


Abstract
There is significant need to find effective, nonaddictive pain medications. κ Opioid receptor (KOPr) agonists have been studied for decades but have recently received increased attention because of their analgesic effects and lack of abuse potential. However, a range of side effects have limited the clinical development of these drugs. There are several strategies currently used to develop safer and more effective KOPr agonists. These strategies include identifying G-protein–biased agonists, developing peripherally restricted KOPr agonists without centrally mediated side effects, and developing mixed opioid agonists, which target multiple receptors at specific ratios to balance side-effect profiles and reduce tolerance. Here, we review the latest developments in research related to KOPr agonists for the treatment of pain.
SIGNIFICANCE STATEMENT This review discusses strategies for developing safer κ opioid receptor (KOPr) agonists with therapeutic potential for the treatment of pain. Although one strategy is to modify selective KOPr agonists to create peripherally restricted or G-protein–biased structures, another approach is to combine KOPr agonists with μ, δ, or nociceptin opioid receptor activation to obtain mixed opioid receptor agonists, therefore negating the adverse effects and retaining the therapeutic effect.
Introduction
The rapid increase in the use of opioid drugs in the United States has been termed the “opioid crisis,” with over 47,000 opioid-related deaths in 2017 (Scholl et al., 2018). Opioids prescribed for treating moderate-to-severe pain act primarily through the μ opioid receptor (MOPr) (Vallejo et al., 2011). Activation of the MOPr stimulates the mesocorticolimbic reward pathway, thereby increasing dopamine levels (Di Chiara and Imperato, 1988), and results in positive reinforcement (Maldonado et al., 1997). In the search for nonaddictive analgesics, κ opioid receptor (KOPr) agonists are a promising alternative. In contrast to MOPr agonists, KOPr agonists play a critical role in regulating the reward system by contributing to the negative feedback of dopamine (Di Chiara and Imperato, 1988), and unlike MOPr agonists, they do not cause respiratory depression (Freye et al., 1983).
κ Opioid Receptor Signaling and the Role in Pain
Activation of the KOPr is associated with regulation of the reward pathway, antinociception, and anxiogenic and stress-related behaviors [reviewed in Wang et al, (2010), Lalanne et al. (2014)]. The KOPr is a class A (rhodopsin-like) γ subfamily of seven-transmembrane G-protein–coupled receptors. Activation of the KOPr leads to conformational changes and dissociation of the pertussis toxin–sensitive G-protein subunits, thereby activating G-protein–gated inwardly rectifying potassium channels (Grudt and Williams, 1993) and inhibiting voltage-gated calcium ion channels (Rusin et al., 1997), which leads to the hyperpolarization of the neuron. The Gα subunit inhibits adenylyl cyclase activity, leading to a decrease in cAMP (Taussig et al., 1993) and phosphorylation of c-Jun N-terminal kinase and extracellular signal-regulated kinase 1 and 2 (ERK1/2) (Belcheva et al., 2005). KOPrs inhibit pain signals in the spinal cord and the brain stem (Porreca et al., 1984; Ruda et al., 1988; Simonin et al., 1995).
KOPr activation also activates a β-arrestin–dependent signaling cascade. The C-terminal intracellular domain is phosphorylated by G-protein receptor kinase 3, and the β-arrestin scaffolding proteins are recruited, leading to the phosphorylation of p38 mitogen-activated protein kinase and activation of the transcription factor cAMP response element binding protein (McLaughlin et al., 2003; Bruchas et al., 2006). Prodepressive and aversive effects of KOPr agonists have been attributed to the phosphorylation of p38 mitogen-activated protein kinase (Bruchas et al., 2007, 2011; Ehrich et al., 2015) and cAMP response element binding protein activity (Pliakas et al., 2001; Mague et al., 2003). Phosphorylation of KOPr leads to the internalization of the receptor (Schulz et al., 2002), contributing to KOPr agonist tolerance (McLaughlin et al., 2004; Chiu et al., 2017).
KOPr knockout mice were generated by Simonin et al. (1998). These animals have been tested in a range of behavioral models of pain, showing increased sensitivity in the acetic acid writhing test, thus indicating the KOPr system is involved in the perception of visceral pain. The endogenous agonists for the KOPr are the dynorphin class of opioid peptides (Goldstein et al., 1979; Chavkin et al., 1982). The natural peptide has 17 amino acids (dynorphin A1–17); however, the shortened 13-amino-acid fragment (dynorphin A1–13) is often used in biologic studies (Chou et al., 1996). Intrathecal injection of dynorphin A1–13 in the rat spinal cord had an antinociceptive effect 6–10 times more potent than morphine on a molar basis (Han and Xie, 1982), and morphine in combination with dynorphin A1–13 produced a synergistic antinociceptive effect in the tail-withdrawal assay (Ren et al., 1985).
In the reward centers, such as the nucleus accumbens, KOPr activation regulates dopamine release and increases uptake by the dopamine transporter (Di Chiara and Imperato, 1988; Kivell et al., 2014). This mechanism is responsible for the antiaddiction effects of KOPr agonism; however, aversive effects can also be induced (Wee and Koob, 2010), and drug-taking can be escalated due to KOPr activation (Schlosburg et al., 2013). During pain, negative affect states may be mediated by recruitment of dynorphin neurons and action through the KOPr system in the nucleus accumbens (Massaly et al., 2019). In a spinal nerve ligation model, experiments using prodynorphin knockout mice showed dynorphin may be required for the maintenance of neuropathic pain (Wang et al., 2001). Further studies have shown that the KOPr system is involved in the aversive component of neuropathic pain (Liu et al., 2019b; Meade et al., 2020).
