


Abstract
The practice of prescribing β-blockers to lower blood pressure and mitigate perioperative cardiovascular events has been questioned because of reports of an increased risk of stroke. The benefit of β-blocker therapy primarily relies on preventing activation of cardiac β1-adrenergic receptors (ARs). However, we reported that β1ARs also mediate vasodilator responses of rat cerebral arteries (CAs), implying that β-blockers may impair cerebral blood flow under some conditions. Here, we defined the impact of metoprolol (MET), a widely prescribed β1AR-selective antagonist, on adrenergic-elicited diameter responses of rat CAs ex vivo and in vivo. MET (1–10 µmol/l) prevented β1AR-mediated increases in diameter elicited by dobutamine in cannulated rat CAs. The β1AR-mediated dilation elicited by the endogenous adrenergic agonist norepinephrine (NE) was reversed to a sustained constriction by MET. Acute oral administration of MET (30 mg/kg) to rats in doses that attenuated resting heart rate and dobutamine-induced tachycardia also blunted β1AR-mediated dilation of CAs. In the same animals, NE-induced dilation of CAs was reversed to sustained constriction. Administration of MET for 2 weeks in drinking water (2 mg/ml) or subcutaneously (15 mg/kg per day) also resulted in NE-induced constriction of CAs in vivo. Thus, doses of MET that protect the heart from adrenergic stimulation also prevent β1AR-mediated dilation of CAs and favor anomalous adrenergic constriction. Our findings raise the possibility that the increased risk of ischemic stroke in patients on β-blockers relates in part to adrenergic dysregulation of cerebrovascular tone.
SIGNIFICANCE STATEMENT β-Blocker therapy using second-generation, cardioselective β-blockers is associated with an increased risk of stroke, but the responsible mechanisms are unclear. Here, we report that either acute or chronic systemic administration of a cardioselective β-blocker, metoprolol, mitigates adrenergic stimulation of the heart as an intended beneficial action. However, metoprolol concomitantly eliminates vasodilator responses to adrenergic stimuli of rat cerebral arteries in vivo as a potential cause of dysregulated cerebral blood flow predisposing to ischemic stroke.
Introduction
β-Adrenergic receptor (AR) antagonists, commonly referred to as β-blockers, are a mainstay therapy for the treatment of hypertension, heart failure, and angina (Frishman, 2003). β-Blocker therapy mitigates sympathetic activation of the heart by antagonizing the binding of norepinephrine (NE) to β1-ARs on cardiac myocytes to lower heart rate and blood pressure and reduce ventricular workload (Frishman, 2003). The first generation of β-blockers with propranolol as the prototype failed to discriminate between β1AR and β2AR subtypes, thereby compromising β2AR-mediated airway dilation to cause unwanted bronchoconstriction. Subsequently, metoprolol (MET) and other second-generation β-blockers were introduced to selectively antagonize β1AR and avoid the off-target effects of nonselective β-blockers (Frishman, 2008). In 2013, more than 83 million prescriptions were filled in the United States for MET, which is the most widely prescribed β1AR-selective antagonist (Aitken et al., 2014). MET use steadily increased and accounted for 89 million prescriptions in 2017, ranking fourth in the nation for total volume of prescribed medications (IQVIA, 2018).
The widespread use of β-blockers persists even though new guidelines in 2014 by the Eighth Joint National Committee of the U.S. Centers for Disease Control and Prevention removed β-blockers as a first-line antihypertensive medication, citing mounting evidence for an increased risk of stroke (James et al., 2014). As early as 2002, the Losartan Intervention for Endpoint Reduction trial reported a significant increased stroke risk associated with antihypertensive therapy using the β-blocker atenolol compared with losartan, an angiotensin receptor blocker (Dahlöf et al., 2002). In 2005, the Lindholm meta-analysis reported a 16%–26% higher risk of stroke for patients taking β-blockers to control hypertension compared with other classes of antihypertensive drugs (Lindholm et al., 2005). Finally, the results of the Perioperative Ischemic Evaluation trial reported in 2008 indicated a 2-fold higher risk of stroke in patients of non–cardiac surgery treated perioperatively with MET compared with placebo (Devereaux et al., 2008). The latter finding resulted in new 2009 guidelines by the American College of Cardiology Foundation and American Heart Association that recommended limited perioperative use of β-blockers (Fleischmann et al., 2009).
The positive correlation between antagonism of β1AR by β-blockers and increased risk of stroke has evaded a mechanistic explanation. However, there had been indications of a potential beneficial contribution of β1AR to maintenance of cerebral blood flow and prevention of cerebral ischemia. In the peripheral vascular beds, the role of β1AR in regulating blood flow is assumed to be minimal because sympathetic stimulation elicits α1AR-mediated vasoconstriction as the predominant adrenergic response, masking mainly β2AR-mediated dilation (Leech and Faber, 1996; Docherty, 2010). However, a different pattern of adrenergic responsiveness appears to characterize the cerebral circulation, which also is populated by sympathetic nerve endings (Edvinsson et al., 1975; Cipolla et al., 2004; Moore et al., 2015). Furthermore, unlike many other vascular beds that are responsive to circulating catecholamines, the cerebral circulation under normal physiologic conditions is largely unaffected by levels of circulating epinephrine because of their exclusion by the blood-brain barrier (Weil-Malherbe et al., 1959; Myburgh et al., 2002). Although β-blockers do not impair cerebral blood flow in healthy individuals (Heinke et al., 2005), β blockade in the presence of a cardiovascular challenge, such as stroke, reduces cerebral blood flow (Stirling Meyer et al., 1974). Indirect measurements of cerebral vasoreactivity in hypertensive or perioperative rat models indicate that βAR may exert a tonic vasodilator influence under these challenges since systemic administration of either propranolol or MET, a prototypic nonselective and a β1AR-selective β-blocker, respectively, attenuated cerebral blood flow (Ooboshi et al., 1990; Ragoonanan et al., 2009). A similar conclusion was reached by a study in nonhuman primates under conditions of induced hypertension (Aqyagi et al., 1976). However, only more recently has the effect of β-blockers on cerebral vasoreactivity in vivo been assessed directly using video imaging through cranial windows. Using this method, Gorshkova et al. (2011) infused NE intravenously to adrenergically activate the cerebral circulation while stabilizing blood pressure. Against a background of nonselective βAR blockade established by propranolol, NE exerted opposing effects on different segments of pial arteries consisting of either vasodilation or vasoconstriction. Later, our laboratory (Moore et al., 2015) topically suffused small-molecule inhibitors of either β1AR or β2AR into cranial windows to demonstrate that β1AR mediate the vasodilator responses of rat middle cerebral arteries to isoproterenol (ISO) and NE. Although these latter findings infer that β1AR can influence the adrenergic responsiveness of cerebral arteries, they do not address the question of whether systemically administered doses of clinical β1AR-selective blockers that confer intended beneficial cardiac effects concomitantly dysregulate adrenergically driven cerebral vasodilator responses as an off-target effect.
