


Abstract
Although there has been an increasing appreciation for functional differences between the dorsal (dH) and ventral (vH) hippocampal sectors, there is a lack of information characterizing the cholinergic and noncholinergic mechanisms of acetylcholinesterase inhibitors on synaptic transmission along the hippocampal dorsoventral axis. Diisopropylfluorophosphate (DFP) is an organophosphate (OP) that is commonly employed as a nerve agent surrogate in vitro as well as in rodent models of disease states, such as Gulf War Illness. The present study investigated the cholinergic and noncholinergic mechanisms responsible for the effects of acute DFP exposure on dH and vH synaptic transmission in a hippocampal slice preparation. A paired-pulse extracellular recording protocol was used to monitor the population spike (PS) amplitude as well as the PS paired-pulse ratio (PS-PPR) in the CA1 subfield of the dH and the vH. We observed that DFP-induced PS1 inhibition was produced by a cholinergic mechanism in the dH, whereas a noncholinergic mechanism was indispensable in mediating the inhibitory effect of DFP on the PS1 in the vH. PS-PPR in both dH and vH sectors was increased by acute DFP exposure, an effect that was blocked by an N-methyl-D-aspartate receptor antagonist but not by cholinergic antagonists. Clinical reports have indicated dorsoventral-specific hippocampal abnormalities in cases of OP intoxications. Therefore, the observed dorsoventral-specific noncholinergic mechanisms underlying the effects of DFP on hippocampal synaptic transmission may have important implications for the treatment of OP overexposures.
SIGNIFICANCE STATEMENT It is unknown if acetylcholinesterase inhibitors differentially impact dorsal and ventral hippocampal synaptic transmission. The data in the present study show that an organophosphate, diisopropylfluorophosphate, impacts glutamatergic transmission along the dorsoventral axis in a hippocampal slice preparation via distinct cholinergic and noncholinergic mechanisms. These findings may provide insight into investigations of therapeutic agents that target noncholinergic mechanisms in cases of organophosphate overexposures.
Introduction
The hippocampus is a bilateral structure located in the medial temporal lobe of mammalian organisms and plays a fundamental role in learning and memory. Over the past decade, there has been increasing appreciation for differential functionality between the posterior [dorsal (dH)] and anterior [ventral (vH)] hippocampal sectors (Bannerman et al., 2004; Fanselow and Dong, 2010). For instance, the dH is associated with spatial memory, whereas the vH is linked to emotional and stress-related responses (Bannerman et al., 2004). In both sectors, hippocampal-dependent behavior is regulated by synaptic plasticity and transmission (Whitlock et al., 2006). Hippocampal synaptic transmission is driven by excitatory neurotransmitters like glutamate and shaped by inhibitory neurotransmitters such as γ-aminobutyric acid (GABA) (Watkins and Evans, 1981). Another neurotransmitter, acetylcholine (ACh), modulates hippocampal pyramidal cell activity (Benardo and Prince, 1982a,b; Madison et al., 1987), consequently influencing hippocampal memory functions (Hasselmo, 2006). For instance, cholinergic projections from the medial septum and diagonal band to the hippocampal formation are involved in controlling hippocampal theta rhythms (Stewart and Fox, 1990; Vertes and Kocsis, 1997; Hasselmo, 2006). Although there is some evidence for differential cholinergic projection (Milner et al., 1983; Amaral and Kurz, 1985) and receptor density (Garcia Ruiz et al., 1993; Jung et al., 1994; Huang and Winzer-Serhan, 2006; Kenney et al., 2012) in the dH compared with the vH, there is a paucity of reports investigating the distinct role of ACh in synaptic transmission along the hippocampal dorsoventral axis.
Organophosphates (OPs) are potent neurotoxicants that are frequently employed in agricultural and military settings (Naughton and Terry, 2018). OPs phosphorylate the serine hydroxyl group in the catalytic triad of acetylcholinesterase (AChE), irreversibly inhibiting the enzyme (Abou-Donia, 2003). Subsequent enhancement of synaptic ACh levels leads to cholinergic modulation of hippocampal glutamatergic transmission via activation of metabotropic muscarinic (mAChR) and ionotropic nicotinic (nAChR) ACh receptors. If not aggressively treated, acute OP-induced cholinergic hyperactivity precipitates the presentation of cholinergic crisis followed by glutamatergic-mediated persistence of seizures and, ultimately, death (Abou-Donia et al., 2016).
Exposure to OPs also induces persisting toxicity via multiple noncholinergic mechanisms, i.e., by affecting oxidant formation, mitochondrial energetics, axoplasmic transport, immune system functionality, and brain endocannabinoid homeostasis (Casida and Quistad, 2004; Pope et al., 2005; Mense et al., 2006; Nallapaneni et al., 2006; Kaur et al., 2007; Grigoryan et al., 2008; Nallapaneni et al., 2008; Grigoryan et al., 2009; Jiang et al., 2010; Middlemore-Risher et al., 2010; Wright et al., 2010; Rohlman et al., 2011; Banks and Lein, 2012; O’Callaghan et al., 2015; Locker et al., 2017; Miller et al., 2018; Naughton et al., 2018; Naughton and Terry, 2018; Michalovicz et al., 2019). Nevertheless, it is unknown if such noncholinergic effects differentially impact synaptic transmission along the hippocampal dorsoventral axis.
Low-level exposure to OPs in a variety of settings has been consistently associated with learning and memory impairments in humans. Repeated subacute OP insecticide exposure in agricultural settings has been linked with impairments in short-term memory (Kaplan et al., 1993; Eaton et al., 2008; Rohlman et al., 2011; Ross et al., 2013). Aerotoxic syndrome, an occupational disease linked to exposure to OPs circulating in unfiltered aircraft cabin air, is associated with cognitive deficits (Michaelis et al., 2017). Additionally, civilians exposed to OP nerve agents in the Tokyo metro terror attacks (Hood, 2001; Nishiwaki et al., 2001; Miyaki et al., 2005) and in Kurdistan during the Iran-Iraq war (Talabani et al., 2018) present persisting neurologic impairments, including cognitive deficits. Moreover, US veterans of the 1990 to 1991 Persian Gulf War exhibit dose-dependent associations between OP exposure and impaired performance on neurobehavioral tasks (Proctor et al., 2006) as well as decreased white matter volume (Heaton et al., 2007). Notably, the performance of veterans with Gulf War Illness and predicted sarin exposure in a learning and memory task is negatively correlated with estimated exposure levels (Chao et al., 2017).