Traditional Arylacetamide κ Opioid Receptor Agonists
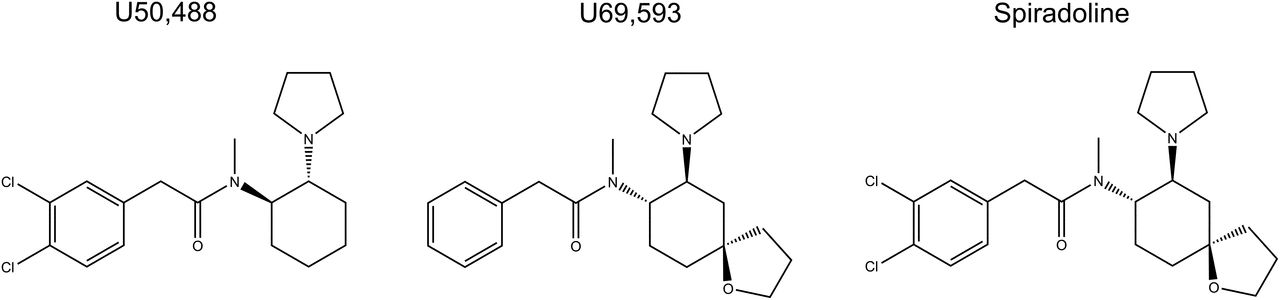
The prototypical arylacetamide KOPr agonists include U50,488 (Von voigtlander and Lewis, 1982), U69,593 (Lahti et al., 1985), and spiradoline (also known as U62,066E; Fig. 1) (Vonvoigtlander and Lewis, 1988). The KOPr agonists have demonstrated antinociceptive effects in several thermal, inflammatory, and neuropathic pain models (Vonvoigtlander et al., 1983; Calcagnetti et al., 1988; La Regina et al., 1988; Vonvoigtlander and Lewis, 1988; Kunihara et al., 1989; Pelissier et al., 1990; France et al., 1994; Wilson et al., 1996; Bartok and Craft, 1997; Catheline et al., 1998; Binder et al., 2001; Bileviciute-Ljungar and Spetea, 2004; Bileviciute-Ljungar et al., 2006; Gallantine and Meert, 2008; Negus et al., 2008; Auh and Ro, 2012). Unfortunately, these KOPr agonists also have side effects. U50,488 and U69,593 produce prodepressive effects (Mague et al., 2003; Zhang et al., 2015), aversion (Mucha and Herz, 1985; Suzuki et al., 1992; Bals-Kubik et al., 1993; Skoubis et al., 2001; Ehrich et al., 2015), anxiety (Privette and Terrian, 1995; Kudryavtseva et al., 2004; Vunck et al., 2011; Wang et al., 2016), muscle weakness, and sedation (Dykstra et al., 1987; Zhang et al., 2015). In clinical studies, spiradoline produced diuresis, sedation, and dysphoria (Ur et al., 1997; Wadenberg, 2003). Because of these side effects, these arylacetamide KOPr agonists have not been developed further in the clinical setting.

Chemical structure of the traditional arylacetamide κ opioid receptor agonists, U50,488, U69,593, and Spiradoline.
Strategies to Develop κ Opioid Receptor Agonists with Reduced Side Effects
There are several strategies used to develop novel, safer, and more effective KOPr agonists. These strategies include identifying G-protein–biased agonists, developing peripherally restricted KOPr agonists without centrally mediated side effects, and developing mixed opioid agonists to target multiple receptors at specific ratios to balance side-effect profiles and reduce tolerance.
G-Protein–Biased κ Opioid Receptor Agonists
Evidence suggests that many KOPr side effects are mediated through β-arrestin–dependent pathways (Bruchas and Chavkin, 2010). For instance, in mice lacking G-protein receptor kinase 3, aversion is absent (Bruchas et al., 2007). Therefore, developing biased KOPr agonists with preferential activation of the G-protein pathway has received significant attention [reviewed by Mores et al. (2019)]. Biased agonism is calculated by evaluating activation in both G-protein and β-arrestin signaling pathways and comparing affinity and efficacy to a reference agonist with balanced signaling properties. The biased factors of common KOPr agonists are presented in Table 1.
κ Opioid receptor agonists with G-protein bias
1) White et al., 2014. 2) Dunn et al., 2019. 3) Dunn et al., 2018. 4) DiMattio et al., 2015. 5) White et al., 2015. 6) Liu et al., 2019a. 7) Schattauer et al., 2017. 8) Kaski et al., 2019. 9) Kivell et al., 2018. 10) Zhou et al., 2013. 11) Lovell et al., 2015. 12) Brust et al., 2016. 13) Ho et al., 2018. 14) Schmid et al., 2013. 15) Stahl et al., 2015. 16) Spetea et al., 2017. 17) Bedini et al., 2020.
However, application of the theoretical idea of biased agonism has proven more difficult than anticipated (Michel and Charlton, 2018). When comparing studies, there is a lack of consistency in signaling assays, cell types, and choice of reference ligands, including use of U50,488 (Schattauer et al., 2017; Kivell et al., 2018), U69,593 (Ho et al., 2018; Dunn et al., 2019), Salvinorin A (SalA) (White et al., 2015), or the endogenous peptide dynorphin A1–17 (DiMattio et al., 2015). In addition, receptors from different species have different signaling properties. DiMattio et al. (2015) calculated the bias factor in N2A cells stably transfected with either the human or mouse KOPr. SalA was significantly β-arrestin–biased for the human receptor; however, the mouse KOPr had similar activation of both pathways. On the contrary, U50,488 was β-arrestin–biased at the mouse receptor but was unbiased at the human KOPr.