The objective of the present study was to use cerebral window video imaging to directly answer this question. The debate over the risks and benefits of β-blocker therapy continues without insight into the mechanisms of increased stroke risk (Dahlöf et al., 2002; Lindholm et al., 2005; Devereaux et al., 2008). Thus, the design of the present study included different dosing protocols intended to recapitulate the dual clinical use of MET as either a single-dose perioperative medication or a long-term therapy for cardiovascular conditions.
Materials and Methods
Animals.
All experiments in this study were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. Male Sprague-Dawley (SD) rats (Harlan/Envigo Laboratories, Indianapolis, IN) were used at 10–12 weeks of age for all experiments.
Diameter Measurements.
Measurements of diameter in isolated superior cerebral arteries (CAs) were performed as described earlier (Moore et al., 2014). Briefly, CAs were isolated, cannulated, and pressurized at an intraluminal pressure of 80 mm Hg and then equilibrated at 37°C for 1 hour during superfusion with physiologic salt solution (PSS) bubbled with a 7% CO2–93% O2 gas mixture to maintain a pH of ∼7.4. CAs that failed to develop spontaneous tone after 1-hour equilibration or failed to constrict to 60 mmol/l KCl were omitted from studies. External diameters were continuously measured and recorded by DMT software (Danish Myo Technology, Aarhus, Denmark). Although the internal diameter is a more accurate determinant of resistance, internal and external diameters correlate closely with each other in an isolated vessel (Supplemental Fig. 1). At the end of each study, arteries were superfused with Ca2+-free PSS solution to obtain maximal diameter. In preliminary studies, we identified the concentration of MET required to fully block β1AR-mediated dilations in isolated CAs. Vasodilation was elicited either by the β1AR agonist dobutamine (DOB) or the mixed β1/α1AR agonist NE prior to and then during incubation of the vessel with increasing concentrations of MET.
Acute and Chronic Administration of MET.
Acute doses of (±) MET (+)-tartrate (10 or 30 mg/kg) were dissolved in water (10 or 30 mg/ml) and administered to rats 30 minutes before the start of cranial window surgery. For chronic treatment, rats either were provided ad libitum access for 2 weeks to MET in drinking water (0.3 or 2.0 mg/ml to provide an average dose of 35 or 130 mg/kg per day, respectively) or were implanted with MET-filled subcutaneous osmotic mini-pumps (Model 2002; Alzet, Cupertino, CA) to administer 5 or 15 mg/kg per day. Water bottles containing MET were light-protected to prevent photocatalytic degradation.
DOB Challenge.
DOB was administered intravenously to anesthetized rats to confirm effective antagonism of β1AR by systemic doses of MET. A DOB dosing regimen was chosen that would increase heart rate in control animals to approximately 85% of maximal heart rate (Bolter and Atkinson, 1988; Plante et al., 2005; Hazari et al., 2012), similar to DOB stress tests in patients (Henzlova et al., 2016). Rats were anesthetized with 1.5%–2.0% isoflurane, and the heart rate was monitored by a three-lead electrocardiogram (ADInstruments, Colorado Springs, CO). Body temperature was monitored with a rectal probe and maintained at 37°C with a direct feedback heating pad (Physitemp). Control or DOB-containing normal saline solution was administered by a syringe pump (Harvard Apparatus, Cambridge, MA) via an intravenous line (SV-S25BL; Terumo, Tokyo, Japan) into the lateral tail vein. DOB was prepared as a 33-µg/ml solution per 0.3 kg b.wt. An initial 2-minute infusion of normal saline was used to establish a baseline heart rate. Subsequently, DOB was administered by three progressive infusion rates (3.6, 11, and 18 µg/kg per minute). Heart rate was averaged for 2 minutes before DOB infusion to obtain a baseline value and then measured during the final 40 seconds of DOB infusion to obtain average values for analysis. Experiments were completed within the 4-hour window after the dark period of the rats’ light/dark cycle to match the time of day of the cranial window experiments.
Craniectomy and In Vivo Imaging.
Craniectomy surgeries and fitting of a custom-made cranial window containing a drug port were performed as described earlier (Moore et al., 2014). The tissue surface underneath the cranial window was suffused with 37°C PSS bubbled with a 7% CO2–93% O2 gas mixture to maintain pH of ∼7.4. Branches of the middle cerebral artery were imaged using an HDR-PJ580 camera (Sony, Tokyo, Japan) and analyzed using an automated IPLab script (Scanalytics, Milwaukee, WI). Drugs including ISO, NE, CG920712, and sodium nitroprusside (SNP) were suffused through ports on the surface of the cranial window. The concentrations of ISO, NE, and CGP20712 were chosen based on earlier studies by our laboratory (Moore et al., 2015). All drugs were prepared in PSS. Sodium nitroprusside (100 μmol/l) was suffused at the end of experiments to verify that arteries retained reactivity and, hence, viability.
Drugs.
All drugs and chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise. The following drugs were prepared as stock solutions in water: CGP20712 10 mmol/l (Tocris, Bristol, UK); ISO 20 mmol/l; NE 20 mmol/l; DOB 20 mmol/l (Hospira, Lake Forest, IL); MET 10 mmol/l; and SNP 100 mmol/l. For ex vivo diameter studies, drugs were diluted first in PSS when appropriate and added directly in 1:1000 dilutions to the perfusion chamber. Drug concentrations in the text refer to final bath concentrations. For cranial window experiments, drugs were diluted in PSS to their final concentration and directly suffused through ports of the cranial window.
Statistical Analysis.
