infused intravenously to conscious restrained rats. An exteriorized arterial and venous catheter for arterial pressure measurement and arterial blood sampling and venous infusions was used for these acute studies. The rats were placed in restrainers, and the measurements were taken after the rats were acclimated and quietly resting in the restrainer. The carotid catheter was connected to a Deltran pressure transducer (Utah Medical), and mean arterial pressure in control and treated groups was measured over 4 hours. Data were collected using an amplifier and PowerLab from ADInstruments.
Renal Ischemia-Reperfusion Model.
Male SD rats (∼250–300 g) were anesthetized with ketamine (100 g/kg) and xylazine (5 mg/kg) and subjected to bilateral ischemia-reperfusion injury by clamping the renal pedicle for 30 minutes, similar to procedures previously described (Phillips et al., 2010). M-DDAH or vehicle was first administered by intravenous injection at the time of reperfusion and 3 hours after the first administration. Renal function was determined by measuring plasma creatinine using a Pointe Scientific Analyzer. Blood pH and analytes were measured with a GEM Premier 4000 (Instrumentation Laboratories). Animals were euthanized with Fatal Plus, and kidneys were removed and processed for immune cells and histology.
Immune Cells Analysis.
Harvested kidneys from the ischemia-reperfusion study were minced and digested in Liberase (2 μg/ml) (Roche, Switzerland) for 15 minutes at 37°C with the help of gentleMACS (Miltenyli, Germany). The digested tissue was filtered through a 100-μm filter mesh and washed with tissue culture medium. The lymphocytes were separated by Percoll (Sigma-Aldrich) and counted by hemocytometer. To evaluate T lymphocytes, the cells were stained with antibodies against rat CD4 (Phycoerythrin-Cyanine7) and CD8a (Alexa 647). To evaluate the cytokines secreted by T cells, the cells were stained for CD4 surface marker and then permeabilized and stained with antibodies against rat Interleukin-17 (Fluorescein isothiocyanate). Macrophages were stained using anti-CD11b/c (Phycoerythrin). All antibodies were obtained from BD Biosciences. Cells were scanned using flow cytometry (FACSCalibur; BD Biosciences), and scans were analyzed using FlowJo software (Tree Star). Representative gating for cell populations identified by flow cytometry was exactly as described previously (Mehrotra et al., 2015). The data are expressed as the presence of total number of a specific cell population per gram of kidney tissue.
Histologic Studies.
One half of the kidney from the ischemia-reperfusion study was fixed in 10% formalin and embedded in paraffin, and 5-μm sections were prepared. Sections were stained with hematoxylin and eosin and imaged with a Nikon Optiphot-2 microscope using a 20× objective, equipped with a Leica DMC 2900 camera using Lieca LAS software. At least three random images through the renal outer medulla were obtained from each animal. Sections were assessed for tubular injury as described previously (Basile et al., 2013). The degree of injury is expressed as the percentage of damaged tubules that display cellular necrosis, loss of brush border, cast formation, and tubular dilation. Both the individual who acquired the images and the individual who scored the slides were blinded to the treatment groups.
Statistical Analysis.
Statistical significance was evaluated by Student’s t test or one-way ANOVA, followed by Dunnett’s test for multiple comparison between the experimental groups. All data are presented as means ± S.D., and P values of <0.05 were considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Cloning, Expression, and Purification of Recombinant DDAH.
The translated DNA sequences of human and PA-DDAH genes were synthesized and cloned in pE-SUMO plasmid and then expressed in E. coli. To facilitate purification, DDAH was expressed as a protein containing His-tag at the C terminus. Recombinant protein was purified using the Ni-Sepharose column. Figure 1A shows that the two-step Ni-Sepharose purification produced a single PA-DDAH band with greater than 90% purity when analyzed by SDS gel electrophoresis. The DDAH activity was determined by hydrolysis of ADMA to citrulline using a colorimetric assay. Previous studies have reported discrepant values of the Michaelis-Menten constant (Km) of PA-DDAH using different assay systems. Stone et al. (2005) reported a Km of 310 ± 20 μM for recombinant PA-DDAH using a less sensitive colorimetric assay, whereas a Km of 39 ± 2 μM was reported by Hong and Fast (2007) using a more sensitive assay. To confirm the Km, we used an HPLC-based sensitive assay to determine ADMA degradation to citrulline. The Km of PA-DDAH was 76 μM (Fig. 1B), a value close to the one previously reported using a sensitive assay. The recombinant PA-DDAH, designated as rDDAH, dose-dependently (Fig. 1C) and rapidly (Fig. 1D) reduced ADMA in plasma when incubated in vitro. These data demonstrated that rDDAH produced as described has the anticipated biologic activity and is a suitable molecule for investigation of its actions in vivo.