System bias (cell background) and observational bias (assay conditions) in experimental design create further complications (Gundry et al., 2017). Observational bias problems include not accounting for intrinsic efficacy and using both amplified and linear signaling measurements (Gillis et al., 2020b). Using in vitro models, there is often high receptor expression, thus commonly producing a ceiling effect within the assay whereby most agonists will reach the maximal effect, therefore not allowing for comparisons of efficacy. When receptor pools are lowered, the intrinsic efficacy of the agonist may be measured. G-protein measurements often use amplified assays (such as cAMP), whereas β-arrestin recruitment assays are not amplified. In this situation, the results are skewed toward G-protein bias. Furthermore, the kinetic context (Klein Herenbrink et al., 2016), receptor occupancy, and receptor conformation (Kenakin, 2014) are not often considered. Most experiments assess an agonist at a single time point, which may be based on the reference ligand. However, this does not allow for the complexity of the binding kinetics and transient signaling responses (Klein Herenbrink et al., 2016). As seen in Table 1, the most commonly used model to calculate bias is the operational model (Black and Leff, 1983); however, for this calculation to be correct, system and observational bias must not be present in the data. Many laboratories are addressing system bias issues and are beginning to evaluate signaling in more relevant cellular models, including primary neurons (Jamshidi et al., 2015; Ho et al., 2018; Ehrlich et al., 2019). A major limiting factor is the inability to evaluate signaling bias in vivo, although advances are ongoing, such as measuring cAMP signaling in mice (Muntean et al., 2018).
The MOPr has been extensively studied [for recent review see Grim et al. (2020)] and may be able to guide the development of G-protein–biased KOPr agonists. Raehal et al. (2005) showed the respiratory depressive effects and constipation caused by morphine were not present in β-arrestin-2 knockout mice. This led to much attention on developing G-protein–biased agonists. However, it was recently shown in three different laboratories that the respiratory depressive effects of morphine were independent of β-arrestin-2 signaling (Kliewer et al., 2020). There is a similar story with the compound PZM21, which was originally shown to have few side effects (Manglik et al., 2016) and has since been shown to have respiratory depression (Hill et al., 2018). Finally, the G-protein–biased MOPr agonist oliceridine (TRV130 or Olinvyk) completed phase III clinical trials; originally, the drug was not approved because of safety concerns (https://www.fda.gov/media/121233/download). Interestingly, Gillis et al. (2020a) found that low intrinsic efficacy rather than G-protein bias could explain the reduced side effects of oliceridine and PZM21, therefore leaving the question as to whether apparent G-protein–biased MOPr agonists need to be reassessed under these experimental conditions. Regardless of mechanism, evaluation of drugs at higher doses in vivo is required to fully assess both safety and side effects.
These discrepancies need to be resolved before conclusions can be drawn on the role of G-protein–biased KOPr agonists for treating pain with fewer side effects. However, several KOPr agonists have been identified with improved side-effect profiles.
Nalfurafine.
Nalfurafine (or TRK-820, Fig. 2) is the first selective KOPr agonist to be clinically approved for medication-resistant pruritus in patients with hemodialysis in Japan (Kumagai et al., 2010). Nalfurafine has antinociceptive and antipruritic effects (Endoh et al., 1999, 2000) and does not produce anhedonia or aversion (Liu et al., 2019a). The success of nalfurafine has demonstrated it is possible to develop KOPr agonists to be used in a clinical setting. Many studies have sought to measure the bias factor to understand the lack of side effects traditionally associated with KOPr agonists; however, there have been mixed results (Table 1). In human embryonic kidney-293 (HEK-293) cells, nalfurafine was a G-protein–biased agonist compared with U50,488 (Kaski et al., 2019), with moderate G-protein bias at the rat KOPr and extreme G-protein bias at the human KOPr (Schattauer et al., 2017). However, a recent paper found nalfurafine was a balanced agonist compared with U50,488 as the reference ligand (Liu et al., 2019a), and another found nalfurafine was β-arrestin-2–biased, with 20-fold–higher β-arrestin signaling than U69,593 in U2OS cells (Dunn et al., 2019).

Chemical structures of the G-protein–biased κ opioid receptor agonists.
Salvinorin A Analogs.
The neo-clerodane diterpene SalA (Fig. 2) is a KOPr agonist derived from Salvia divinorum, a Sage plant native to Mexico (Valdés, 1994; Roth et al., 2002). SalA has antinociceptive effects in thermal (John et al., 2006; McCurdy et al., 2006; Paton et al., 2017; Sherwood et al., 2017), visceral (McCurdy et al., 2006; Fichna et al., 2012), inflammatory (Aviello et al., 2011; Fichna et al., 2012; Guida et al., 2012; Paton et al., 2017), and neuropathic pain models (Coffeen et al., 2018). However, side effects include aversion (Zhang et al., 2005), anxiety (Braida et al., 2009), prodepressive effects (Carlezon et al., 2006), motor incoordination (Fantegrossi et al., 2005), sedation (Butelman et al., 2009), and learning and memory impairments (Braida et al., 2011). SalA also has a short duration of action in vivo (Butelman et al., 2007; Ranganathan et al., 2012). All these effects have limited the clinical usefulness of SalA. However, the structural scaffold of SalA has identified multiple analogs with improvements in metabolic stability and side-effect profile.
Modifications at the carbon-2 position have led to increased potency by adding metabolism-protective groups and removing the hydrolysable ester (Munro et al., 2008). These include β-tetrahydropyran (β-THP) Salvinorin B (SalB), Mesyl SalB, and ethoxymethyl ether (EOM) SalB (Fig. 2). Mesyl SalB and EOM are G-protein–biased at the human KOPr using U50,488 as a reference ligand (Kivell et al., 2018; Kaski et al., 2019). However, EOM SalB was also found to be β-arrestin–biased at the mouse KOPr with dynorphin A1–17 as the reference ligand (DiMattio et al., 2015).