Statistics were calculated using GraphPad Prism 5 (San Diego, CA). Data are expressed as mean ± S.D. Diameter results from vessel perfusion studies are reported as the change in outer vessel diameter as percent of calcium-free dilation from baseline. The results of in vivo cranial window experiments are reported as the percent change in outer vessel diameter from baseline. Data were analyzed using t tests or, when appropriate, using one-way ANOVA or two-way ANOVA with post hoc Bonferroni t tests. Sample sizes were chosen based on our previous experience to yield power of 0.8–0.9; P < 0.05 was considered statistically significant. Sample size (n) represents the number of arteries used in ex vivo studies or the number of animals used in in vivo studies. In box and whisker plots, boxes represent the inner quartile range, and whiskers represent minimum and maximum values.
Results
MET Abolishes β1AR-Induced Dilation Ex Vivo.
Initially, we performed proof-of-principle studies ex vivo to determine whether clinically relevant plasma concentrations of MET antagonize β1AR-induced vasodilator responses in rat CAs. The range of therapeutic plasma concentrations of MET in patients is between 20 and 600 ng/ml, which is comparable to a molar range of 0.07–2.24 µmol/l (Ritscher et al., 2019). Although we have demonstrated that CGP20712, a highly potent and selective small-molecule antagonist of β1AR, prevents β1AR-mediated vasodilation in rat CAs, MET has a less favorable profile as a β1AR-selective antagonist. It is a reversible antagonist unlike CGP20712 and only 2.5- to 30-fold more selective for β1AR than β2AR (Smith and Teitler, 1999; Baker, 2005). Regardless of these shortcomings, we found that MET antagonized the progressive vasodilator responses of cannulated rat CAs to β1AR agonist. For example, increasing log concentrations (10 nmol/l–100 μmol/l) of the β1AR-selective agonist DOB (Fig. 1A; left) were attenuated in a concentration-dependent manner after preincubation of arteries with MET (1, 3, or 10 μmol/l) (Fig. 1A; right). The clinically achievable concentration of 1 µmol/l MET attenuated the maximal dilator response to 100 μmol/l DOB by 45.5% ± 23.4%, whereas higher concentrations of 3 and 10 µmol/l MET resulted in 72.1% ± 23.4% and complete block of DOB-elicited vasodilation (Fig. 1B). The other statistical measures of EC50 and maximal effect are tabulated in Supplemental Table 1.
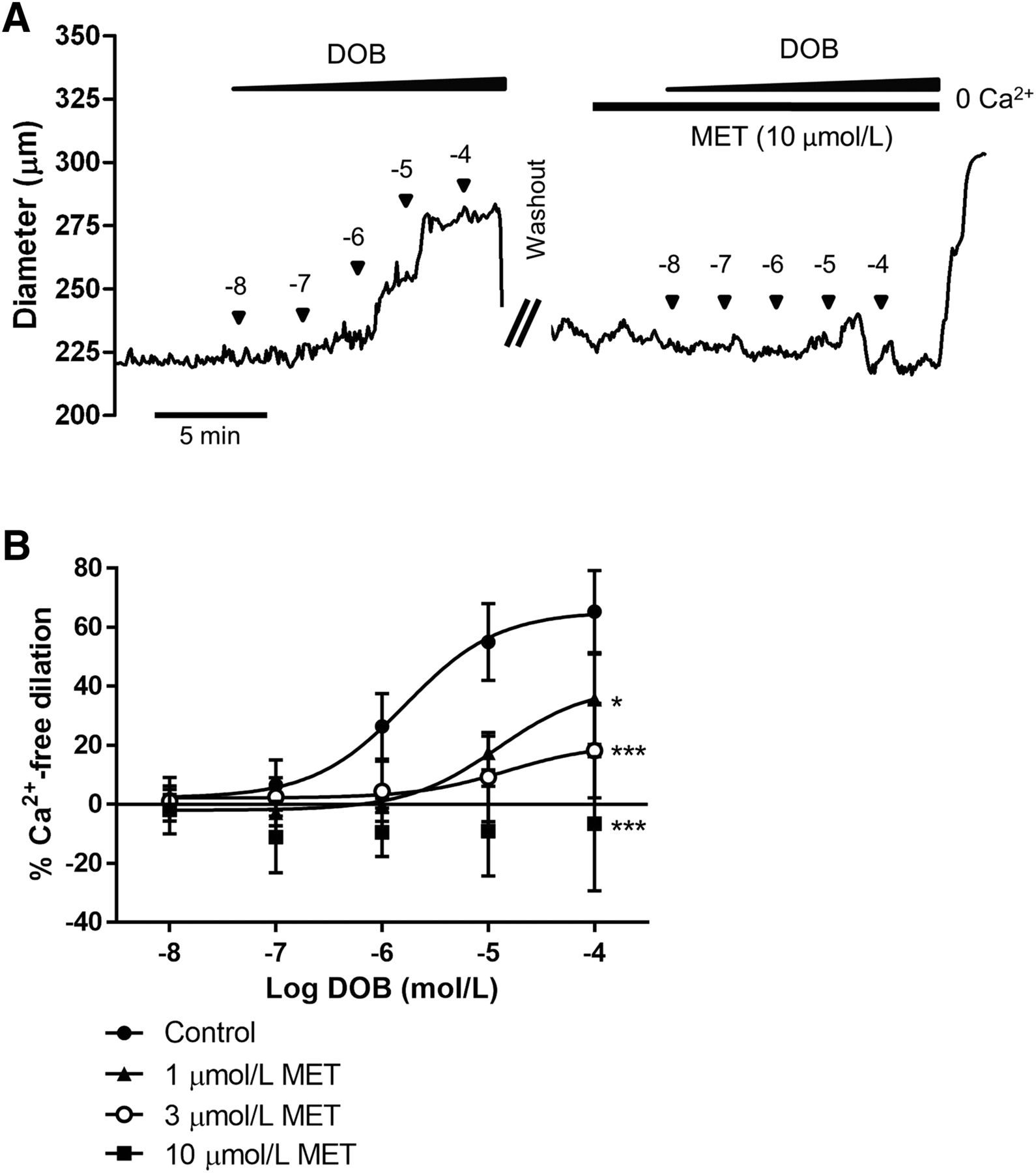
MET inhibits DOB-induced dilation of rat CAs ex vivo. (A) Diameter response of an isolated, pressurized CA to cumulative log concentrations of DOB before (left trace) and 10 minutes after (right trace) superfusion with 10 μmol/l MET. (B) Average diameter responses to DOB in the absence of MET (control) and after 1, 3, or 10 μmol/l MET; n = 6 each. One-way ANOVA at the highest concentration of MET with post hoc Bonferroni t tests; significant difference from control: *P < 0.05; ***P < 0.001.