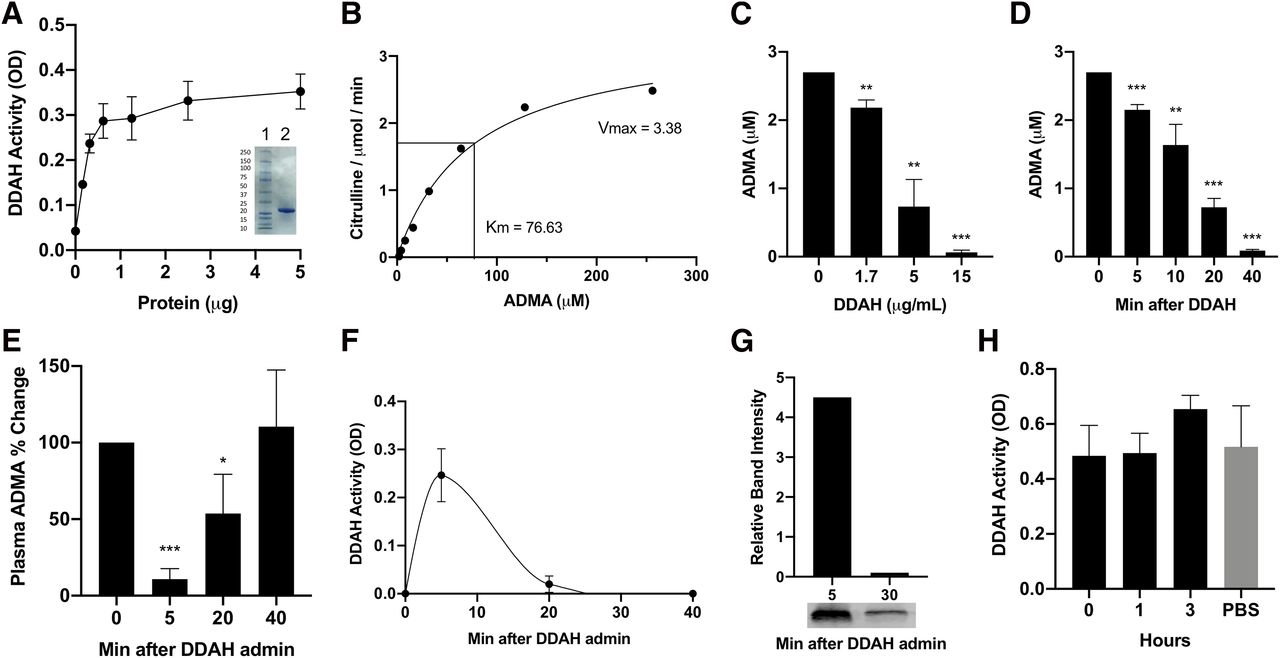
In vitro and in vivo activity of recombinant DDAH. PA-DDAH with His-tag was cloned in pE-SUMO vector and then expressed in E. coli. Purified rDDAH shows activity and purity in SDS gel (inset lane 2) (n = 4) (A). Km determination using high-sensitivity citrulline assay (B). In vitro ADMA lowering in rat plasma containing 2.7 μM ADMA after 1 hour of incubation (C) and ADMA lowering with 10 μg/ml rDDAH incubated for different times (n = 3) (D). In vivo activity of rDDAH after intravenous administration of a single dose of 2 mg/kg of rDDAH to rats. Blood was drawn at the indicated time points and ADMA levels (E), and DDAH activity in plasma (F) was determined (n = 4). Plasma samples collected at 5 and 30 minutes post–rDDAH administration were analyzed by Western blotting to detect DDAH protein (G). Stability in blood was determined by incubation of rDDAH with heparinized blood for the indicated time at room temperature or PBS at 4°C and then assayed for DDAH activity (n = 3) (H). All data are given as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
In Vivo ADMA Lowering by rDDAH.