Compared with SalA, EOM SalB has higher metabolic stability in the rat liver microsome assay (Ewald et al., 2017), and brain concentrations declined at a slower rate in baboons and rats (Hooker et al., 2009). EOM SalB, Mesyl SalB, and β-THP SalB all have a longer duration of action than SalA in the warm-water tail-withdrawal assay in mice (Simonson et al., 2015; Paton et al., 2017). β-THP SalB reduced mechanical and cold allodynia in the chemotherapy-induced neuropathic pain model and significantly reduced formaldehyde-induced pain behaviors (Paton et al., 2017), whereas Mesyl SalB had minimal effects (Kivell et al., 2018). Mesyl SalB produced no aversion, anxiety, or learning and memory impairments (Kivell et al., 2018). EOM SalB did not cause sedation, anxiety, or depressive-like effects in rodents (Ewald et al., 2017). However, in discrimination studies, EOM SalB substituted for both U69,593 (Baker et al., 2009) and SalA in rats (Peet and Baker, 2011).
A semisynthetic analog, RB-64 (22-thiocyanatosalvinorin A, Fig. 2) (Yan et al., 2009), is G-protein–biased compared with SalA as the reference ligand (White et al., 2014, 2015). In C57BL/6J mice, RB-64 had antinociceptive effects in the hot-plate (55°C) assay and did not show anhedonia-like effects in the intracranial self-stimulation test or locomotor deficits in the Rotarod performance assay or novelty-induced locomotion test (White et al., 2015). However, RB-64 did have aversive effects in the conditioned place aversion (CPA) paradigm, an effect previously believed to be β-arrestin–mediated (Bruchas and Chavkin, 2010; White et al., 2015).
Collybolide.
Collybolide (Fig. 2) is a selective KOPr agonist extracted from the mushroom Collybia maculata (Gupta et al., 2016). The chemical structure is similar to SalA in that both are terpene compounds containing a furyl-δ-lactone structure. In HEK-293 cells expressing the human KOPr, displacement of the [3H]U69,593 radioligand showed binding affinity (Ki) values of 40 ± 10 nM for SalA compared with 9 ± 2 nM for Collybolide (Gupta et al., 2016). In mice, Collybolide had similar antinociceptive effects to SalA in tail withdrawal, was not prodepressive in the forced swim test, did not affect locomotor activity in open field tests, and was not anxiogenic in the elevated plus maze; however, there was significant aversion in the CPA test (Gupta et al., 2016). Moreover, Collybolide, unlike SalA, also attenuated chloroquine-mediated pruritus (Gupta et al., 2016).
Triazole 1.1.
A high-throughput screening process was used to identify selective KOPr agonists in a library of 300,000 compounds (Frankowski et al., 2012). Triazole 1.1 (Fig. 2) was identified as a G-protein–biased KOPr agonist compared with U69,593 (Zhou et al., 2013; Lovell et al., 2015). Triazole 1.1 displayed antinociceptive effects in the warm-water tail-withdrawal assay (Zhou et al., 2013) and antipruritic effects in response to chloroquine phosphate (Brust et al., 2016). Moreover, Triazole 1.1 showed an improved side-effect profile. It did not alter locomotor activity nor induce dysphoria in the intracranial self-stimulation assay, did not lower dopamine levels in the nucleus accumbens (Brust et al., 2016), and did not induce sedation or motor impairment in male rhesus monkeys (Huskinson et al., 2020).
6′-Guanidinonaltrindole.
The naltrindole derivative 6′-guanidinonaltrindole (6′-GNTI) (Fig. 2) is a potent partial KOPr agonist in G-protein activation assays, with no activity in β-arrestin recruitment assays (Rives et al., 2012). In fact, 6′-GNTI inhibited both the β-arrestin recruitment and the KOPr internalization actions of the KOPr agonist ethylketocyclazocine (Rives et al., 2012). Furthermore, in striatal neurons, 6′-GNTI induced the phosphorylation of Akt but not ERK1/2 compared with the traditional agonist U69,593, which activated both kinases (Schmid et al., 2013). It appears that the δ opioid receptor (DOPr) has a role in the function of 6′-GNTI, either because of actions on KOPr/DOPr heterodimers or convergence of downstream signaling pathways (Waldhoer et al., 2005; Jacobs et al., 2019). 6′-GNTI has antinociceptive effects in the radiant-heat tail-withdrawal assay in male mice (Waldhoer et al., 2005) and did not display CPA (Zangrandi et al., 2016).
Diphenethyalmine Derivatives.
The diphenethylamine derivatives, HS665 (also known as MCBPHA) and HS666 (Fig. 2), are selective KOPr agonists (Spetea et al., 2012) displaying G-protein bias with U69,593 as the reference agonist (Spetea et al., 2017; Dunn et al., 2018). Both compounds produced dose-dependent antinociception in the warm-water (55°C) tail withdrawal and did not produce motor incoordination in Rotarod assays (Spetea et al., 2017). HS665 also had antinociceptive effects in the acetic acid–induced writhing test in mice (Spetea et al., 2012). HS665 had aversive side effects; however, HS666 did not show either aversion or preference in a counterbalanced conditioned place paradigm (Spetea et al., 2017). Another derivative, N-n-butyl-N-phenylethyl-N-3-hydroxyphenylethyl-amine (BPHA), did not recruit β-arrestin compared with HS665, which showed submaximal β-arrestin signaling, and this was correlated with altered motor coordination for HS665 but not BPHA (Dunn et al., 2018, 2019). Additional diphenethylamine derivatives have been developed with promising antinociceptive effects in the acetic acid–induced writhing assay without causing motor incoordination (Erli et al., 2017).
LOR17.
LOR17 is a novel peptidic KOPr agonist displaying extreme G-protein bias with U50,488 as the reference ligand (Bedini et al., 2020). In male CD-1 mice, LOR17 had similar antinociceptive effects to U50,488 in the warm-water tail withdrawal and acetic acid–induced writhing test, whereas LOR17 was more effective at reducing thermal hypersensitivity in the oxaliplatin-induced neuropathic pain model (Bedini et al., 2020). LOR17 did not affect motor coordination in the Rotarod test or exploratory behavior in the hole-board test and did not have prodepressant effects in the forced swim test (Bedini et al., 2020).