The prototypical alkylphosphate OP diisopropylfluorophosphate (DFP) was developed for the study of chemical weapons (Saunders, 1957). DFP is structurally homologous, but significantly less volatile, than nerve agents such as sarin, making DFP useful for investigating the mechanisms of sarin and other highly toxic OPs. Accordingly, DFP is one of the most widely studied OP alkylphosphates in the laboratory (Prendergast et al., 1997, 1998; Pope et al., 2005; Grigoryan et al., 2008; Nallapaneni et al., 2008; Wright et al., 2010; Terry et al., 2011, 2012; Ferchmin et al., 2015; Naughton et al., 2018; Naughton and Terry, 2018). Moreover, DFP is currently employed as an initiator of status epilepticus in seizure models (Deshpande et al., 2010; Todorovic et al., 2012; Pouliot et al., 2016) and a nerve agent surrogate in models of Gulf War Illness (O’Callaghan et al., 2015; Phillips et al., 2019). Nonetheless, there is a lack of information characterizing the cholinergic and noncholinergic mechanisms of Ops, such as DFP on hippocampal synaptic transmission. Though there is evidence of OPs inducing dorsoventral-specific pathology in the hippocampus (Apland et al., 2010), there are no reports in the literature investigating dorsoventral-specific effects of AChE inhibitors on hippocampal synaptic transmission. It is also unknown if dorsoventral-specific effects of AChE inhibitors are dependent on cholinergic and noncholinergic mechanisms resulting from the functional division of the two hippocampal sectors. Therefore, using a hippocampal slice preparation, the aim of the present work was to investigate the cholinergic and noncholinergic mechanisms that mediate the effects of DFP exposure on synaptic transmission in the dH and the vH.
Methods
Animal Maintenance.
Male C57BL/6J (Jackson Laboratory, Bar Harbor, ME) mice, aged 7–13 weeks, were housed in groups of five. Animals were acclimated to their home cages for at least 1 week after arrival and were maintained on a 12-hour light/dark cycle (lights on from 0700 to 1900 hours). Food and water were provided ad libitum. All experiments were performed in compliance with the University of Georgia Animal Care and Use Committee guidelines.
Slice Preparation and Electrophysiology.
Hippocampal slice preparation and electrophysiology experiments were conducted as previously described (Keralapurath et al., 2017; Preston et al., 2019). Briefly, mice were sacrificed following deep anesthetization with 2-bromo-2-chloro-1,1,1-trifluoroethane (halothane). Brains were removed and quickly submerged in oxygenated (95% O2/5% CO2) ice-cold dissection artificial cerebrospinal fluid (120 mM NaCl, 3 mM KCl, 4 mM MgCl2, 1 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose). A mid-sagittal cut was made, and half of the brain was mounted on its caudal pole and sectioned along the horizontal plane with a vibratome to obtain 400-µm slices containing the dH. The other half of the brain was mounted on its rostral pole and sectioned along the horizontal plane with a vibratome to acquire 400-µm slices containing the vH. The dH and vH were subdissected free from the rest of the slice, and the CA3 subfield was removed. These slices were then quickly placed in a submersion-type chamber and perfused with oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) (120 mM NaCl, 3 mM KCl, 1.5 mM MgCl2, 1 mM NaH2PO4, 2.5 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose). Slices were continuously perfused with ACSF (1 ml/min) during a 45-minute recovery period at room temperature followed by a 45-minute recovery period at the recording temperature of 30°C. Schaffer collateral fibers were stimulated by placing a bipolar electrode (Kopf Instruments, Tujunga, CA) in the stratum radiatum of the CA1 subfield, whereas a 1.0 MΩ tungsten recording microelectrode (World Precision Instruments, Sarasota, FL) was inserted in the stratum pyramidale of CA1. During recording experiments, a stimulus pulse was applied once every minute to elicit responses from Schaffer Collateral-CA1 synapses.
A graphical representation of the experimental protocol is shown in Fig. 1A. A stimulus-response curve (I/O curve) was acquired at the beginning of each experiment by incrementally delivering single 20 µA stimulus pulses ranging from 40 to 200 µA in intensity and measuring the population spike (PS1) response (millivolts) from PS trough to PS peak. The stimulus intensity was adjusted to elicit ss that did not receive antagonist pretreatmen baseline responses of 75% of the maximal PS1 amplitude. Baseline paired-pulse ratio (PS-PPR) was then monitored each minute for 30 minutes via paired-pulse stimulation with an interpulse interval of 10 ms. Baseline responses were recorded for 40 minutes during antagonist wash-in experiments. Antagonists were present for the entire duration of the experiment following 20 minutes of antagonist-free baseline recording. After establishing the baseline response, DFP was washed in to the bath for 30 minutes, and an I/O curve was immediately recorded at the end of the wash-in period. Slice PS-PPR was monitored for the following 1 hour. A final I/O curve was measured to determine the persisting effects of DFP exposure on basal synaptic transmission.

The effect of diisopropylfluorophosphate (DFP) wash-in on the population spike (PS1) and AChE activity in the dorsal (dH) and the ventral (vH) hippocampus. (A) Timeline of DFP wash-in experiments. (B and C) Results of dose-response curve experiments investigating the effect of DFP on the PS1 in the dH (B) and the vH (C). Slices were exposed to DFP as indicated by the arrow at 30 minutes. Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. Representative traces of the baseline PS1 (gray) and the PS1 following 60 minutes 30 μM DFP wash-out (black) are shown. (D and E) Acetylcholinesterase (AChE) activity in dH and vH slices exposed to DFP for 30 minutes and washed with ACSF for 80 minutes (IC50 = 2.70 μM). Each data point represents the average AChE activity from 12 slices (collected from the dH and the vH). * denotes p < 0.05. Data are expressed as mean ± S.E.M. I/O curve, input-output curve.
Determination of AChE Activity.
Briefly, each assay was completed using 400 µm individual dH or vH slices superfused with 1 ml/min ACSF ± DFP for 4.75 hours ± 15 minutes at 37°C. Slices were immediately stored at −80°C following completion of electrophysiology experiments. On the day of the assay, slices were kept frozen on dry ice until homogenized with Branson Sonifier 250 (Branson Ultrasonics, Danbury, CT) in ice-cold 0.05 M Tris-HCl (pH 7.4) buffer. Sixty slices (30 from the dH and 30 from the vH) were randomly selected for homogenization. Six slices from each sector (dH or vH) and treatment level (0, 3, 10, 30, and 100 µM DFP) were prepared for the activity assay. AChE activity of each slice homogenate was measured in triplicate wells (three replicates/slice homogenate).
AChE activity was measured spectrophotometrically using a modification (Chambers et al., 1988; Chambers and Chambers, 1989) of Ellman et al. (1961), with acetylthiocholine as the substrate and 5,5′-dithio-bis(nitrobenzoic acid) as the chromogen. The method from Chambers et al. (1988) was scaled to a microplate format. Following 20-minute incubation of each slice homogenate with the reaction mixture described in Chambers et al. (1988), absorbance was measured via BioTek Epoch Microplate spectrophotometer at 412 nM. An AChE standard curve was included alongside the samples at the following concentrations: 0, 1, 3, 10, 30, 100, 300, and 1000 mU/ml [diluted in 0.05 M Tris-HCl (pH 7.4) buffer; AChE source: Electrophorus electricus (Sigma-Aldrich, St. Louis, MO)]. Physostigmine hemisulfate (10 μM) was incubated with a subset of control samples to serve as the positive control. AChE enzyme activity was normalized on a per–milligram of tissue protein basis. Protein content in the homogenized samples was quantified via the Bradford protein assay method (Bradford, 1976) in a 96-well plate using Bradford reagent from Bio-Rad (Hercules, CA). Bovine serum albumin (Fisher Scientific, Pittsburgh, PA) was used as the standard for the protein quantification assay.