The effect of rDDAH on ADMA in vivo was determined by administration of a single dose (2.0 mg/kg) intravenously to normal rats. Blood was collected at various times for measurement of ADMA and DDAH activity in plasma. Figure 1E shows that ADMA in plasma was reduced within 5 minutes of rDDAH administration but returned to baseline levels by 40 minutes. Measurement of rDDAH in plasma showed that DDAH activity in the circulation was rapidly reduced (Fig. 1F). Western blotting confirmed that rDDAH protein was similarly reduced in plasma within 30 minutes (Fig. 1G). These data suggested that exogenous native rDDAH was rapidly eliminated from the blood. To test whether the blood constituents inactivated or sequestered DDAH, rDDAH was incubated with whole blood for varying lengths of time. As shown in Fig. 1H, the DDAH activity was stable in blood, suggesting that in vivo loss of rDDAH was not simply due to its inactivation or sequestration in the blood. These data also suggested that continuous presence of DDAH activity is required to achieve sustained ADMA lowering.
Generation of Long-Acting M-DDAH.
Several approaches to increase the duration of rDDAH activity in vivo and to achieve extended reduction of ADMA were investigated. We first tested whether filtration by the kidney can be reduced by increasing the molecular size using site-specific PEGylation of a mutant rDDAH. We cloned and expressed PA-DDAH mutants in which lysine residues (residues 3, 25, 103 or 159) were individually replaced with cysteine. Adding a single cysteine allows site-specific PEGylation using mPEG40K-maleimide reaction with the cysteine residue. As shown in Fig. 2A, all Lys-Cys mutants were active in vitro with varying potency. Based on the solubility characteristics, enzyme activity, and PEGylation efficiency, the 159Lys-Cys mutant was selected for further studies. The 159Lys-Cys mutant was PEGylated using mPEG40K-maleimide. As shown in Fig. 2B, the 159Lys-Cys PEGylated protein with a molecular mass of 75 kDa was active in metabolizing ADMA. However, the 159Lys-Cys PEGylated protein was also rapidly reduced in circulation (Fig. 2C). Since a protein with a molecular mass of ∼75 kDa likely filtered only minimally in the glomerulus, these data suggest that the clearance of the PEGylated 159Lys-Cys via a different mechanism and a different approach to enhance the in vivo duration of DDAH activity would be required.

Generation of site-specific PEGylated and long-acting DDAH. DDAH mutants were generated by site-directed replacement of Lys 3, 25, 103, or 159 with cysteine. Mutant genes were cloned and expressed in E. coli. Purified proteins were assayed for activity of rDDAH (●), 3Lys-Cys (■), 25Lys-Cys (▲), 103Lys-Cys (▼), and 159Lys-Cys (◆) mutants (n = 3) (A). Mutant 159Lys-Cys was PEGylated at cysteine using mPEG40K-maleimide. Activity of 159Lys-Cys mutant before (●) and after PEGylation (■) and SDS gels comparing molecular size (inset gel 1 before and gel 2 after PEGylation) are shown (n = 3) (B). In vivo activity of PEGylated 75-kDa protein after a single administration to rats (C). Long-acting M-DDAH was generated by maximum PEGylation protocol using mPEG10K-NHS (n = 4). Shown are in vitro activity of rDDAH before (●) and after PEGylation (M-DDAH) (■), and SDS gels (inset lane 2 before and lane 3 after PEGylation) with 250-kDa protein (D). In vivo activity of rDDAH-1 (●), M-DDAH (■), (E) and ADMA lowering in rat plasma after M-DDAH administration (n = 7) (F). All data are given as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
We then PEGylated the rDDAH using mPEG10k-NHS, which conjugated at the lysine residues. The PEGylation conditions were optimized to PEGylate all available lysine residues to produce an rDDAH with an average molecular mass of 250 kDa. The maximum PEGylated rDDAH, designated as M-DDAH, maintained 90% activity in vitro (Fig. 2D). Intravenous administration to rats showed that the activity of M-DDAH remained in circulation significantly longer as compared with rDDAH (Fig. 2E). Consistent with the prolonged activity in vivo, a significant reduction in plasma ADMA was observed for up to 6 hours (Fig. 2F). These data showed that, as compared with the native rDDAH, M-DDAH exhibited more than 15-fold duration of activity in vivo, suggesting that it is suitable for pharmacological studies in animal models.