Peripherally Restricted κ Opioid Receptor Agonists
Another strategy to remove the centrally mediated side effects is to create peripherally restricted KOPr agonists that do not cross the blood-brain barrier. This strategy relies on the premise that activation of peripheral KOPrs alone can produce a meaningful analgesic effect. Peripherally restricted compounds can be beneficial to patients suffering from visceral or neuropathic pain. However, many of the compounds tested only have moderate antinociceptive effects. One of the first developed was ICI 204,448 (Fig. 3) (Shaw et al., 1989). In a rat model of neuropathic pain, local injection of ICI 204,448 reduced antinociceptive behaviors after sciatic nerve constriction injury (Keïta et al., 1995). However, ICI 204,448 had minimal effects in the formaldehyde-induced inflammatory pain model in mice when administered via intraperitoneal injection (1–2 mg/kg) (Paton et al., 2017) and minimal antinociceptive activity after oral administration (Barber et al., 1994).

Chemical structures of the peripherally restricted κ opioid receptor agonists.
Another KOPr agonist with low central penetration is asimadoline (Fig. 3; also known as EMD 61753) (Barber et al., 1994; Gottschlich et al., 1995). Asimadoline is orally active, although it had more potent antinociceptive activity after subcutaneous administration (Barber et al., 1994). When it advanced into human testing, oral administration of asimadoline (7.5 mg) had no effect in hyperalgesia models using radiant heat and mechanical stimuli (Bickel et al., 1998). Furthermore, when tested in patients with postoperative pain, some of those administered 10 mg orally reported increased pain levels, and therefore, asimadoline was considered to be less tolerable than placebo (Machelska et al., 1999). Asimadoline is currently in development by Tioga Pharmaceuticals for the treatment of atopic dermatitis [reviewed in Abels and Soeberdt (2019)].
CR845 (also known as FE-202845 or Difelikefalin) and CR665 (also known as FE-200665 or JNJ-38488502) are peripherally restricted tetrapeptide KOPr agonists (Fig. 3) in development by Cara Therapeutics (Olesen et al., 2013). CR845 is in phase III clinical trials as a treatment of postoperative pain and uremic pruritus (Beck et al., 2019a). There are promising results in animal models, with CR845 reducing writhing behaviors, abdominal pain, inflammatory pain, and mechanical allodynia in a spinal nerve ligation model of neuropathic pain without inducing gastrointestinal side effects (Gardell et al., 2008). However, there is a lack of peer-reviewed papers on the clinical efficacy of this compound (Hesselink, 2017). CR665 has antinociceptive effects in the complete Freund’s adjuvant-induced model of inflammatory pain (Binder et al., 2001) and the acetic acid–induced writhing test in mice, with a 548-fold–higher dose required to induce centrally mediated motor incoordination effects, indicating it does not readily cross the blood-brain barrier (Vanderah et al., 2008). Intravenous CR665 (0.36 mg/kg) was effective at reducing visceral pain in a human model of esophageal distension (Arendt-Nielsen et al., 2009); however, CR665 (0.42 mg/kg, i.v.) did not reduce pain in a colonic distension model (Floyd et al., 2009).
CR665 is not orally active, and therefore, structural alterations have been made to improve the oral bioavailability. One of the derivatives (compound 9) reduced acetic acid–induced writhing behaviors in male Sprague-Dawley rats with no effect on centrally mediated hot-plate pain after oral administration (30 mg/kg), indicating a peripheral site of action (Hughes et al., 2013). This compound has been renamed JT09 (Fig. 3) and is currently in development by JT Pharmaceuticals. In the follow-up paper, the acetic acid writhing test was repeated, showing that 20 mg/kg JT09 via oral gavage had the same effect as 10 mg/kg morphine (Beck et al., 2019b). JT09 was not self-administered by rats and showed no CPA, indicating no rewarding effects (Beck et al., 2019b). Finally, there were no prodepressive effects in the forced swim test model, nor sedative effects in the spontaneous locomotor test (Beck et al., 2019b). The authors state that further work is planned to understand the pharmacodynamics of JT09 and to assess the antinociceptive effects in chronic pain models.
Peripherally restricted derivatives of nalfurafine are also in development by Toray Industries (Suzuki et al., 2017). There are a range of compounds with an increased number of hydrogen bond donors, including 17-hydroxy-cyclopropylmethyl (compound 8) and 10α-hydroxy (compound 10) (Fig. 3), yielding promising results. The two compounds are highly selective for the KOPr over the MOPr; in fact, compound 8 had >5,200,000 times greater selectivity for the KOPr (Suzuki et al., 2017). To assess brain penetration, the brain-plasma concentration ratio (Kp,brain) was calculated 15 minutes after intravenous injection in male ICR mice. Nalfurafine had a ratio of 0.41, and the novel compounds had lower brain penetrations; compound 8 was 0.11, and compound 10 was 0.07 (Suzuki et al., 2017). Finally, the compounds were tested in the acetic acid writhing model in mice, with both producing dose-dependent antiallodynic effects (Suzuki et al., 2017). These hydroxy nalfurafine compounds have encouraging results so far; however, further in vivo experiments are required to fully evaluate the antinociceptive potential and confirm there are no centrally mediated side effects.
Despite the number of compounds that have been tested, there has been some criticism of this strategy. One limitation with developing peripherally restricted pain medications is that the blood-brain barrier must be intact; however, the blood-brain barrier becomes more permeable in some chronic pain conditions (DosSantos et al., 2014). Furthermore, peripherally restricted KOPr agonists can reduce pain-related stimulation of behavior; however, there is a lack of evidence that these compounds block pain-related depression of behavior (Negus, 2019). In Sprague-Dawley rats, two peripherally restricted KOPr agonists, ICI 204,448 and the tetrapeptide ffir, had weak and no antinociceptive effect, respectively, in a lactic-acid depressed intracranial self-stimulation assay (Negus et al., 2012), whereas the nonsteroidal anti-inflammatory drug, ketoprofen, had significant effect in this assay (Negus et al., 2012). Further studies are required to understand the effect of peripherally restricted KOPr agonists in these assays of pain-related depression of behavior.