Subsequently, we used a similar protocol to demonstrate that MET also effectively antagonizes the dilator response of cannulated rat CAs to the endogenous adrenergic agonist NE. In these studies, increasing log concentrations of NE (10 nmol/l–100 μmol/l) resulted in progressively larger diameters that ultimately reached 40.7% ± 19.0% of the maximal dilator response to Ca2+-free PSS (Fig. 2A, left). Induction of β1AR blockade by 10 μmol/l MET resulted in a reversal of the progressive NE-induced dilation to a sustained constriction (Fig. 2A, right), which represented a 24.7% ± 19.3% reduction in resting diameter (Fig. 2B).

NE-induced dilation of isolated CAs is reversed to constriction by MET. (A) Diameter response of an isolated, pressurized rat CA to cumulative log concentrations of NE before (left trace) and 10 minutes after (right trace) superfusion with 10 μmol/l MET. (B) Avg. diameter responses to NE in the absence of MET (control) and after 10 μmol/l MET; n = 6 (control), n = 5 (MET). Two-way ANOVA with post hoc Bonferroni t tests; significant difference from control: ***P < 0.001.
A Single Oral Dose of MET Acutely Inhibits β1AR-Mediated Dilation.
The perioperative oral administration of MET to confer cardiac protection is correlated with an increased risk of stroke (Devereaux et al., 2008). We sought to establish a rat model that mimicked the clinical scenario of perioperative MET administration to determine its impact on cerebrovascular reactivity. Based on earlier pharmacokinetic and pharmacodynamic analyses of MET in a variety of species, which we adapted to the body weight of our animal subjects, we estimated that oral MET doses of 10 and 30 mg/kg may exert acute β1AR antagonism in rats (Freireich et al., 1966; Höcht et al., 2005; Komura and Iwaki, 2005; Reagan-Shaw et al., 2008; El Beheiry et al., 2011; Yoon et al., 2011). To verify these doses of MET acutely mitigated the heart rate response to β1AR agonism, rats were subjected to a DOB challenge, which consisted of progressive infusion of three doses (3.6, 11, and 18 μg/kg per minute) of DOB (Fig. 3A). Heart rates at baseline and in response to an increasing infusion rate of DOB were compared between control rats and similar animals gavaged with a single dose of 10 or 30 mg/kg MET. In control animals, DOB maximally increased heart rate from 328 ± 25 to 407 ± 25 bpm (Fig. 3B). Values for resting heart rate and DOB-elicited heart rate in animals treated with 10 mg/kg MET were not significantly different from the control, averaging 318 ± 23 and 396 ± 21 bpm, respectively. However, rats gavaged with 30 mg/kg MET showed significantly lower resting heart rate and DOB-elicited heart rate, averaging 298 ± 19 and 375 ± 32 bpm, respectively (Fig. 3B). Mean arterial pressure in anesthetized rats recorded by femoral catheter was 94.7 ± 6.5 mm Hg at baseline and was reduced to 72.9 ± 10.8 mm Hg by 30 minutes after oral gavage of 30 mg/kg MET (Supplemental Fig. 2, A and B).

A single oral dose of MET attenuates the heart rate response to DOB challenge. (A) Sample plot of the control heart rate response to infusion of three cumulative doses (3.6, 11, and 18 µg/kg per minute) of the β1AR agonist, DOB, which was administered by a syringe pump into the tail vein. Initiation of a new dose of DOB is indicated by ▼. (B) Avg. heart rate at baseline and at the highest dose of 18.4 µg/kg per minute DOB in control rats (Control) and in rats gavaged 30 minutes before the DOB challenge with a single dose of 10 or 30 mg/kg MET. (- - -) indicates 85% of estimated maximal heart rate; n = 9 (control), n = 10 (MET). One-way ANOVA with post hoc Bonferroni t tests; significant difference from control: *P < 0.05.
Subsequently, we evaluated whether the oral doses of 10 or 30 mg/kg MET disrupted the in vivo vasodilator responses of rat CAs to adrenergic agonists, ISO and NE. In these studies, rats were administered 10 or 30 mg/kg MET by oral gavage 30 minutes prior to surgery for installation of a cranial window (Moore et al., 2014). Agonists were infused topically through a port in the window to avoid confounding systemic cardiovascular effects (Moore et al., 2015). We reported earlier that increasing log concentrations of ISO (10 nmol/l–10 μmol/l) progressively dilate CAs by a maximum of 19.4% ± 5.6% in untreated animals (Moore et al., 2015). Animals given a single oral dose of 10 mg/kg MET showed a maximal dilation of 20.2% ± 4.0%, which was similar to untreated animals (Fig. 4A). However, the maximal dilator response to ISO was significantly reduced to 10.2% ± 3.1% in animals given a single dose of 30 mg/kg MET (Fig. 4A). Thus, the effective oral dose of 30 mg/kg MET that mitigates β1AR-driven tachycardia (Fig. 3B) also disrupts the in vivo cerebral vasodilator responses to the adrenergic agonist ISO. At this effective oral dose of 30 mg/kg MET, CAs failed to exhibit the expected β1AR-mediated dilation in response to topical NE (Moore et al., 2015) and instead exhibited a stepwise diameter loss reaching −9.3% ± 11.2% (Fig. 4B). Thus, a single oral administration of MET that confers cardiac protection disrupts the β1AR-mediated dilator responses to the endogenous neurotransmitter NE in vivo and unmasks α1AR-mediated constriction (Moore et al., 2015). Basal diameter as percent of the maximal diameter elicited by SNP was 63.4% ± 1.2% and 62.1% ± 1.9% in rats gavaged with 10 and 30 mg/kg MET, respectively. These values were not significantly different from the value of 61.3% ± 2.1% obtained in control rats (Supplemental Fig. 2C).