Effect of M-DDAH on Endothelial Function.
Plasma ADMA levels in human subjects are strongly correlated with endothelial dysfunction (Cooke, 2000). DDAH-1 gene deletion in mice resulted in high plasma ADMA, endothelial dysfunction, and increased blood pressure (Leiper et al., 2007). We first determined whether M-DDAH treatment reduced the intracellular levels of ADMA in endothelial cells. Figure 3A shows that incubation of cells with ADMA resulted in a 3-fold increase in intracellular ADMA, which was reduced by the addition of M-DDAH. Thus, extracellular M-DDAH can effectively reduce ADMA levels in the cells. We then determined the effect of M-DDAH on inhibition of NO responses induced by ADMA. HUVECs were treated with ADMA or ADMA + M-DDAH, and the effect on cGMP, a marker of NO pathway, was determined. Figure 3B shows that ADMA treatment significantly reduced cGMP levels in HUVECs, which was reversed by treatment with M-DDAH. These data demonstrate that extracellular M-DDAH attenuated inhibition of NO synthesis within the cells by reducing ADMA.

Effect of M-DDAH on endothelial function. Effect of M-DDAH on cellular ADMA in endothelial cells was determined using HUVEC culture (A). Triplicate wells were treated with vehicle or 10 μM ADMA. After 30 minutes, cells were washed to remove exogenous ADMA and then incubated in the absence or presence of 100 μg/ml of M-DDAH for 1 hour. Medium was removed, and cells were extracted for measuring ADMA remaining in the cells using HPLC (area under the curve) (n = 3). Effect on cGMP production in HUVECs was determined in cells treated with control, ADMA, and ADMA + M-DDAH (B). Triplicate wells were treated with 0.5 mM IBMX (control) or 0.5 mM IBMX + 100 μM ADMA for 5 minutes. The medium was aspirated, and cells were incubated in fresh medium containing 0.5 mM IBMX or 0.5 mM IBMX + 10 μg/ml M-DDAH for 60 minutes. Cells were collected, and cGMP was analyzed using enzyme immunoassay kit (n = 3). In vitro angiogenesis was determined by incubation of HUVECs in Matrigel in the presence of control, ADMA, M-DDAH, or ADMA + M-DDAH (C) (n = 3 wells per treatment). Angiogenesis was quantified using ImageJ and expressed as number of nodes, segments, and mesh (n = 3 in each figure) (D). All data are given as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
M-DDAH Promotes In Vitro Angiogenesis.
Effect of M-DDAH on endothelial function was assessed using an in vitro angiogenesis assay. HUVECs in Matrigel were treated with ADMA in the presence or absence of M-DDAH, and branch formation was determined by image analysis (Fig. 3C). Figure 3D shows that treatment of HUVEC with M-DDAH without addition of exogenous ADMA significantly enhanced angiogenesis measured by number of nodes, segments, and mesh. Addition of ADMA significantly inhibited in vitro angiogenesis, and treatment with M-DDAH reversed the effect of ADMA (Fig. 3, C and D). These results suggest that reduction of ADMA by M-DDAH may improve endothelial function and their regenerative function.
Effect of M-DDAH on Blood Pressure.
Because ADMA levels are associated with elevation in blood pressure, we investigated whether reductions in ADMA by M-DDAH might directly lower blood pressure in an animal model of hypertension. Dahl salt-sensitive (DSS) hypertensive rats have been shown to exhibit higher levels of ADMA and spontaneous hypertension with age (Rapp and Garrett, 2019). Single intravenous administration of M-DDAH to DSS hypertensive rats showed sustained enzymatic activity in plasma (Fig. 4A) and produced a significant reduction in ADMA (Fig. 4B) and blood pressure for up to 4 hours postinjection (Fig. 4C). These data suggest that a targeted reduction of ADMA by M-DDAH manifests physiologically relevant blood pressure lowering in a model of hypertension.