Mixed κ Opioid Receptor Agonists
The final strategy used to overcome the undesirable effects of KOPr agonism is to target multiple opioid receptors simultaneously. The four classes of opioid receptors MOPr, KOPr, DOPr, and nociceptin opioid receptors (NOPrs) all modulate pain (McDonald and Lambert, 2005; Dietis et al., 2011; Darcq and Kieffer, 2018). Mixed opioid receptor agonists may be a viable strategy to develop analgesics with reduced side effects (Balboni et al., 2002; Váradi et al., 2016; Majumdar and Devi, 2018). Mixed agonists could maintain antinociceptive effects, with the KOPr-mediated dysphoric or aversive-like effects balanced by the euphoric properties of MOPr or DOPr activation.
Mixed KOPr/MOPr Compounds
KOPr/MOPr Full Agonism.
8-Carboxamidocyclazocine (8-CAC) is a full agonist at both KOPr and MOPr, with potent, long-acting antinociceptive effects in male ICR mice at nanomolar doses in warm-water tail-withdrawal and acetic acid–induced writhing tests (ED50 = 0.21 nmol, i.c.v.) (Bidlack et al., 2002). In addition, 8-CAC produced antinociception that lasted for 15 hours in the writhing test (Bidlack et al., 2002). Acute administration of 8-CAC blocked cocaine-maintained responding and decreased food–maintained responding. In contrast, chronic administration increased cocaine self-administration in rhesus monkeys (Stevenson et al., 2004), indicating that 8-CAC may have abuse potential when administered chronically.
Combined KOPr Agonism and Partial MOPr Agonism.
Butorphan (MCL-101), is a KOPr agonist with partial MOPr agonist actions that reduced the rewarding effects of cocaine in rats (Provencher et al., 2013) and rhesus monkeys (Bowen et al., 2003). Neumeyer et al. (2000) found that Butorphan produced potent antinociceptive effects in male ICR mice in warm-water tail-withdrawal (ED50 = 7.3 nmol, i.c.v.) and acetic acid–induced writhing tests (ED50 = 0.79 nmol, i.c.v., Table 2).
i.pl., intraplantar; SNL, spinal nerve ligation; STZ, streptozotocin; CFA, Complete Freund's adjuvant; p.o., per os. Analgesic and antinociceptive effects of mixed KOPr agonists
1) Bidlack et al., 2002; 2) Neumeyer et al., 2000; 3) Aldrich et al., 2013; 4) Brice-Tutt et al., 2020; 5) Pick et al. 1992; 6) Patrick et al., 1999; 7) Wong and Wai, 1984; 8) Ortiz et al., 2007; 9).Yoa-Pu et al., 1998; 10) Kshirsagar et al., 2008; 11) Wang et al., 2009; 12) Sun et al., 2010; 13) Fillingim et al., 2004; 14) Lotsch et al., 1997; 15) Codd et al., 1995; 16) Paul et al., 1991; 17) Lasagna and Beecher, 1954; 18) Keats and Telford, 1956; 19) Stevens, 1996; 20) Sibille et al., 2011; 21) Yadav et al., 2018; 22) Dönselmann Im Sande et al., 2017; 23) Singh et al., 2017; 24) Williams et al., 2016; 25) Zimmerman et al., 1987; 26) Yekkirala et al., 2011; 27) Tang et al., 2010; 28) Waldhoer et al., 2005; 29) Jacobs et al., 2018; 30) Daniels et al., 2005; 31) Váradi et al., 2015; 32) Ulker et al., 2020; 33) Glazebrook, 1952; 34) Morrison et al., 1971; 35) Rowbotham et al., 2003; 36) Le Rouzic et al., 2019; 37) Lattanzi et al., 2018; 38) Gringauz et al., 2001; 39) Linz et al., 2017; 40) Schiene et al., 2018; 41) Christoph et al., 2018; 42) Linz et al., 2014; 43) Schunk et al., 2014; 44) Dahan et al., 2017; 45) Scholz et al., 2018; 46) Christoph et al., 2017; 47) Koch et al., 2019.
The macrocyclic tetrapeptide CJ‐15,208 (cyclo [Phe‐D‐Pro‐Phe‐Trp]) is a natural product with centrally acting multifunctional KOPr/MOPr agonist and KOPr antagonist actions. CJ‐15,208 produced antinociception without displaying hypolocomotor effects in the Rotarod test in mice after oral administration (Ross et al., 2010; Aldrich et al., 2013). Its analog, cyclo [Pro‐Sar‐Phe‐D‐Phe] also displayed similar antinociceptive effects with reduced side effects compared with morphine (Brice-Tutt et al., 2020; Ferracane et al., 2020). Of interest, the alanine analogs of [D-Trp]CJ-15,208 displayed pharmacological profiles in vivo that were distinctly different from the parent compound. Although the analogs exhibited varying opioid receptor activities in vitro, they produced potent opioid receptor–mediated antinociception (ED50 = 0.28–4.19 nmol, i.c.v.) in vivo in mice (Aldrich et al., 2014).
Discrepancies between in vitro binding affinity and in vivo efficacy and potency are not uncommon, and it is possible that metabolism may account for many of these observed differences. Another possibility is allosteric modulation. For example, BMS-986122 is a positive allosteric modulator for MOPr, and BMS-986187 is a structurally distinct positive allosteric modulator for DOPr (with 100-fold selectivity in promoting DOPr over MOPr). Livingston et al. (2018) also provide evidence that selective allosteric modulators may enhance signaling bias.