Drugs.
AFDX-116(11-[[2-[(Diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one), atropine sulfate, acetylthiocholine iodide, acetylcholinesterase, 5,5′-dithio- bis(nitrobenzoic acid), and diisopropylfluorophosphate (Purity: 99%, Lot: MKCD1107) were purchased from Sigma-Aldrich. Dantrolene-sodium, D-(-)-2-Amino-5- phosphonopentanoic acid (D-APV), mecamylamine HCl, and physostigmine hemisulfate were purchased from Tocris Bioscience (Minneapolis, MN). Pirenzepine was purchased from Alfa Aesar (Tewksbury, MA). A total of 2.5 mM AFDX-116 and 20 mM dantrolene (DAN) stock solutions were prepared in DMSO. Then, 0.1% DMSO vehicle control experiments were conducted to confirm that DMSO did not induce a significant effect on population spike responses.
Data Analysis.
Electrophysiology data were digitized at 10 kHz, low-pass filtered at 1 kHz, and analyzed with pCLAMP 10.2 software (Axon Instruments, Sunnyvale, CA). Synaptic responses of the first PS (PS1) were normalized to one using the average PS1 amplitude of the last 5 minutes of baseline. Thirty-minute DFP wash-in and 60-minute DFP wash-out PS1 values were calculated using the average PS1 amplitude values 26–30 minutes after beginning DFP wash in and 56–60 minutes into DFP wash-out, respectively. The effect of an antagonist on the PS1 was evaluated by comparing the average PS1 amplitude of the last 5 minutes prior to antagonist wash-in (16–20 minutes) compared with the average PS1 amplitude of the last 5 minutes prior to DFP wash-in (36–40 minutes). PS-PPR values were obtained by pairing a conditioning stimulus, which evoked PS1, with a second stimulus, evoking PS2, and calculating the percent change of PS2 with respect to PS1 (PS-PPR = PS2/PS1).
Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. All statistical analyses and graphic production were completed using R version 3.3.2 (The R Foundation for Statistical Computing, Vienna, Austria). Statistical analysis was completed by using the aov() function for analysis of variance (ANOVA) and the t.test function for the Student’s t test. Graphic production was completed via R package “ggplot2.” DFP dose-response curve statistical analysis was conducted via one-way repeated measures ANOVA followed by Dunnett’s multiple comparison test. The Student’s t test (one-tailed) was used for all other pairwise comparisons of data. An α level of 0.05 was used as the criterion for statistical significance. All data are presented as mean ± S.E.M.
Results
Population Spike Inhibition and Concurrent Population Spike Paired-Pulse Facilitation Were Observed in Dorsal and Ventral Hippocampal Slices Following DFP Wash-In.
A significant main effect of DFP exposure on the PS1 amplitude was observed in the dH (F4,39 = 38.2, P < 0.001) and the vH (F4,38 = 46.6, P < 0.001; Fig. 1, B and C, respectively). A Dunnett’s multiple comparison test revealed significant inhibition of the PS1 amplitude in dH (P < 0.05) and vH (P < 0.001) slices exposed to 30 μM DFP compared with unexposed slices. Additionally, Dunnett’s test revealed that 100 µM DFP wash-in induced significant PS1 inhibition in dH (P < 0.001) and vH slices (P < 0.001) compared with unexposed slices. Interestingly, marginal (nonsignificant) recovery of the PS1 amplitude was observed in both dH and vH slices treated with 100 µM DFP. dH and vH slices that were not exposed to DFP displayed no significant change in PS1 amplitude approximately 2 hours after establishing baseline. A one-way ANOVA (DFP treatment as a factor) revealed a significant main effect of DFP on AChE activity, indicating that dH and vH slices exposed to DFP exhibited a significant reduction in AChE activity (F4,55 = 15.2, P < 0.001; IC50 = 2.70 μM; Fig. 1, D and E). dH and vH slices were combined in AChE activity assay analyses, as a two-way ANOVA (sector and DFP treatment as factors) revealed no significant main effect of hippocampal sector on AChE activity (F1,50 = 0.0220, P = 0.88). Based on these results, 30 µM DFP was used as the DFP concentration for all remaining experiments throughout this study.
Nonselective nAChR Antagonism Enhances DFP-Mediated Population Spike Inhibition in the Dorsal and the Ventral Hippocampus.
The nonselective, noncompetitive nAChR antagonist mecamylamine (MEC) was employed to investigate the role of the nAChR in DFP-mediated PS1 inhibition. Pretreatment with 30 µM MEC significantly enhanced DFP-mediated PS1 inhibition in the dH [t(17) = 2.00, P = 0.031] and the vH [t(15) = 1.86, P = 0.042] compared with 30 µM DFP alone (Fig. 2, A and B, respectively). Furthermore, significantly enhanced PS1 inhibition in the dH [t(17) = 2.77, P = 0.0066] and the vH [t(15) = 4.26, P < 0.001] was observed following 60 minutes DFP wash-out. The magnitude of PS1 inhibition in dH slices treated with DFP and MEC was significantly less than the inhibition observed in the vH after 30 minutes DFP wash-in [t(18) = 2.74, P = 0.0068] and 60 minutes DFP wash-out [t(18) = 2.82, P = 0.0057]. MEC alone did not induce a significant effect on PS1 amplitude in the dH or the vH.

Cholinergic antagonist pretreatment prevents DFP-mediated PS1 inhibition in the dH but not in the vH. (A and B) The effect of 30 μM mecamylamine (MEC) pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (A) and the vH (B). (C and D) The effect of 3 μM atropine (ATR) pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (C) and the vH (D). (E and F) The effect of 100 nM pirenzepine (PZP) pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (E) and the vH (F). (G and H) The effect of 300 nM AFDX-116 pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (G) and the vH (H). In antagonist experiments, slices were pretreated with antagonist(s) following 20 minutes of baseline recording. Baseline was then monitored for an additional 20 minutes (40 minutes total baseline). Slices that did not receive antagonist pretreatment (□) and slices that received antagonist pretreatment (■) were exposed to DFP as indicated by the arrow at 40 minutes. Representative traces are shown for antagonist experiments in which the baseline PS1 (gray) and the PS1 following 60 minutes DFP wash-out (black) are shown. Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. Data are expressed as mean ± S.E.M.
Nonselective mAChR Blockade Prevents DFP-Mediated Population Spike Inhibition in the Dorsal Hippocampus but Not in the Ventral Hippocampus.
The nonselective mAChR antagonist atropine (ATR) was selected to investigate the role of the mAChR in DFP-mediated PS1 inhibition. Pre-exposure to 3 µM ATR prevented DFP-mediated PS1 inhibition the dH [t(13) = 3.32, P = 0.0027; Fig. 2C]. Interestingly, PS1 in the dH was enhanced following 60 minutes DFP wash-out [t(13) = 4.11, P < 0.001]. Conversely, ATR pretreatment prevented DFP-induced PS1 inhibition in the vH after 30-minute application of DFP [t(15) = 2.20, P = 0.022], but this effect did not persist following 60 minutes DFP wash-out [t(15) = 0.0941, P = 0.46; Fig. 2D]. The magnitude of PS1 inhibition between dH and vH slices treated with DFP and ATR was significantly different after 30 minutes DFP wash-in [t(14) = 3.41, P = 0.0021] and 60 minutes DFP wash-out [t(14) = 2.64, P = 0.0097]. ATR alone did not induce a significant effect on PS1 amplitude in the dH or the vH.
Selective M2 mAChR, but Not M1 mAChR, Blockade Prevents DFP-Mediated Population Spike Inhibition in the Dorsal Hippocampus but Not in the Ventral Hippocampus.
Pretreatment of slices with 100 nM of the M1 mAChR selective antagonist pirenzepine (PZP) did not prevent DFP-mediated PS1 inhibition in either the dH [t(12) = 0.645, P = 0.27] or the vH [t(13) = 0.126, P = 0.45; Fig. 2, E and F, respectively]. Pretreatment with 300 nM AFDX-116, a selective M2 mAChR (M2R) antagonist, prevented DFP-mediated PS1 inhibition in the dH [t(17) = 2.01, P = 0.030; Fig. 2G]. Strikingly, PS1 in the dH quantitatively resembled the enhancement observed in ATR experiments following 60 minutes DFP wash-out [t(17) = 3.08, P = 0.0034]. Conversely, AFDX-116 pretreatment had no significant effect on DFP-induced PS1 inhibition in the vH after 30-minute application of DFP [t(14) = 1.02, P = 0.16] nor after 60 minutes DFP wash-out [t(14) = 0.277, P = 0.39; Fig. 2H]. The magnitude of PS1 inhibition in dH and vH slices treated with DFP and AFDX-116 was significantly different following 30 minutes DFP wash-in [t(17) = 2.27, P = 0.018] and 60 minutes DFP wash-out [t(17) = 3.02, P = 0.0039]. Neither PZP nor AFDX-116 alone induced a significant effect on PS1 amplitude in the dH or the vH.
Concurrent Nonselective mAChR and nAChR Blockade Prevents DFP-Mediated Population Spike Inhibition in the Dorsal Hippocampus but Not in the Ventral Hippocampus.
Coapplication of 3 μM ATR and 30 μM MEC to dH slices prior to wash-in of DFP prevented the DFP-mediated PS1 inhibition after 30 minutes of DFP application [t(12) = 2.18, P = 0.025] and 60 minutes of DFP wash-out [t(12) = 2.16, P = 0.026; Fig. 3A]. The DFP-mediated PS1 inhibition was significantly prevented in the vH following 30 minutes of DFP application [t(13) = 2.09, P = 0.028], but the effect of cholinergic antagonist pretreatment did not persist following a 60-minute DFP wash-out period [t(13) = 0.649, P = 0.26; Fig. 3B]. The dorsoventral-specific effect of DFP-mediated PS1 inhibition observed in previous cholinergic antagonist experiments was also observed in ATR and MEC coapplication experiments, in which dH PS1 was significantly greater than vH PS1 following 30 minutes DFP wash-in [t(11) = 1.80, P = 0.049] and 60 minutes DFP wash-out [t(11) = 2.96, P = 0.0065]. Coapplication of ATR and MEC did not induce a significant effect on PS1 amplitude in the dH or the vH.

Either cholinergic or NMDAR blockade is sufficient in preventing DFP-mediated PS1 inhibition in the dH, but NMDAR blockade is indispensable in the vH. (A and B) The effect of 3 μM atropine (ATR) and 30 μM mecamylamine (MEC) pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (A) and the vH (B). (C and D) The effect of 300 nM AFDX-116, 30 μM MEC, and 25 μM D-APV pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (C) and the vH (D). (E and F) The effect of 25 μM D-APV pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (E) and the vH (F). (G and H) The effect of 20 μM dantrolene (DAN) pretreatment on 30 μM DFP-mediated PS1 inhibition in the dH (G) and the vH (H). Slices that did not receive antagonist pretreatment (□) and slices that received antagonist pretreatment (■) were exposed to DFP as indicated by the arrow at 40 minutes. Representative traces are shown for antagonist experiments in which the baseline PS1 (gray) and the PS1 following 60 minutes DFP wash-out (black) are shown. Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. Data are expressed as mean ± S.E.M.
M2R, N-methyl-D-aspartate receptor, and Nonselective nAChR Blockade Prevents DFP-Mediated Population Spike Inhibition in the Dorsal and the Ventral Hippocampus.
Coapplication of 300 nM AFDX-116, 25 µM D-APV [an N-methyl-D-aspartate receptor (NMDAR) competitive antagonist], and 30 µM MEC prior to wash-in of DFP resulted in no observable effect of DFP on the PS1 in the dH [t(14) = 1.89, P = 0.040] or the vH [t(15) = 2.86, P = 0.0059; Fig. 3, C and D, respectively]. Prevention of DFP-mediated PS1 inhibition persisted in the dH [t(14) = 4.42, P < 0.001] and the vH [t(15) = 2.12, P = 0.026] following 60 minutes of DFP wash-out. The dorsoventral-specific effect of DFP-mediated PS1 inhibition that was present in cholinergic antagonist pretreatment experiments was not observed in this triple antagonist wash-in experiment following 30 minutes DFP wash-in [t(15) = 0.808, P = 0.22] and 60 minutes DFP wash-out [t(15) = 0.124, P = 0.45]. Coapplication of AFDX-116, D-APV, and MEC did not induce a significant effect on PS1 amplitude in the dH or the vH. These data and those previously shown suggest that DFP-induced PS1 inhibition is mediated by the M2R in the dH, whereas the effect of DFP is mediated by both cholinergic receptors and the NMDAR in the vH.
NMDAR Antagonism Prevents DFP-Mediated Population Spike Inhibition in the Dorsal and the Ventral Hippocampus.
Pretreatment of dH slices with 25 µM D-APV did not significantly prevent DFP-mediated PS1 inhibition following 30 minutes DFP wash-in [t(15) = 1.33, P = 0.10]; however, a significant reduction in PS1 inhibition was observed following 60 minutes DFP wash-out [t(15) = 3.12, P = 0.0035; Fig. 3E]. D-APV pretreatment of vH slices did not significantly prevent DFP-mediated PS1 inhibition compared with slices that received DFP alone following 30 minutes DFP wash-in [t(12) = 1.24, P = 0.12] or 60 minutes DFP wash-out [t(12) = 1.05, P = 0.15; Fig. 3F]; however, vH slices pretreated with D-APV did not display a significantly different PS1 amplitude compared with control vH slices at the 30-minute DFP wash-in [t(15) = 1.69, P = 0.56] or the 60-minute DFP wash-out time point [t(15) = 0.421, P = 0.34]. Interestingly, the magnitude of DFP-induced PS1 inhibition was not significantly different in the dH compared with the vH following 30 minutes DFP wash-in [t(13) = 1.56, P = 0.072] and 60 minutes DFP wash-out [t(13) = 0.692, P = 0.25] when D-APV was present in the bath. Prior to DFP application, D-APV alone did not induce a significant effect on PS1 amplitude in the dH or the vH (Supplemental Fig. 2, A and B, respectively).
Ryanodine Receptor Antagonism Enhances DFP-Mediated Population Spike Inhibition in the Dorsal and the Ventral Hippocampus.
As the NMDAR is involved in hippocampal Ca2+-induced Ca2+ release, we investigated the role of intracellular Ca2+ ([Ca2+]i) stores in DFP-mediated PS1 inhibition by blocking ryanodine receptors (RyRs). Pretreatment with 20 µM DAN significantly enhanced DFP-mediated PS1 inhibition in the dH [t(14) = 4.88, P < 0.001] and the vH [t(12) = 2.44, P = 0.016] compared with 30 µM DFP alone (Fig. 3, G and H, respectively). Furthermore, inhibition of the PS1 in the dH [t(14) = 8.11, P < 0.001] and the vH [t(12) = 4.33, P < 0.001] was significantly enhanced following 60 minutes DFP wash-out. There was no significant difference in PS1 inhibition in dH and vH slices pretreated with DAN after 30 minutes DFP wash-in [t(12) = 1.07, P = 0.15] and 60 minutes DFP wash-out [t(12) = 0.117, P = 0.45]. DAN alone did not induce a significant effect on PS1 amplitude in the dH or the vH.
Population Spike Paired-Pulse Facilitation Observed in Dorsal and Ventral Hippocampus Slices Exposed to DFP Is Mediated by a Noncholinergic Mechanism.
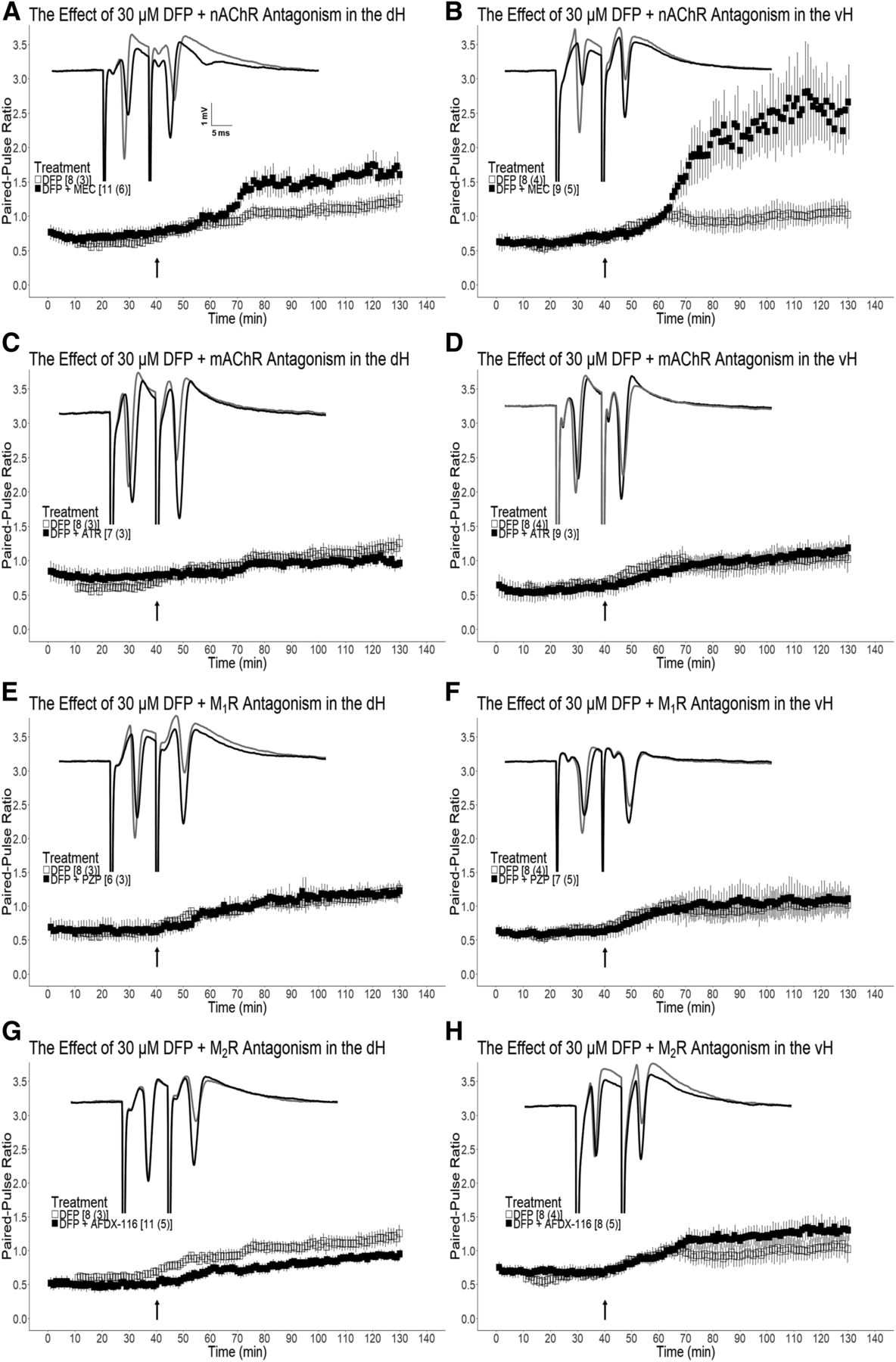
The PS-PPR recorded from control dH and vH slices initially exhibit PS paired-pulse inhibition (PS-PPI, ratio <1.0), an indicator of functional recurrent inhibition (RI) in the CA1 subfield (Supplemental Fig. 1, A and B). Following 30 µM DFP treatment, PS paired-pulse facilitation (PS-PPF, ratio >1.0) in the dH [t(7) = 3.00, P = 0.010] and the vH [t(7) = 3.29, P = 0.0067] was observed (Fig. 4, A and B). Persisting PS-PPF was observed in the dH [t(7) = 4.26, P = 0.0019] and the vH [t(7) = 3.20, P = 0.0075] following DFP wash-out. DH [t(6) = 0.380, P = 0.36] and vH [t(10) = 1.34, P = 0.11] slices that were not exposed to DFP displayed no significant change in PS-PPR approximately 2 hours after establishing baseline (data not shown). Pretreatment of dH slices with the nAChR antagonist MEC resulted in significantly enhanced PS-PPR compared with slices that received DFP alone [t(17) = 1.84 P = 0.040; Fig. 4A]. Likewise, pretreatment of vH slices with MEC resulted in significantly enhanced PS-PPR compared with slices that received DFP alone [t(15) = 2.60 P = 0.010; Fig. 4B]. DH slices pretreated with the mAChR antagonist ATR did not exhibit significantly different PS-PPR [t(13) = 0.958, P = 0.18] from slices that received only DFP (Fig. 4C). Moreover, we did not observe a significant difference in PS-PPR in vH slices pretreated with ATR compared with those that received DFP alone [t(15) = 1.38, P = 0.09; Fig. 4D]. Pre-exposure of dH slices to the M1 mAChR selective antagonist PZP did not prevent the significant increase of PS-PPR observed following DFP wash-in [t(12) = 0.0937, P = 0.46; Fig. 4E]. Likewise, pre-exposure of vH slices to PZP did not prevent the significant increase of PS-PPR after DFP wash-in [t(13) = 0.0622, P = 0.48; Fig. 4F]. However, dH slices that received pre-exposure to the M2R selective antagonist AFDX-116 exhibited significantly reduced PS-PPR compared with slices that received DFP alone [t(17) = 1.89, P = 0.038; Fig. 4G], whereas pretreatment of vH slices with AFDX-116 had no effect on PS-PPR following DFP wash-in [t(14) = 0.372, P = 0.36; Fig. 4H]. Coexposure of dH [t(12) = 0.488, P = 0.32] and vH [t(13) = 1.12, P = 0.14] slices to ATR and MEC did not result in a significant reduction in the PS-PPR that was observed following DFP wash-in (Fig. 5, A and B, respectively). Together, these data suggest that cholinergic receptors do not mediate the PS-PPF that is observed following exposure of dH and vH slices to 30 μM DFP.

Cholinergic antagonist pretreatment does not preserve population spike paired- pulse inhibition (PS-PPI) in either dH or vH slices exposed to DFP. (A and B) The effect of 30 μM mecamylamine (MEC) pretreatment on 30 μM DFP-mediated population spike paired-pulse ratio (PS-PPR) modulation in the dH (A) and the vH (B). (C and D) The effect of 3 μM atropine (ATR) pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (C) and the vH (D). (E and F) The effect of 100 nM pirenzepine (PZP) pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (E) and the vH (F). (G and H) The effect of 300 nM AFDX-116 pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (G) and the vH (H). Slices that did not receive antagonist pretreatment (□) and slices that received antagonist pretreatment (■) were exposed to DFP as indicated by the arrow at 40 minutes. Representative traces are shown for antagonist experiments in which the baseline PS-PPR (gray) and the PS-PPR following 60 minutes DFP wash-out (black) are shown. Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. PS-PPR < 1.0 = paired-pulse inhibition (PS-PPI); PS-PPR > 1.0 = paired-pulse facilitation (PS-PPF). Data are expressed as mean ± S.E.M.

NMDAR antagonist pretreatment preserves PS-PPI in dH and vH slices exposed to DFP. (A and B) The effect of 3 μM atropine (ATR) and 30 μM mecamylamine (MEC) pretreatment on 30 μM DFP-mediated population spike paired-pulse ratio (PS-PPR) modulation in the dH (A) and the vH (B). (C and D) The effect of 300 nM AFDX-116, 30 μM MEC, and 25 μM D-APV pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (C) and the vH (D). (E and F) The effect of 25 μM D-APV pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (E) and the vH (F). (G and H) The effect of 20 μM dantrolene (DAN) pretreatment on 30 μM DFP-mediated PS-PPR modulation in the dH (G) and the vH (H). Slices that did not receive antagonist pretreatment (□) and slices that received antagonist pretreatment (■) were exposed to DFP as indicated by the arrow at 40 minutes. Representative traces are shown for antagonist experiments in which the baseline PS-PPR (gray) and the PS-PPR following 60 minutes DFP wash-out (black) are shown. Reported n values [x(y)] indicate the number of slices (x) and the number of mice (y) assessed. PS-PPR < 1.0 = paired-pulse inhibition (PS-PPI); PS-PPR > 1.0 = paired-pulse facilitation (PS-PPF). Data are expressed as mean ± S.E.M.
NMDAR Blockade Prevents Population Spike Paired-Pulse Facilitation that Is Observed in Dorsal and Ventral Hippocampus Slices Exposed to DFP.
Interestingly, PS-PPI was preserved in both dH [t(14) = 1.82, P = 0.045] and vH [t(15) = 2.24, P = 0.020] slices treated with AFDX-116, MEC, and the NMDAR antagonist D-APV prior to DFP exposure (Fig. 5, C and D, respectively). PS-PPI persisted in dH [t(14) = 2.98, P = 0.0049] and vH [t(15) = 2.21, P = 0.021] slices pre-exposed to the three antagonists after 60 minutes DFP wash-out. No significant difference in PS-PPR was observed between dH and vH slices that received triple antagonist pretreatment. Additionally, 20 minutes of exposure to only D-APV resulted in a significant reduction in PS-PPR in dH [t(8) = 2.17, P = 0.031] and vH [t(5) = 2.19, P = 0.040] slices (Supplemental Fig. 2, C and D, respectively). D-APV pretreatment alone preserved PS-PPI in the dH [t(15) = 3.24, P = 0.0028] and the vH [t(12) = 2.28, P = 0.021] following DFP wash-in (Fig. 5, E and F, respectively). When D-APV was the only antagonist present in the bath, PS-PPI persisted in the dH and vH following 60 minutes DFP wash-out [t(15) = 3.59, P = 0.0013; t(12) = 2.68, P = 0.010; respectively]. No significant difference in PS-PPR was observed between dH and vH slices pretreated with D-APV following DFP wash-in [t(13) = 0.436, P = 0.33] and 60 minutes DFP wash-out [t(13) = 0.720, P = 0.24]. Additionally, pretreatment with the RyR antagonist DAN resulted in a significant enhancement of PS-PPF observed in the dH [t(14) = 3.22, P = 0.0031] and the vH [t(12) = 2.42, P = 0.016] following DFP exposure (Fig. 5, G and H, respectively). Collectively, these data suggest that the NMDAR plays an indispensable role in mediating the PS-PPF that is observed in dH and vH slices exposed to 30 μM DFP.
Discussion
The data in the present report demonstrate that DFP-induced PS1 inhibition in the dH and vH CA1 subfield is mediated by cholinergic receptors and NMDARs. Increased PS-PPR observed in dH and vH slices exposed to DFP is prevented by an NMDAR antagonist. To the best of our knowledge, this is the first report to comparatively characterize both cholinergic and noncholinergic effects of OP exposure on dH and vH synaptic transmission in a hippocampal slice preparation. Additionally, our findings of a DFP-NMDAR interaction affecting synaptic transmission are novel. Our results provide insight into the circuit-level role of the NMDAR in maintaining functional RI in the dH and the vH following exposure to an OP. The observed dorsoventral-specific effects may provide insight into OP-induced deficits in cognition and mood. A circuitry schematic containing the minimal components needed to explain the proposed dorsoventral-specific effects is presented in Fig. 6.

Proposed schematic illustrating the effects of DFP on dorsal (dH) and ventral (vH) hippocampal neurocircuitry in relation to the mAChR, the nAChR, and the NMDAR. DFP induces irreversible AChE inhibition, leading to M2R-mediated PS1 inhibition in the dH (1, 3; gray). DFP induces PS1 inhibition through an M2R, nAChR, and NMDAR-dependent mechanism in the vH (1, 3, 4; black). Following DFP exposure, PS-PPI shifts to PS-PPF in the dH and the vH. PS-PPI can be partially preserved by M2R blockade in the dH (2) or completely preserved by NMDAR blockade in the dH or the vH (4). SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum.
DFP has previously been employed to investigate the acute effects of an OP on the PS1, a response variable that can be directly correlated to the number of functional pyramidal cells in the stratum pyramidale of CA1 (Williamson and Sarvey, 1985; Jones et al., 1990; Ferchmin et al., 2015). Our results are consistent with these prior reports indicating that acute DFP exposure inhibits CA1 glutamatergic transmission and that a substantial component of this inhibition is mediated via cholinergic receptors. More specifically, following irreversible inhibition of AChE, enhanced synaptic ACh leads to hyperactivation of mAChRs, a well-established mechanism of OP-induced neurotoxicity (Abou-Donia et al., 2016). Previous studies have shown that mAChR blockade via ATR pretreatment partially prevents DFP-induced PS1 inhibition (Ferchmin et al., 2015). The present study showed that ATR pretreatment not only prevented DFP-mediated inhibition of the dH PS1 amplitude but also that the amplitude was in fact significantly enhanced by DFP application. Similarly, dH slices pretreated with AFDX-116 quantitatively resembled ATR experiments, suggesting that DFP-mediated PS1 inhibition is due to activation of M2Rs located in dH CA1. These receptors may be located at the Schaffer Collateral-CA1 synapse and somata of pyramidal cells (Vilaró et al., 1992; Auerbach and Segal, 1996; Seeger and Alzheimer, 2001; Seeger et al., 2004), where activation of these M2Rs enhances K+ conductance, hyperpolarizing principal cells and decreasing excitatory glutamatergic transmission (Drever et al., 2011). In support of these findings, prior evidence indicates OPs preferentially act as indirect agonists via M2Rs (Liu et al., 2002).
ACh-mediated activation of nAChRs on pyramidal cells enhances cation conduction, facilitating principal cell depolarization (Albuquerque et al., 2009). Interestingly, nAChR blockade enhanced DFP-mediated PS1 inhibition in both the dH and the vH. Prior evidence indicates that acute OP exposure accelerates the rate of nAChR desensitization (Tattersall, 1990), an effect that may contribute to the enhanced PS1 inhibition observed in the present study. Specifically, this enhanced inhibition may be a result of reducing the excitatory tone of α7-like nAChRs located on stratum pyramidale principal cells (Castro and Albuquerque, 1993; Mike et al., 2000). Additionally, reducing the activity of CA1 interneuron-selective interneurons following the blockade of α4β2-like nAChRs would also decrease pyramidal cell excitability, reducing the PS1 amplitude (Bell et al., 2015).
Direct interactions of OPs with cholinergic receptors are well established (Pope, 1999; Pope et al., 2005). A number of reports have indicated that OPs and other AChE inhibitors directly compete at the agonist site of M2Rs (Bakry et al., 1988; Silveira et al., 1990; Fitzgerald and Costa, 1992; Ward et al., 1993; Ward and Mundy, 1996; Rocha et al., 1999; Santos et al., 2003) or directly phosphorylate (Bomser and Casida, 2001) M2Rs. Moreover, OPs have been shown to preferentially interact with M2/M4 mAChRs and activate their Gα/i-coupled pathways (Ward et al., 1993; Ward and Mundy, 1996). We postulate that AChE-independent mechanisms may underlie some of the results observed in the present study. In particular, no increase in AChE inhibition was observed at DFP concentrations greater than 10 μM, yet progressively greater inhibition of the PS1 was observed at higher concentrations. Furthermore, we observed a noteworthy recovery of the PS1 amplitude in dose-response experiments utilizing 100 µM DFP. We attribute this recovery to wash-out of DFP following direct interaction with nAChRs on pyramidal cells. This interpretation is supported by previous reports of DFP directly inducing nAChR desensitization at concentrations near 100 µM (Albuquerque et al., 1984; Bakry et al., 1988; Eldefrawi et al., 1988).
A number of noncholinergic mechanisms mediating OP effects have been characterized. DFP and other OPs covalently interact with lysine and tyrosine residues of structural proteins such as tubulin, actin, and kinesin, resulting in axonal transport deficits (Grigoryan et al., 2008, 2009; Jiang et al., 2010). A recent report demonstrated that subacute DFP exposure leads to structural disruptions of myelinated axons, a putative result of axonal transport deficits (Naughton et al., 2018). AChE-independent decreases in mitochondrial number and energetics following OP exposure have been described (Kaur et al., 2007; Middlemore-Risher et al., 2010). Moreover, a series of reports have emphasized the role of the NMDAR in initiating Ca2+-induced Ca2+ release mechanisms after OP exposure (Deshpande et al., 2010; Phillips et al., 2019). Interestingly, DFP has been shown to directly interact with the glutamate-binding pocket of the NMDAR, irreversibly inhibiting the receptor (Johnson and Michaelis, 1992). In the present report, we observed a partial reduction of DFP-mediated pyramidal cell inhibition in dH and vH slices pretreated with an NMDAR competitive antagonist. Experiments in which slices were pre-exposed to AFDX-116, D-APV, and MEC completely prevented DFP-mediated pyramidal cell inhibition. There is evidence that bath application of an NMDAR antagonist disinhibits glutamatergic transmission (Čapek and Esplin, 1991; Grunze et al., 1996), which may contribute to preventing the DFP-mediated PS1 inhibition. Blockade of RyRs, receptors involved in Ca2+-induced Ca2+ release, enhanced DFP-mediated PS1 inhibition in the dH and the vH, an effect that may involve decreased pyramidal cell excitability because of disruption of [Ca2+]i homeostasis. There is also evidence suggesting OPs directly interact with receptors involved in Ca2+-induced Ca2+ release (Sun et al., 2000), an interaction that may further contribute to the disruption of [Ca2+]i homeostasis. Noncholinergic mechanisms unexplored in the present study, such as endocannabinoid signaling, promote OP-induced modulations in the hippocampus (Nallapaneni et al., 2006, 2008; Wright et al., 2010; Hoffman et al., 2019). Future studies investigating such noncholinergic mechanisms may be fruitful, particularly because there is a paucity of reports characterizing the effects of OPs on cannabinoid signaling along the hippocampal dorsoventral axis.
Recurrent inhibition modulates principal cell excitability and network functionality by ensuring a sufficient GABAergic inhibitory tone is present in the CA1 subfield (Andersen et al., 1963; Papatheodoropoulos and Kostopoulos, 1998). When using the double orthodromic stimulation protocol to monitor RI in the CA1 subfield of healthy hippocampal tissue (Michelson and Lothman, 1989; Sloviter, 1991), a paired-pulse interpulse interval of 10 ms results in fast-acting, ionotropic GABAA receptor-mediated PS PS-PPI (PS2/PS1 <1) (Papatheodoropoulos et al., 2002; Petrides et al., 2007). It has been noted that a linear increase in PPR is observed as GABAA receptor-mediated inhibition decreases (Lopes da Silva et al., 1995). Therefore, we monitored the PS-PPR as a measurement of functional synaptic inhibition in CA1, where it would be expected that a decrease in RI would result in an increase in PS-PPR. This was observed in dH and vH slices treated with 30 µM DFP, in which an initial PS-PPI response was shifted to PS-PPF. Interestingly, individual or coapplication of mAChR and nAChR antagonists had no significant effect on the PS-PPF that was observed following DFP application. Nonetheless, we observed a marginal preservation of PS-PPI when slices were pretreated with AFDX-116. A previous report demonstrated that activation of mAChRs reduced the frequency of tetrodotoxin-insensitive miniature inhibitory postsynaptic currents as well as monosynaptically evoked inhibitory postsynaptic currents, resulting in weakened RI (Behrends and ten Bruggencate, 1993). M2Rs are frequently expressed on parvalbumin-positive interneurons, and blocking these receptors increases GABA release from perisomatic parvalbumin-positive cells (Hájos et al., 1998; Fukudome et al., 2004; Szabó et al., 2010) as illustrated in Fig. 6, a result consistent with our AFDX-116 experiments.
Strikingly, NMDAR antagonist pretreatment alone or combined with cholinergic antagonists preserved PS-PPI following DFP treatment. Previous reports have shown that GABAergic input onto CA1 principal neurons is modulated by NMDAR-dependent transmission, where bath application of D-APV reduces CA1 pyramidal cell RI postsynaptic potential by more than 30% (Grunze et al., 1996). The unique subunit composition and receptor density of NMDAR-expressing interneurons facilitate enhanced sensitivity to D-APV compared with NMDAR-expressing principal cells (Monyer et al., 1994; Martina et al., 2003, 2013; Nyiri et al., 2003; Avignone et al., 2005). Therefore, we postulate that the preservation of PS-PPI in D-APV pretreatment experiments is a result of the modulatory role of NMDAR-expressing interneurons. Specifically, blockade of the NMDAR results in disinhibition of pyramidal cells, thereby promoting excitation of perisomatic interneurons mediating RI and preserving PS-PPI. We also observed a significant reduction of dH and vH PS-PPR during D-APV application, which is consistent with our NMDAR-mediated disinhibition hypothesis. As this is the first report of the NMDAR modulating the PS-PPR in the presence of an AChE inhibitor, further investigation into this mechanism is warranted.
The long-range in vivo projections comprising the septohippocampal system are complex (Teles-Grilo Ruivo and Mellor, 2013), making the hippocampal slice preparation a sufficient but limited model for investigating cholinergic and noncholinergic mechanisms that mediate the effects of OPs on synaptic transmission along the hippocampal dorsoventral axis. At the behavioral level, the dH is associated with spatial memory, whereas the vH facilitates emotional-related responses (Floresco et al., 1997; Moser and Moser, 1998; Bannerman et al., 2004; Pothuizen et al., 2004; Tamura et al., 2017). Receptor densities differ between the dH and vH sectors, resulting in intrinsic synaptic distinctions (Pandis et al., 2006; Petrides et al., 2007; Segal et al., 2010; Papatheodoropoulos, 2018). In the context of hippocampal cholinergic transmission, differential cholinergic projection (Milner et al., 1983; Amaral and Kurz, 1985) and receptor densities (Garcia Ruiz et al., 1993; Jung et al., 1994; Huang and Winzer-Serhan, 2006; Kenney et al., 2012) are found along the hippocampal dorsoventral axis. In light of these previous reports, accounting for dorsoventral-specific effects of OP exposure may be critical in interpreting the impact of exposure to these agents. For instance, we observed a reduction of DFP-mediated pyramidal cell inhibition in dH and vH slices pretreated with an NMDAR antagonist, an effect that was statistically significant in the dH but not in the vH. These statistical differences may be attributed to significantly greater expression of GluN2A and GluN2B subunits in the dH CA1 compared with the vH CA1 (Pandis et al., 2006). This may be particularly relevant to OP exposure in a low-level exposure setting such as the 1990–1991 Gulf War, in which OP nerve agent exposure has been associated with a dose-dependent impairment of cognition and structural hippocampal abnormalities (Heaton et al., 2007; Chao et al., 2017). Additionally, cognitive impairments following low-level exposure to OP pesticides in agricultural settings have been reported (Rohlman et al., 2011). Given the present evidence for NMDAR involvement of DFP-mediated effects on synaptic transmission, our findings underscore the value of future investigations of therapeutic agents that target noncholinergic mechanisms in cases of low-level OP exposures that lead to persisting cognitive impairments.
Acknowledgments
We thank Caroline Grant for the anatomical illustration of the hippocampus in Fig. 6.
Authorship Contributions
Participated in research design: Brown, Filipov, Wagner.
Conducted experiments: Brown.
Contributed reagents or analytic tools: Filipov, Wagner.
Performed data analysis: Brown.
Wrote or contributed to the writing of the manuscript: Brown, Filipov, Wagner.
Footnotes
- Received October 3, 2019.
- Accepted December 16, 2019.
This research was supported by the Department of Defense Congressionally Directed Medical Research Programs [Grant W81XWH-16-1-0586] to N.M.F. K.A.B. received support from the Interdisciplinary Toxicology Program at the University of Georgia.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ACh
- acetylcholine
- AChE
- acetylcholinesterase
- AFDX-116(11-[[2-[(Diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one),ATR
- atropine
- DAN
- dantrolene
- DFP
- diisopropylfluorophosphate
- D-APV
- D-(-)-2-Amino-5- phosphonopentanoic acid
- dH
- dorsal hippocampus
- I/O curve
- input-output curve
- mAChR
- metabotropic muscarinic
- MEC
- mecamylamine
- nAChR
- ionotropic nicotinic
- NMDAR
- N-methyl-D-aspartate receptor
- OP
- organophosphate
- PS
- population spike
- PS-PPF
- population spike paired-pulse facilitation
- PS-PPI
- population spike paired-pulse inhibition
- PS-PPR
- population spike paired-pulse ratio
- PZP
- pirenzepine
- RI
- recurrent inhibition
- RyR
- ryanodine receptor
- vH
- ventral hippocampus
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}