Effect of M-DDAH on blood pressure. M-DDAH (1 mg/kg) (■) or vehicle (●) was intravenously administered to conscious, restrained Dahl salt-sensitive hypertensive female rats (n = 4). Blood was collected at the indicated time points, and plasma DDAH activity (A) and ADMA levels (B) were determined. Mean arterial blood pressure was determined by Deltran pressure transducer (Utah Medical) connected to the catheter in the carotid artery (C). Data were collected using an amplifier and PowerLab from ADInstruments. All data are given as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
M-DDAH Protects Kidney Function in Response to Ischemia-Reperfusion Injury.
ADMA plays an important role in the kidney by inducing microcirculatory dysfunction, which may contribute to accelerated renal disease. Ischemia-reperfusion (I/R) reduces DDAH, resulting in the accumulation of ADMA in the target organ (Nakayama et al., 2014). To assess the effect of M-DDAH on I/R injury, we first determined the effect of I/R on DDAH expression and ADMA using a model of renal I/R injury. Consistent with previous data (Nakayama et al., 2014), renal I/R produced a time-dependent reduction in DDAH-1 mRNA (Supplemental Fig. 1A) and increased ADMA in the kidney (Supplemental Fig. 1B). To test the effect of M-DDAH, rats were subjected to 30 minutes of ischemia followed by reperfusion and then treated with vehicle or M-DDAH. The renal function, immune response, and injury score were assessed after 24 hours of reperfusion. Rats treated with M-DDAH were significantly protected from I/R injury, as determined by serum creatinine, which was significantly lower in M-DDAH–treated animals (Fig. 5A). M-DDAH treatment significantly reduced the number of inflammatory cells in the kidney (Fig. 5B) and tubular injury (Figs. 5, C and D) in renal outer medulla. Metabolic acidosis due to lowered bicarbonate levels was also observed in the setting after I/R. Postischemic rats showed significantly lower levels of serum bicarbonate and pH relative to the postischemic M-DDAH–treated animals (Supplemental Table 1). These results showed that reduction of ADMA by M-DDAH produced a significant reduction in inflammatory response and injury score in the kidney and improvement in renal function in response to I/R injury.

Effect of M-DDAH on renal ischemia-reperfusion injury. Sprague-Dawley rats were subjected to 30 minutes of bilateral ischemia by clamping the renal arteries and then reperfusion. M-DDAH (2 mg/kg) or vehicle was administered by intravenous injection at the time of reperfusion and a second injection 3 hours after reperfusion. Plasma and kidney samples were collected after 24 hours (n = 5). Renal function was determined by measuring plasma creatinine (A). Inflammatory cells were identified by flow cytometry, and the number of CD11/b/c-positive cells, IL-17–positive cells, CD4-positive cells, and CD8-positive cells was calculated per gram kidney weight for vehicle-treated (black bar) or M-DDAH–treated rats (gray bar) (B). Representative hematoxylin and eosin–stained sections through renal outer medulla demonstrating significant necrosis in vehicle-treated group (arrows) versus minimal tubular damage in M-DDAH–treated group. The higher-magnification image is shown in inset (C). Quantification of percent damaged tubules (D). All data are given as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
In this first study of its kind, we have developed a novel, long-acting, modified DDAH (M-DDAH) and investigated the physiologic and therapeutic significance of modulation of ADMA in vitro, ex vivo, and in vivo. The major findings of this study are that 1) therapeutic levels of M-DDAH can lower plasma ADMA in vivo, and it is suitable for pharmacological studies in animal models; 2) M-DDAH can reverse ADMA-induced inhibition of cGMP generation; 3) lowering of ADMA can increase angiogenic function in endothelial cells; 4) reduction in ADMA by M-DDAH lowers blood pressure in hypertensive rodents; and 5) reduction of ADMA by M-DDAH attenuates inflammatory response and kidney injury and improves renal function after ischemia-reperfusion injury.
Multiple clinical studies have established an association of plasma ADMA with hypertension, cardiovascular and renal disease, and mortality. Whether ADMA is a bystander, a biomarker, or a contributor to the mechanism of disease is yet to be established. The evidence for the physiologic role of ADMA and its metabolizing enzyme DDAH has been largely derived from genetic studies in rodents. DDAH-1 gene deletion in mice increased plasma ADMA, endothelial dysfunction, and blood pressure (Leiper et al., 2007). DDAH-1 gene overexpression improved angiogenesis in the ischemic hind limb model (Jacobi et al., 2005) and protected from myocardial and renal ischemia-reperfusion injury (Stuhlinger et al., 2007). However, the results of other genetic studies have been contradictory. For example, DDAH-1 gene deletion did not affect the development of diabetic nephropathy in the streptozotocin-induced diabetes mellitus model (Rodionov et al., 2019), whereas adenoviral-mediated DDAH-1 gene delivery demonstrated improvement in diabetic nephropathy in the streptozotocin-induced diabetes mellitus model (Wetzel et al., 2020). In another study, DDAH-1 gene deletion in the proximal tubule of mouse kidney increased tubular ADMA but was protective in kidney injury, collagen deposition, and profibrotic cytokine expression (Tomlinson et al., 2015). These results were contrary to the reported protection of kidney injury in DDAH-1 transgenics (Nakayama et al., 2014). The results of gene manipulation can be confounded by developmental compensatory or redundant mechanisms. Since two isoforms of DDAH that catalyze ADMA degradation exist (Leiper and Nandi, 2011), the complexity of genetic models are particularly relevant to the ADMA metabolism pathway. In addition, glyoxylate aminotransferase 2 can also play a role in metabolizing ADMA (Rodionov et al., 2014). Therefore, single DDAH gene deletion or overexpression may not produce a clear phenotype. Therefore, specific modulation of ADMA by a pharmacological molecule can provide a unique opportunity to investigate its pathophysiological role and determine whether ADMA can be a target for therapy.
To specifically lower ADMA, we have developed a novel pharmacologically viable DDAH. rDDAH expressed and purified from E. coli effectively reduced ADMA in plasma in vitro even while the concentration of ADMA was 100- to 200-fold below its Km. rDDAH rapidly lowered ADMA in rat plasma in vivo. However, the ADMA lowering was short-lived because of the elimination of rDDAH from the circulation. Further, PEGylated DDAH mutant with a higher molecular mass (75 kDa) was also short-lived in vivo, suggesting that kidney filtration may not be the mechanism of DDAH clearance. A DDAH molecule produced by multiple PEGylation at the lysine residues (M-DDAH) demonstrated more than 15-fold longer duration of activity in vivo. M-DDAH reduced ADMA levels in rat plasma by greater than 80% within 5 minutes after administration. The rapid and large magnitude of ADMA reduction was unexpected. First, the Km of rDDAH (76 μM) is about 150-fold greater than the ADMA concentration in rat plasma. Second, previous studies have shown that ADMA remains bound to plasma proteins and is not removable by dialysis (Kielstein et al., 2004). Based on the DDAH protein structure and localization of the active site within the three-dimensional structure (Frey et al., 2006), it is unlikely that the protein-bound ADMA is accessible to the active site for catalysis. The rapid and effective lowering of ADMA by M-DDAH suggests that there may be an efficient dynamic equilibrium between protein-bound and free ADMA such that the bound ADMA molecules can readily dissociate and become available for hydrolysis. These studies showed that M-DDAH is highly suitable for specific modulation of ADMA in vivo; therefore, it offers a unique opportunity to investigate the physiologic impact of ADMA lowering in disease states.
The effect of M-DDAH was investigated in vitro and in vivo under conditions involving high ADMA. In vitro, addition of M-DDAH to endothelial cells reduced intracellular ADMA. Concordantly, M-DDAH treatment increased NO pathway responses as measured by increase in cGMP in endothelial cells. The improvement in endothelial function was further validated by the reversal of the inhibitory effects of ADMA on in vitro angiogenesis. Importantly, reduction in endogenous ADMA by M-DDAH enhanced endothelial activity to undergo branching. Collectively, these results showed that specific reduction of ADMA by M-DDAH significantly improved endothelial function. The abrogation of ADMA effects on cGMP suggest that improvement in NO bioavailability is an important contributing mechanism to its effects on endothelial function. Since ADMA has been shown to induce other cellular responses, including the generation of reactive oxygen species by nitric oxide synthases uncoupling, the use of M-DDAH may also allow investigation of the other potential mechanisms contributing to the cellular actions of ADMA.
Two different disease models were used for the investigation of the physiologic response to ADMA lowering. The DSS hypertensive rats are known to exhibit high levels of circulating ADMA and endothelial dysfunction (Matsuoka et al., 1997). Administration of M-DDAH to DSS hypertensive rats significantly reduced ADMA in plasma and reduced blood pressure. These data showed that ADMA plays a pathologic role in inducing high blood pressure and that M-DDAH produces a positive therapeutic outcome. These studies have also provided direct evidence that ADMA is not simply a biomarker. Rather, ADMA acts as a pathologic molecule, and its lowering may provide a new therapeutic approach to manage hypertension.
I/R injury is another pathologic condition in which high ADMA levels have been implicated. In models of I/R injury to the heart (Stuhlinger et al., 2007) and kidney (Nakayama et al., 2014), ADMA levels are significantly increased, and DDAH expression and activity are reduced. The renal I/R injury may parallel with the acute kidney injury (AKI) that commonly occurs during the cardiopulmonary bypass and aortic crossclamping (Thakar et al., 2005; Lannemyr et al., 2017). In the rat model of renal I/R injury, M-DDAH significantly attenuated the loss of renal function as measured by reduced serum creatinine. Concurrently, M-DDAH significantly improved serum bicarbonate and acidosis and reduction in inflammatory cells in the kidney. These effects may be due to the improved renal perfusion and endothelial function, which play a critical role in AKI. The modulation of injury response by M-DDAH is particularly relevant to cardiac surgery–associated AKI in which high levels of ADMA are produced (Plicner et al., 2014). Cardiac surgery–associated AKI is the second most common cause of kidney injury in hospitalized patients (Uchino, 2006). In addition, contrast media are associated with a high incidence of AKI (Hennessy et al., 2010). Thus, M-DDAH may represent an important therapeutic molecule for the preservation of microcirculatory flow and prevention and treatment of postoperative AKI.
A limitation of the current M-DDAH molecule is the duration of its in vivo activity. ADMA lowering by DDAH may be suitable for investigating its efficacy in acute settings, such as during the cardiothoracic and heart valve replacement surgery. However, the duration of M-DDAH action is still not adequate for chronic diseases. Also, in the current studies, intravenous delivery of M-DDAH was used. Further development of a molecule that can be delivered by subcutaneous administration could provide a more convenient method of treatment. The primary goal of the current studies was to test the hypothesis that specific lowering of ADMA can produce physiologic response. Our experimental approach has clearly demonstrated efficacy of ADMA lowering, which could be clinically relevant for patients undergoing cardiac surgery. Future studies will focus on improving the duration of DDAH action in vivo, which will enable testing of prevention as well as treatment approaches for acute and chronic conditions.
Acknowledgments
The authors thank Michael Garrett for providing Dahl salt-sensitive rats for use in this study and Shenique Ries for supporting ex vivo studies on vascular reactivity.
Authorship Contributions
Participated in research design: Lee, Mehrotra, Basile, Skill, Sasser, Shekhar, J. Singh.
Conducted experiments: Lee, Mehrotra, Ullah, A. Singh, Skill, Younes.
Contributed new reagents or analytic tools: Lee, A. Singh, J. Singh.
Performed data analysis: Lee, Mehrotra, Ullah, Skill, Younes, Sasser.
Wrote or contributed to the writing of the manuscript: Lee, Basile, J. Singh.
Footnotes
- Received July 13, 2020.
- Accepted November 13, 2020.
This project was supported in part with a grant from the Indiana Clinical and Translational Sciences Institute’s Advancing Innovative Medicine (AIMS) and National Center for Advancing Translational Science at the National Institutes of Health [Grant UL1TR001108]. The project was also supported in part by the Funding Opportunities for Research Commercialization and Economic Success (FORCES) grant from Indiana University Purdue University. David Basile research and Michael Garrett (maintenance of Dahl SS/jr colony) were supported by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01-DK0631140] and National Heart, Lung, and Blood Institute [Grant R01-HL137673].
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. No author has an actual or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ADMA
- asymmetric dimethylarginine
- AKI
- acute kidney injury
- DDAH
- dimethylarginine dimethylaminohydrolase
- DSS
- Dahl salt-sensitive
- HUVEC
- human umbilical vein endothelial cell
- I/R
- ischemia-reperfusion
- M-DDAH
- modified DDAH by PEGylation
- NO
- nitric oxide
- OD
- optical density
- PA
- Pseudomonas aeruginosa
- PEG
- poly(ethylene glycol)
- rDDAH
- recombinant dimethylarginine dimethylaminohydrolase
- SD
- Sprague-Dawley
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}