KOPr Agonism with MOPr Antagonism.
The mixed KOPr agonist and partial MOPr antagonist nalbuphine has potent antinociceptive effects in male (Pick et al., 1992; Patrick et al., 1999; Ortiz et al., 2007) and female mice (Wong and Wai, 1984), rabbits (Yoa-Pu et al., 1998), and humans (Kshirsagar et al., 2008) in a wide range of pain models (Table 2). Moreover, nalbuphine attenuates cocaine abuse–related effects in men (Mello et al., 2005) with lower respiratory depression and fewer psychomimetic side effects compared with other narcotic analgesics, such as nalorphine or pentazocine (Schmidt et al., 1985). Similarly, 3-amino-thiazolo [5,4-b]-N-cyclopropylmethylmorphinan hydrochloride (ATPM) attenuated heroin self-administration in male Sprague-Dawley rats (Wang et al., 2009) and, along with the analog [(−)-3-N-ethyl]-ATPM, showed antinociceptive effects and inhibited morphine-induced antinociceptive tolerance in mice (Wang et al., 2009; Sun et al., 2010) (Table 2).
Although mixed KOPr agonists and partial MOPr antagonists have been proposed as nonaddictive analgesics, some have abuse potential and adverse side effects. For example, pentazocine has analgesic effects in moderate-to-severe pain in humans with mild respiratory depressive effects and did not induce nalorphine-like psychoactive or morphine-like reward behaviors (Sadove et al., 1964). Pentazocine was also effective in patients that underwent a nasal irritation pain model (Lotsch et al., 1997) and in thermal and pressure pain assays (Fillingim et al., 2004) (Table 2). However, the analgesic effects of pentazocine were not as potent as morphine and produced side effects, including hallucinations, disorientation, respiratory depression (Miller, 1975), and abuse potential (Pawar et al., 2015).
Similarly, levallorphan is a KOPr agonist and MOPr antagonist that blocked the euphoric effects of morphine while retaining antinociceptive effects via KOPr meditation in male CD1 mice (Codd et al., 1995). Moreover, it was found to be protective against respiratory depression (Pawar et al., 2015). However, levallorphan, because of actions at KOPr, can produce hallucinations, dissociation, and other psychotomimetic effects (Hall, 2012).
Partial KOPr Agonism with Partial MOPr Agonism/Antagonism.
Nalmefene, nalorphine, and butorphanol are weak partial KOPr agonists with partial MOPr antagonism and have been studied extensively. A clinical study reported that low dose of nalmefene enhanced morphine analgesia in patients with postsurgical pain (Crain and Shen, 2000). Likewise, nalorphine has potent antinociceptive effects in mice (Paul et al., 1991) and analgesic effects in humans (Lasagna and Beecher, 1954; Keats and Telford, 1956) (Table 2); however, clinical development was ceased because of diuresis (Leander, 1983). Butorphanol is used to treat labor pain in pregnant women (Halder and Agarwal, 2013; Haiying et al., 2018; Yadav et al., 2018). In nondependent heroin-using males, acute administration of butorphanol produced little or no physical dependence compared with morphine; however, it did cause dysphoria, hallucinations, and sedation (Tennant et al., 1976; Greenwald and Stitzer, 1998; Pandya, 2010).
6β-N-Heterocyclic Substituted Naltrexamine Derivative (BNAP) is another example of an MOPr antagonist and partial KOPr agonist. BNAP is a peripherally restricted naltrexamine derivative (Williams et al., 2016) with potent antinociceptive effects in male ICR-CD1 mice (Williams et al., 2016) (Table 2). An example of a mixed partial KOPr/MOPr agonist is proxorphan, which produced antinociceptive effects in male albino mice in the abdominal constriction assay (Zimmerman et al., 1987; Hayes and Birch, 1988) (Table 2).
KOPr/MOPr Heteromer.
N-naphthoyl-β-naltrexamine is a highly selective and potent activator of MOPr/KOPr heteromer and had potent antinociceptive effects in male ICR-CD1 mice in the tail-withdrawal assay (Yekkirala et al., 2011) (Table 2).
Mixed KOPr/DOPr Compounds
Miaskowski et al. (1990) found that KOPr (U50,488) and DOPr [D- Pen 2,D- Pen 5]enkephalin agonists administered via intrathecal injection produced a synergistic antinociceptive response. Although DOPr agonists can produce seizures (Comer et al., 1993; Bilsky et al., 1995; Jutkiewicz et al., 2006; Lutz and Kieffer, 2013) and have abuse potential (Shippenberg et al., 2009; Pradhan et al., 2011; Mori et al., 2015), KOPr agonists have anticonvulsant, antiseizure (Zangrandi et al., 2016), and antiaddiction effects (Negus et al., 1997; Mello and Negus, 2000). Therefore, it is hypothesized that mixed KOPr and DOPr agonism could have fewer side effects.
A novel analog of 3-iodobenzoyl naltrexamine called MP1104 has dual KOPr/DOPr agonist actions (Váradi et al., 2015). MP1104 produced potent antinociceptive effects in the radiant-heat tail-withdrawal assay in male CD1 mice (Váradi et al., 2015) and reduced inflammatory pain in the intradermal formalin test in both male and female ICR mice (Ulker et al., 2020) (Table 2). Moreover, MP1104 has anticocaine effects in male Sprague-Dawley rats and showed no anxiogenic, prodepressive, or aversive side effects (Atigari et al., 2019).