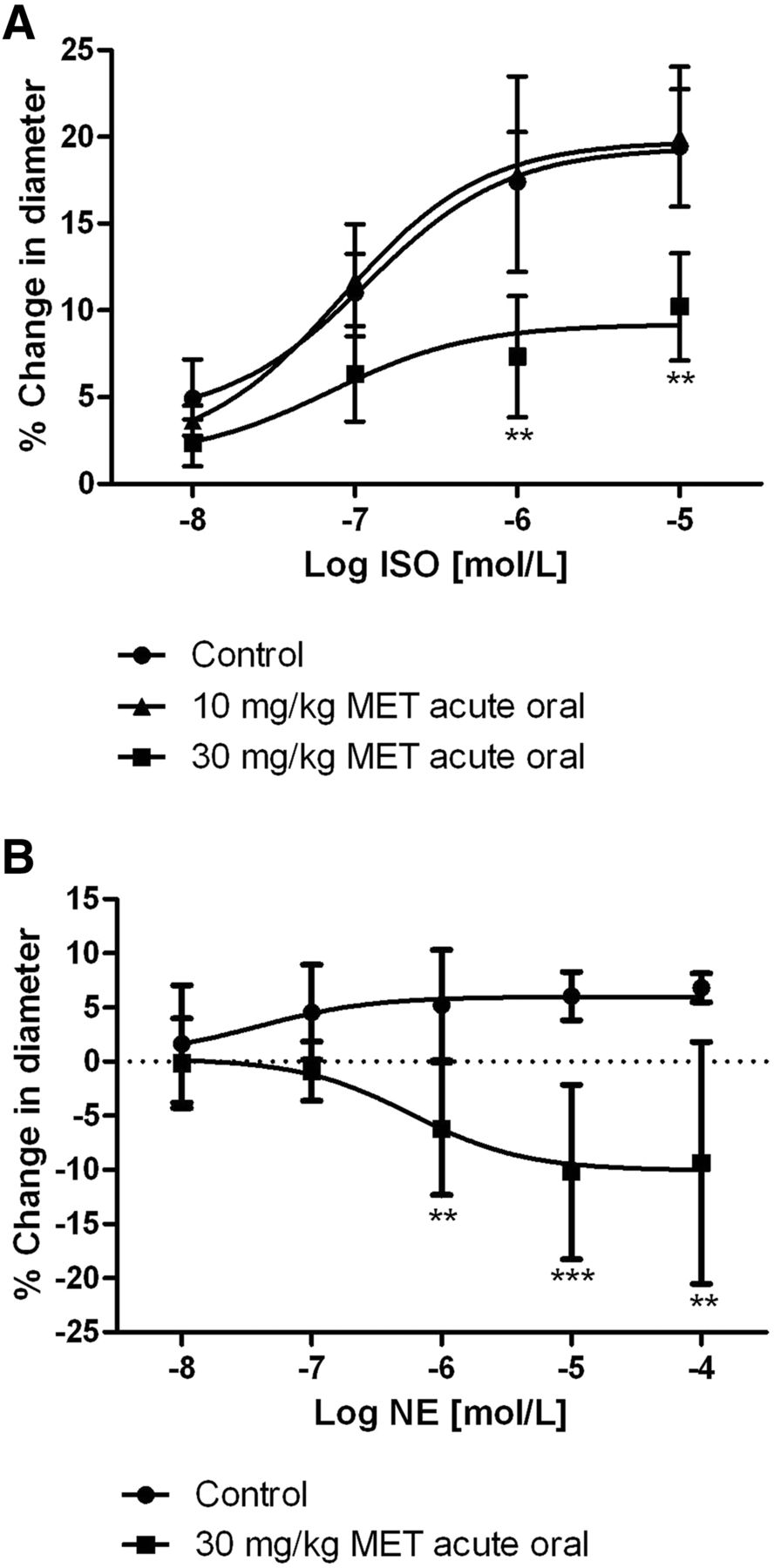
A single oral dose of MET inhibits the dilator response to ISO of rat CAs and reverses NE-induced dilation to constriction in vivo. (A) Avg. diameter response to topical suffusion of cumulative concentrations of ISO. Suffusion was initiated 30 minutes after rats were gavaged with either 10 or 30 mg/kg MET. Vessels were imaged through a cranial window; n = 6 (control), 5 (10 mg/kg MET), 6 (30 mg/kg MET). (B) Avg. diameter response to topical suffusion of cumulative concentrations of NE initiated 30 minutes after rats were gavaged with 30 mg/kg MET; n = 7 (control), 5 (10 mg/kg MET), 6 (30 mg/kg MET). Two-way ANOVA with post hoc Bonferroni t tests; significant difference from control: **P < 0.01; ***P < 0.001. Control data figures modified from Moore et al. (2015) with permission.
Chronic Administration of MET Also Disrupts β1AR-Mediated Dilation.
A final set of studies employed two different 2-week dosing protocols to recapitulate the clinical use of MET as a long-term therapy for chronic cardiovascular diseases, a treatment scenario correlated with increased risk of stroke (Dahlöf et al., 2002; Lindholm et al., 2005; James et al., 2014). As before, our goal was to define the impact of systemically administered MET on β1AR-mediated cerebral vasodilation. We chose our protocols for MET administration after normalizing MET doses used in humans (Reagan-Shaw et al., 2008) to rat body weight and also considering the more rapid metabolism of MET in rats (Yoon et al., 2011). Accordingly, rats were administered MET for 2 weeks in drinking water (0.3 or 2.0 mg/ml) to mimic the oral route of MET administration in patients or administered MET by subcutaneous mini-pump (5 or 15 mg/kg per day) to avoid potential fluctuations in plasma levels of MET caused by diurnal patterns of water consumption. Regardless of the route of administration, all in vivo experiments were conducted in animals within a 4-hour window after the dark period of the light/dark cycle, which is associated with higher animal activity and water consumption.
For both routes of MET administration, DOB challenge was employed again at the end of the 2-week drug treatment to identify MET doses that provided protection from β1AR-driven cardiac stimulation in vivo. Compared with control animals in which DOB maximally increased heart rate from 325 ± 10 to 412 ± 24 bpm, only the higher oral MET dose of 2.0 mg/ml significantly reduced resting heart rate to 299 ± 23 bpm and DOB-induced heart rate to 383 ± 26 bpm (Fig. 5A). The lower oral MET dose of 0.3 mg/ml did not significantly reduce resting (312 ± 27 bpm) or DOB-induced heart rate (397 ± 6 bpm). In animals administered MET by mini-pump infusion, resting heart rate in rats administered the lower subcutaneous dose of 5 mg/kg per day was significantly lower than that of control animals (296 ± 12 and 325 ± 10 bpm, respectively), but the tachycardia caused by DOB challenge was not significantly different from that of untreated animals (385 ± 8 bpm and 412 ± 23 bpm, respectively). Only the higher MET dose of 15 mg/kg per day significantly lowered both resting heart rate (273 ± 9 bpm) and DOB-induced tachycardia (328 ± 26 bpm) (Fig. 5B). Based on observed daily water consumption, the two oral doses of MET, 0.3 and 2.0 mg/ml in drinking water, were estimated to be 34 ± 10 and 130 ± 20 mg/kg per day of MET, respectively (Supplemental Fig. 3A). Neither the 0.3 nor 2.0 mg/ml oral dosing of MET significantly altered systolic blood pressure after 1 week (3.5 ± 15.2 or −1.0 ± 8.8 mm Hg, respectively) or 2 weeks (0.0 ± 14.1 or −7.1 ± 14.4 mm Hg, respectively) of treatment relative to pretreatment levels (Supplemental Fig. 3B).
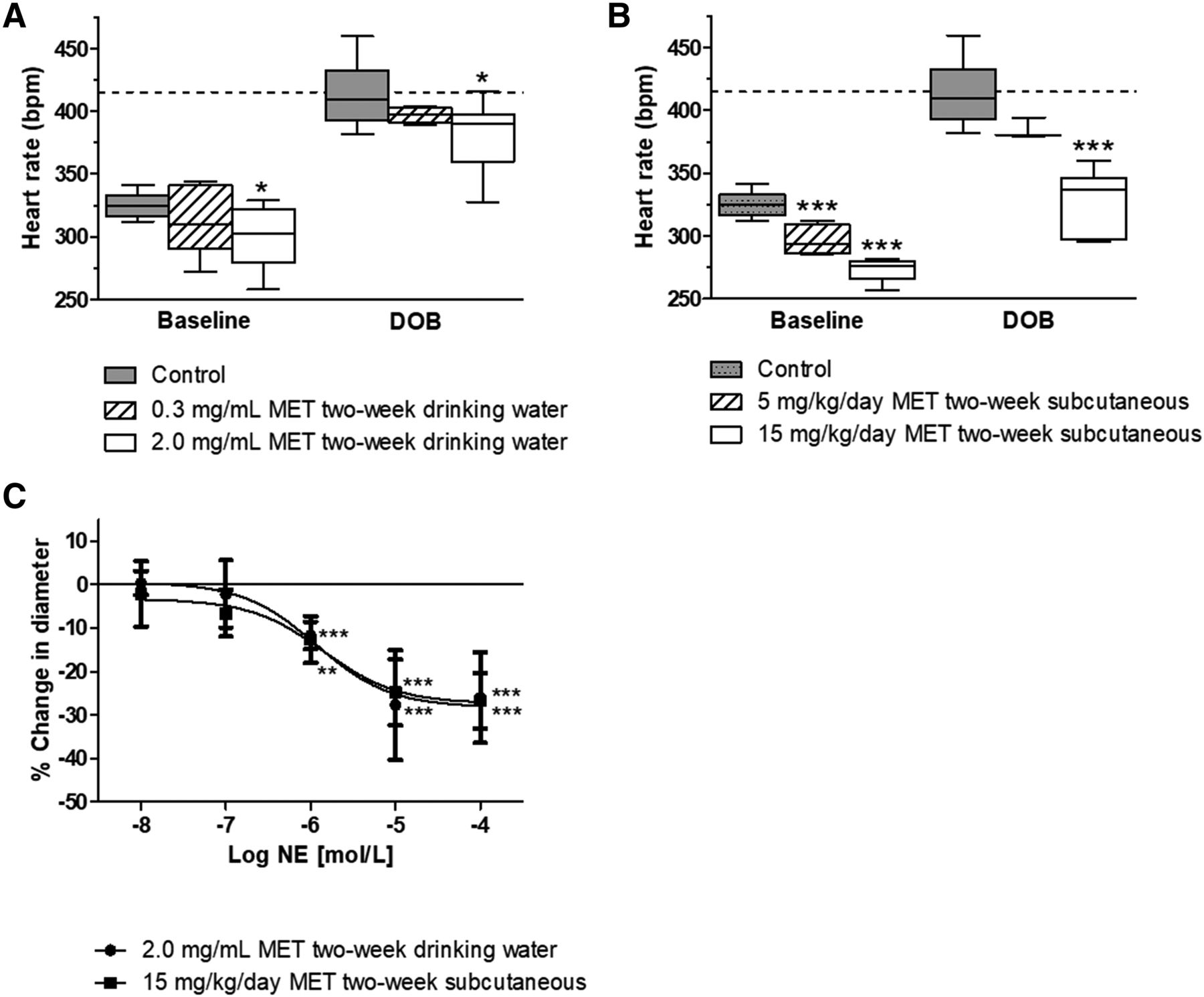
Systemic administration of MET for 2 weeks attenuates the heart rate response to DOB and results in a cerebral vasoconstrictor response to topical NE in vivo. Heart rate at baseline and in response to a DOB challenge after 2 weeks of either (A) MET administration in drinking water (0.3 or 2.0 mg/ml) or (B) MET administration by subcutaneous mini-pump (5 or 15 mg/kg per day). Data are shown only for the highest dose of DOB (18.4 μg/kg per minute) administered during the challenge; n = 11 (control), n = 11 (2.0 mg/ml MET), n = 6 (0.3 mg/ml MET), n = 6 (15 mg/kg per day MET), n = 4 (5 mg/kg per day MET). (- - -) indicates 85% of estimated maximal heart rate. One-way ANOVA with post hoc Bonferroni t tests; significant difference from control: *P < 0.05; ***P < 0.001. (C) Constriction response of middle CA branches in cranial window to suffused NE after 2-week MET treatment; n = 7 (control), n = 8 each MET. Two-way ANOVA: no differences between routes of administration (P > 0.05). Bonferroni t tests; significant difference from control: **P < 0.01; ***P < 0.001. Control data in (C) are modified from Moore et al. (2015) with permission.
Subsequently, rats dosed for 2 weeks with MET in drinking water (2.0 mg/ml) or by osmotic mini-pump (15 mg/kg per day) were subjected to cranial windows to assess the impact of 2-week MET administration on the cerebrovascular response to NE. Topical application of increasing concentrations of NE resulted in maximal constriction of cerebral arteries in vivo averaging 26.0% ± 10.4% and 26.8% ± 6.4% in rats treated with MET orally or by subcutaneous mini-pump, respectively, in contrast to a mild dilation of CAs in untreated control rats reported previously (Moore et al., 2015) (Fig. 5C). Isolated CAs from rats treated with 2.0 mg/ml MET for 2 weeks showed similar vasodilator responses to NE ex vivo as CAs from untreated control rats (Supplemental Fig. 4A), presumably reflecting the return of β1AR-mediated vasodilation after washout of MET in the perfusion chamber. Resting and Ca2+-free diameters at 80 mm Hg also were not significantly different between CAs isolated from control and MET-treated rats (Supplemental Fig. 4B). Finally, the resting diameter of CAs in cranial windows in vivo expressed as percent of the maximal dilator response to SNP was not significantly different between untreated rats and rats treated with 2.0 mg/ml MET for 2 weeks (Supplemental Fig. 4C).
Discussion
Reports of an increased risk of stroke associated with the perioperative or long-term use of β-blockers resulted in revised guidelines by the AHA/ACC and Eighth Joint National Committee that advocated to limit the use of these medications (Fleischmann et al., 2009; James et al., 2014). However, the number of prescriptions for the β1AR-selective blocker MET continues to increase each year in the United States (Aitken et al., 2014; IQVIA, 2018) despite cautionary guidelines. This dichotomy may relate to the fact that we lack a mechanistic explanation for the positive correlation between β-blockers and stroke. In this regard, the findings of the present study that acute and longer-term administration of MET disrupts β1AR-mediated cerebral vasodilation may provide clues to a mechanistic link. It is possible that the loss of β1AR-mediated cerebral vasodilation resulting from β-blocker therapy predisposes to deficits in cerebral blood flow during times of stress when circulating levels of catecholamines and tissue oxygen demands are at their highest (Myers et al., 1981; Cechetto et al., 1989; Ragoonanan et al., 2009; Akıl et al., 2015).
Our recent preclinical studies describe a powerful β1AR-dependent vasodilator pathway in rat CAs (Moore et al., 2015). Importantly, earlier studies also provide evidence that β1AR-dependent vasodilation is a feature of the human cerebral circulation. Pharmacological studies (Edvinsson et al., 1976) reported prominent β1AR-mediated relaxations in strips of human pial artery. Radioligand-binding studies (Nakai et al., 1986; Tsukahara et al., 1986) suggested coexpression of β1AR with β2AR in human basilar and middle cerebral arteries. Collectively, these studies indicate the presence of a β1AR-mediated vasodilator pathway in the human cerebral circulation, which potentially could be compromised by systemic administration of β-blockers.
Our initial experiments verified that clinically relevant plasma concentrations of MET (Ritscher et al., 2019) can antagonize β1AR-mediated vasodilation in isolated CAs, which were removed from native influences that may confound interpretation of changes in cerebrovascular reactivity in vivo (Gorshkova et al., 2011). Notably, these studies also revealed that antagonism of β1AR by MET reverses NE-elicited vasodilation to a vasoconstrictor response. A similar “NE reversal” in isolated rat CAs has been attributed to the experimental β1AR blocker CGP20712 (Moore et al., 2015) and to the nonselective β-blocker propranolol in bovine caudal CAs ex vivo (Ayajiki and Toda, 1990). This anomalous vasoconstriction to adrenergic stimulation in the presence of β-blockers may create an environment of risk for individuals predisposed to cerebral ischemia. Indeed, hemodilution studies in rats suggest that MET is associated with reduced cerebral tissue oxygen tension (Myers et al., 1981; Cechetto et al., 1989; Ragoonanan et al., 2009; Akıl et al., 2015).
Next, we designed in vivo experiments to mimic clinical MET uses as either acute or chronic therapies. Interestingly, in perioperative use of β-blockers, the risk of stroke is greatest in patients who are healthy and otherwise low-risk undergoing noncardiac surgery in which high fixed doses of β-blockers are used on the day of surgery (Fleischmann et al., 2009; Poldermans et al., 2009). Starting from standardized conversion of doses from humans to rats (Reagan-Shaw et al., 2008; Yoon et al., 2011), we evaluated two acute oral doses of MET to blunt baseline heart rate and DOB-induced tachycardia. The effective dose of MET (30 mg/kg) that antagonized cardiac β1ARs and lowered heart rate also effectively prevented ISO-induced dilation of CAs in vivo. The inhibition of CA dilation by 30 mg/kg MET was similar to that produced by a topical application of the highly selective β1AR blocker CGP20712 in our previous study (Moore et al., 2015), suggesting MET mainly acts on β1AR to limit adrenergic-mediated dilation of CAs. Under these conditions, a concomitant NE-induced contraction mediated by αAR becomes apparent, similar to our findings in isolated CAs ex vivo. Although mean arterial pressure in anesthetized rats was significantly less after an acute oral dose of 30 mg/kg MET, the resting diameters of CAs in vivo were not significantly different from untreated control rats.
In accordance with the new guidelines from American College of Cardiology Foundation and American Heart Association that limit acute perioperative use of β-blockers, these medications are now initiated at least 2 weeks before surgery rather than acutely in the preoperative period (Devereaux et al., 2018). Furthermore, MET is taken chronically for several cardiovascular conditions (Lindholm et al., 2005; Frishman, 2008; Fleischmann et al., 2009). Thus, our second set of MET dosing involved 2-week administration using two different routes: oral ad libitum in drinking water and by subcutaneous osmotic mini-pump. Although rats do most of their drinking during their nocturnal waking hours (Johnson and Johnson, 1991), the timing in individual rats in our study between the last drink of MET and the start of cranial window experiments was undoubtedly variable, presumably mimicking the situation of outpatients on oral MET, who present to clinics at different time points after their last dose of MET. On the other hand, delivery by subcutaneous osmotic mini-pumps allows for a consistent exposure to a known dose of MET, thereby eliminating the time-of-day variations in drug plasma concentration analogous to intravenous infusion of MET in inpatient settings. Unlike a single acute oral dose, prolonged use of MET is associated with upregulation of β1AR in the cardiac tissue of patients with heart failure (Heilbrunn et al., 1989). Despite this confounding factor, we again relied on the DOB challenge to find effective doses of MET that would blunt resting heart rate and DOB-induced tachycardia. At the doses of 2.0 mg/ml in drinking water or 15 mg/kg per day subcutaneously at which MET lowered resting heart rate and blunted the DOB-induced tachycardia, it also converted the mild vasodilator response to NE of CAs in vivo to a marked vasoconstriction. These results suggest that even chronic MET doses that blunt cardiac β1AR stimulation as their intended therapeutic effect concomitantly block β1AR in CAs as an off-target effect, thereby unmasking a vasoconstrictive response to adrenergic stimulation.
Roughly 80% of primary stroke events are associated with thrombosis and embolism (Albers, 1995). In this regard, MET-facilitated CA constriction by adrenergic agonists may increase the risk and/or severity of stroke in multiple ways. By reversing a physiologic vasodilation response to vasoconstriction, it may significantly elevate arterial resistance in situations wherein more blood flow to the brain is needed. Notably, a 10% change in diameter leads to >40% change in arterial resistance (Cassot et al., 1995). In the presence of an existing partial obstruction of CAs, vasoconstriction can result in a complete or near-complete blockage of blood flow causing a direct hypoperfusion or ischemia and also may significantly damage the arterial wall from turbulent blood flow. If larger CAs, such as the middle cerebral artery, are constricted, small emboli that would otherwise cause small infarcts in higher-order branches with no significant symptoms may get lodged in lower-order arteries depriving a larger area of adequate cerebral blood flow (Traverse et al., 1995). Notably, in SD rats, it has been reported that βAR density is at its highest in lower-order branches of middle cerebral arteries and shows significant loss of dilation to intravenous NE by propranolol only in the large distributive branches of pial arteries (Gorshkova et al., 2011).
There are a number of limitations in the present study. First, because of high variability in the diameters of CAs observed in cerebral windows, we could not compare resting diameters between untreated and MET-treated rats. This raised the question of whether differences in adrenergic responsiveness to NE were influenced MET treatment. Indeed, we attempted to answer this question by measuring the diameter of CA in a single animal before and after acute oral doses of MET. However, the delicacy of the brain exposed in cranial windows did not allow repositioning of the animal in the stereotaxic apparatus for oral dosing postcraniectomy. Instead, we compared resting diameters as percent of the maximal vessel diameter attained by SNP-induced vasodilation. Using this strategy, we found no statistical difference between untreated and MET-treated groups. This finding concurs with our previous observation that application of topical β-blockers to the cranial window chamber does not alter the resting diameter of rat CAs under anesthesia (Moore et al., 2015). Second, the adrenergic stimulation was pharmacologically simulated with a topical application of exogenous NE rather than direct neuronal stimulations. However, it is interesting to note that constriction of CAs occurred at 1–10 μmol/l of NE comparable with NE levels measured in the brain during stroke in rat models (Cechetto et al., 1989) or during exercise or acute illness in humans (Silverberg et al., 1978). Third, our study only examined the effect of MET treatment on cerebral adrenergic responsiveness in male rats. Potential interactions between estrogen and β1AR-signaling pathways (Machuki et al., 2018) may add an extra layer of complexity that is beyond the scope of the current studies and require additional research. Fourth, we only examined the effect of MET administration on NE-induced diameter responses of CAs. Under normal physiologic conditions, epinephrine plays a minimal role in determining cerebral arterial diameter because of its exclusion by the blood-brain barrier (Weil-Malherbe et al., 1959; Myburgh et al., 2002). However, after the onset of a pathologic state, such as after ischemic stroke, plasma epinephrine concentrations acutely increase (Meloux et al., 2018), and epinephrine may gain access to cerebral arteries because of increased blood-brain barrier permeability (Liebner et al., 2018). Thus, the effects of MET on the adrenergic responsiveness of CAs may potentially be different in disease states associated with a disrupted blood-brain barrier. This possibility draws attention to the fact that our study explored the effect of MET treatment only in young, healthy SD rats rather than rats with high blood pressure or in a rat model of stroke.
It is important to acknowledge that the findings of our study are specific to second-generation, β1AR-selective blockers, such as MET, and should not be generalized to all forms of β-blocker therapies. The newer third-generation β-blockers include drugs that exhibit pharmacological properties additional to βAR blockade, such as nitric oxide production, β2AR agonism, α1AR antagonism, Ca2+ channel blockade, and antioxidant activity (Brunton et al., 2011). Importantly, these third-generation “vasodilating” β-blockers have shown a better stroke risk profile compared with second-generation “cardioselective” β1AR blockers (Poole-Wilson et al., 2003; Remme et al., 2007a,b). However, MET continues to rank the highest of all β-blockers in prescription volume in the United States, whereas no third-generation β-blockers were included in the top 20 (IQVIA, 2018). Investigation of the effects of vasodilating β-blockers on the cerebral arteries represents an important future direction.
In conclusion, we have provided evidence that systemic administration of MET at clinically relevant doses directly blunts β1AR-mediated dilation of rat CAs resulting in abnormal vasoconstriction to adrenergic stimulation by NE. These results suggest β1AR-mediated vasodilation in CAs represents a significant off-target site of action for “cardioselective” β1AR blockers. This effect offers a possible mechanism to account for how conventional β-blocker therapy may increase the risk of stroke as observed in clinical settings.
Authorship Contributions
Participated in research design: Moore, Henry, Rusch, Rhee.
Conducted experiments: Moore, Henry, McClenahan, Ball.
Performed data analysis: Moore, Henry, McClenahan, Ball, Rhee.
Wrote or contributed to the writing of the manuscript: Moore, Henry, Rusch, Rhee.
Footnotes
- Received June 19, 2020.
- Accepted October 16, 2020.
↵1 C.L.M. and D.S.H. contributed equally to this work.
↵2 Current affiliation: Vanderbilt University, Nashville, Tennessee.
This work was supported in part by National Institutes of Health National Heart, Lung, and Blood Institute [R01-HL97107] and American Heart Association [Grant 17GRNT33670970], [Grant 19TPA34880019] (S.W.R.), and [Grant 13PRE17070035] (C.L.M.).
The authors declare no conflicts of interest.
Portions of this work were previously presented in: Moore CL (2015) The role of postsynaptic density-95 scaffolding in cerebral vasodilation: Implications for stroke in beta-blocker therapy. Doctoral dissertation. Figures 4, A and B and 5C modified from Moore CL, McClenahan SJ, Hanvey HM, Jang DS, Nelson PL, Joseph BK, and Rhee SW (2015) J Cereb Blood Flow Metab 35:1537–1546. Reprinted by Permission of SAGE Publications, Ltd.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AR
- adrenergic receptor
- CA
- cerebral artery
- DOB
- dobutamine
- ISO
- isoproterenol
- MET
- metoprolol
- NE
- norepinephrine
- PSS
- physiological salt solution
- SD
- Sprague-Dawley
- SNP
- sodium nitroprusside
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}