There are also compounds created to target KOPr/DOPr heterodimers. These include KDA-16 (also known as ICI199,441), which showed spinal antinociception via selective activation of KOPr-DOPr heterodimers in mice (Tang et al., 2010). KDAN-18, which links KOPr agonist ICI199,441 and DOPr antagonist naltrindole, produced antinociceptive effects in tail-withdrawal assays in male mice (Daniels et al., 2005). As mentioned previously, 6′-GNTI selectively activated DOPr/KOPr heterodimers but not KOPr or DOPr homomers (Waldhoer et al., 2005). Together, this suggests that opioid receptor heterodimers are distinct functional signaling units and could provide a target for the development of tissue-selective analgesics with reduced side effects.
Mixed KOPr/DOPr/MOPr Compounds
Levorphanol is a full agonist at both MOPr and DOPr and a partial KOPr agonist (Le Rouzic et al., 2019). Levorphanol was effective in treating chronic pain resulting from malignancies and bone or joint disease (Glazebrook, 1952) and postoperative pain after abdominal surgery (Morrison et al., 1971). In addition, as a strong N-methyl-D-aspartate receptor antagonist (Pham et al., 2015), levorphanol has inhibitory effects on the uptake of serotonin and norepinephrine, which makes it suitable to be used for the treatment of neuropathic pain (Rowbotham et al., 2003; Zorn and Fudin, 2011). Levorphanol has been investigated for potential clinical uses for treating chronic pain (McNulty, 2007) and opioid-induced hyperalgesia [Stringer et al., 2000; for full review of levorphanol see Gudin et al. (2016)]. Recently, it was shown that the antinociceptive actions of levorphanol were mediated via G-protein–biased MOPr agonism (Le Rouzic et al., 2019). Furthermore, levorphanol produced significantly less respiratory depression than morphine at equal doses (Le Rouzic et al., 2019).
Cyclorphan has mixed weak partial MOPr agonist and antagonist activity in combination with KOPr and DOPr agonism. Cyclorphan had antinociceptive effects in mice in the hot-plate assay (Gringauz et al., 2001) with long-acting antinociceptive effects; however, adverse psychomimetic effects prevented its clinical development (Varghese and Hudlicky, 2014).
Recently, a potent mixed MOPr/DOPr/KOPr agonist called 14-O-phenylpropyloxymorphone was synthesized by modifying the structure of the MOPr agonist 14-O-methyloxymorphone (Lattanzi et al., 2018). 14-O-phenylpropyloxymorphone produced potent antinociceptive effects in the acute hot-plate assay in mice compared with reduced constipation compared with morphine (Lattanzi et al., 2018). Although 14-O-phenylpropyloxymorphone has high affinity toward all three opioid receptors, the antinociceptive effects were found to be mediated via MOPr only (Lattanzi et al., 2018).
Mixed KOPr/DOPr/MOPr/NOPr Compounds
Cebranopadol (also known as GRT-60005) is a full agonist at MOPr and DOPr and a partial agonist at KOPr and NOPr and has potent antinociceptive effects in thermal, inflammatory, chronic neuropathic, and bone cancer pain models in male and female rats with a favorable side-effect profile (Linz et al., 2014). Other studies in rodents have reported potent antinociceptive effects in thermal (Schunk et al., 2014; Linz et al., 2017), arthritic (Schiene et al., 2018), and neuropathic pain models (Schunk et al., 2014; Christoph et al., 2018). Cebranopadol is currently in clinical trials for several indications, including treatment of severe chronic nociceptive (Dahan et al., 2017), postoperative (Scholz et al., 2018), chronic lower back (Christoph et al., 2017), cancer (Koch et al., 2019), and neuropathic pain (Schunk et al., 2014; Lambert et al., 2015) (Table 2).
Conclusions
There are hurdles to overcome in the development of KOPr agonists for the treatment of pain. The KOPr activation can drive the negative affective state during inflammatory pain (Massaly et al., 2019) and the aversive component of neuropathic pain (Liu et al., 2019b; Meade et al., 2020). However, this review provides evidence that several KOPr agonists have been developed with preclinical antinociceptive effects with few side effects. These improvements often correlate to enhanced G-protein signaling over β-arrestin recruitment, although there are clear differences in the determination of signaling bias. An alternative approach to overcome the undesirable effects of selective KOPr agonism is to develop peripherally restricted KOPr agonists or mixed opioid receptor agonists. In particular, a mixed opioid agonist with MOPr and DOPr activation may reduce the aversive-like effects produced by KOPr agonism.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Paton, Atigari, Kaska, Prisinzano, Kivell.
Footnotes
- Received May 28, 2020.
- Accepted August 27, 2020.
↵1 K.F.P. and D.V.A. contributed equally to this work.
This work was supported by the Health Research Council of New Zealand, B.M.K. [Grant 16/646] and National Institutes of Health National Institute on Drug Abuse [Grant RO1: DA018151] (to T.E.P.). K.F.P. and D.V.A. each received a doctoral scholarship from Victoria University of Wellington.
Abbreviations
- ATPM
- 3-amino-thiazolo [5,4-b]-N-cyclopropylmethylmorphinan hydrochloride
- 8-CAC
- 8-carboxamidocyclazocine
- CPA
- conditioned place aversion
- DOPr
- δ opioid receptor
- EOM
- ethoxymethyl ether
- ERK1/2
- extracellular signal-regulated kinase 1 and 2
- 6′-GNTI
- 6′-guanidinonaltrindole
- HEK-293
- human embryonic kidney-293
- KOPr
- κ opioid receptor
- MOPr
- μ opioid receptor
- NOPr
- nociceptin opioid receptor
- SalA
- Salvinorin A
- SalB
- Salvinorin B
- β-THP
- β-tetrahydropyran
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- κ Opioid Receptor Signaling and the Role in Pain
- Traditional Arylacetamide κ Opioid Receptor Agonists
- Strategies to Develop κ Opioid Receptor Agonists with Reduced Side Effects
- G-Protein–Biased κ Opioid Receptor Agonists
- Peripherally Restricted κ Opioid Receptor Agonists
- Mixed κ Opioid Receptor Agonists
- Conclusions
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters