


Abstract
In our drug discovery program, we identified a novel orally available and brain-penetrant phosphodiesterase (PDE) 1 inhibitor, 3-methyl-7-(tetrahydro-2H-pyran-4-yl)-2-{[trans-4-(trifluoromethyl)cyclohexyl]-methoxy}imidazo[5,1-f][1,2,4]triazin-4(3H)-one (DSR-141562). In the present study, we characterized the preclinical profile of DSR-141562. This compound has preferential selectivity for predominantly brain-expressed PDE1B over other PDE1 family members, and high selectivity for the PDE1 family over other PDE families and 65 other tested biologic targets. Oral administration of DSR-141562 at 10 mg/kg slightly elevated the cGMP concentration, and it potently enhanced the increase of cGMP induced by a dopamine D1 receptor agonist in mouse brains. The cGMP level in monkey cerebrospinal fluid was also elevated after treatment with DSR-141562 at 30 and 100 mg/kg and could be used as a translational biomarker. Since PDE1B is believed to regulate dopaminergic and glutamatergic signal transduction, we evaluated the effects of this compound using schizophrenia-related behavioral assays. DSR-141562 at 3–30 mg/kg potently inhibited methamphetamine-induced locomotor hyperactivity in rats, while it had only minimal effects on the spontaneous locomotor activity. Furthermore, DSR-141562 at 1–100 mg/kg did not induce any signs of catalepsy in rats. DSR-141562 at 0.3–3 mg/kg reversed social interaction and novel object recognition deficits induced by repeated treatment with an N-methyl-D-aspartate receptor antagonist, phencyclidine, in mice and rats, respectively. In common marmosets, DSR-141562 at 3 and 30 mg/kg improved the performance in object retrieval with detour tasks. These results suggest that DSR-141562 is a therapeutic candidate for positive, negative, and cognitive symptoms in schizophrenia.
SIGNIFICANCE STATEMENT This is the first paper showing that a phosphodiesterase 1 inhibitor is efficacious in animal models for positive and negative symptoms associated with schizophrenia. Furthermore, we demonstrated that this compound improved cognitive function in the common marmoset, a nonhuman primate.
Introduction
Schizophrenia is a severe psychiatric disorder that consists of positive (e.g., delusions, hallucinations, and thought disorder), negative (e.g., flattened affect, amotivation, and poverty of speech), and cognitive symptoms (e.g., working memory, cognitive flexibility, and attention deficits) (Crow, 1980; Sharma and Harvey, 2000). Although positive symptoms are currently treated with dopamine D2 receptor antagonists, most antipsychotics work within a narrow therapeutic dose range with regard to extrapyramidal side effects (Casey, 1996). Furthermore, the efficacies of current antipsychotics on negative and cognitive symptoms are limited (Woodward et al., 2005; Krause et al., 2018). Thus, there is considerable interest in discovering drug candidates with novel mechanisms for treating positive symptoms without having extrapyramidal side effects or for better treating negative and cognitive symptoms.
Cyclic nucleotide phosphodiesterases (PDEs) are enzymes that regulate intracellular signaling by catalyzing the hydrolysis of the second messenger 3′,5′-cAMP and/or 3′,5′-cGMP (Bender and Beavo, 2006; Omori and Kotera, 2007). There are 11 PDE families according to their sequence homologies and enzymatic properties. The PDE1 family, which consists of PDE1A, PDE1B, and PDE1C isoforms, has the unique feature of being activated by binding Ca2+/calmodulin. Although the PDE1 family is a dual-substrate PDE family, which hydrolyzes both cAMP and cGMP, there are differences in substrate specificities among the three PDE1 isoforms. PDE1A and 1B have a higher affinity for cGMP than cAMP, while PDE1C has a similar affinity for both cAMP and cGMP (Bender and Beavo, 2006; Omori and Kotera, 2007). In addition, three PDE1 isoforms have different tissue distributions. Among the PDE isoforms, PDE1B has the highest expression level in brain, while PDE1A and PDE1C have higher than PDE1B expression levels in peripheral tissues including kidney, thyroid, heart, bladder, and lung (Lakics et al., 2010). The predominance of its distribution in the brain has made PDE1B an attractive drug target for psychologic and neurologic disorders, since peripheral PDE inhibition might cause intolerable adverse effects in humans (Khammy et al., 2017).
The abnormalities of dopamine and glutamate neurotransmission have been hypothesized to be involved in the pathophysiology of schizophrenia (Carlsson et al., 1999). Interestingly, PDE1B is colocalized with dopamine D1 and/or D2 receptors in the striatal and prefrontal cortical neurons in rats and humans (Polli and Kincaid, 1994; Yan et al., 1994; Pekcec et al., 2018). Furthermore, Betolngar et al. (2019) have shown that PDE1 regulates cyclic nucleotide signaling in the context of glutamate and dopamine coincidence. The Ca2+ influx via N-methyl-D-aspartate receptors, which are glutamate-gated Ca2+ channels, transiently activate PDE1B in dopamine D1 or D2 receptor–expressing medium spiny neurons. The theoretical relationship between the function of PDE1B and the pathophysiology of schizophrenia implies the therapeutic potential of PDE1B inhibitors for schizophrenia. Although there are few brain-penetrant compounds with selectivity for PDE1B, a brain-penetrant PDE1 family selective inhibitor—(6aR,9aS)-2-[4-(6-fluoropyridin-2-yl)benzyl]-5-methyl-3-(phenylamino)-5,6a,7,8,9,9a-hexahydrocyclopenta-[4,5]imidazo[1,2-a]pyrazolo[4,3-e]pyrimidin-4-(2H)-one phosphate (ITI-214)—with approximately 10-fold selectivity for PDE1A and PDE1C over PDE1B has recently been identified ((Li et al., 2016a; Snyder et al., 2016)). It was shown that ITI-214 exhibits efficacy for learning and memory in rodent behavioral models (Li et al., 2016a; Snyder et al., 2016). Other research groups have shown that another brain-penetrant PDE1 inhibitor 6-(2-fluoro-4-methoxybenzyl)-9-[(tetrahydro-2H-pyran-4-yl)methyl]-8,9,10,11-tetrahydropyrido[4′,3′:4,5]thieno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidin-5(6H)-one (DNS-0056) improves learning and memory in rodent behavioral models, although its selectivity for PDE1B among PDE1 family members has not been reported (Dyck et al., 2017; McQuown et al., 2019). However, there has been a paucity of research comprehensively assessing the effects of PDE1 inhibitor in animal models relevant to positive and negative symptoms in schizophrenia. In addition, there has been no preclinical evidence demonstrating the efficacy of PDE1 inhibitors in cognitive functions in nonhuman primates (Li et al., 2016a; Snyder et al., 2016; Pekcec et al., 2018). It is important to assess cognitive effects in nonhuman primates with a well-developed prefrontal cortex, since cognitive deficits associated with schizophrenia have been hypothesized to be related to dysfunction of the prefrontal cortex (Lewis and González-Burgos, 2008). In our drug discovery program, we discovered the PDE1 inhibitor 3-methyl-7-(tetrahydro-2H-pyran-4-yl)-2-{[trans-4-(trifluoromethyl)cyclohexyl]-methoxy}imidazo[5,1-f][1,2,4]triazin-4(3H)-one (DSR-141562), which has preferential selectivity for PDE1B over PDE1A and PDE1C. In the present study, we characterized the preclinical profile of DSR-141562 in pharmacological assays in vitro, and in pharmacokinetic and biomarker studies in vivo. Furthermore, we evaluated the potential of DSR-141562 as a therapeutic candidate in animal models for positive, negative, and cognitive symptoms associated with schizophrenia. In the cognitive function, we used not only a learning and memory task in rats but also an executive function task in common marmosets.
Materials and Methods
Animals.
We used male Institute of Cancer Research (ICR) strain mice (Japan SLC Inc., Shizuoka, Japan, or Clea Japan Inc., Tokyo, Japan), male Sprague-Dawley strain rats (Japan SLC Inc. or Charles River Laboratories Japan, Inc., Yokohama, Japan), and male cynomolgus monkeys (Hamri Co., Ltd., Ibaraki, Japan, or Tian Hu Cambodia Animal Breeding Research Center Ltd., Kampong Cham, Cambodia), which are commonly used laboratory animals in pharmacology research. However, female Long-Evans rats were used in the novel object recognition task as described previously (Horiguchi et al., 2013), since they are commonly used for this task. In addition, it is known that there are gender differences in the pharmacological actions and pharmacokinetics of phencyclidine (Nabeshima et al., 1984) or cognitive performance in this task (Sutcliffe et al., 2007). We used four male and six female common marmosets (Clea Japan Inc.) in an object retrieval with detour task, as we described previously (Murai et al., 2013; Baba et al., 2015; Kotani et al., 2016), to obtain sufficient numbers of well-trained marmosets. There were no gender differences in the basal performance of this task (our unpublished data). All animals were housed in a controlled environment under a 12/12 hour light/dark cycle with ad libitum access to water. Rodents had free access to food, and nonhuman primates were given food once daily in the morning. All procedures involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committee of Sumitomo Dainippon Pharma Co., Ltd. (Osaka, Japan) or Shin Nippon Biochemical Laboratories Ltd. (Kagoshima, Japan).
Drugs and Reagents.
DSR-141562, methamphetamine hydrochloride, and phencyclidine hydrochloride were prepared by Sumitomo Dainippon Pharma Co., Ltd. 3-Allyl-6-chloro-1-phenyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine-7,8-diol hydrobromide (SKF-82958 hydrobromide) was purchased from Sigma-Aldrich (St. Louis, MO). As for the route of DSR-141562 administration, we chose the oral route, which would be a potential route in clinical studies, for pharmacological experiments not involving c-fos expression measurement. DSR-141562 was subcutaneously administered to achieve the high plasma drug concentrations needed for c-fos expression measurement. In the pharmacokinetic experiments, the compounds were orally and intravenously administered to calculate bioavailability. Doses of DSR-141562 were chosen based on the in vitro IC50 value for PDE1B, our pharmacokinetic results, and results of pilot pharmacological experiments. DSR-141562 was suspended in 0.5% (w/v) methylcellulose or 10% (w/v) hydroxypropyl-β-cyclodextrin for oral administration, and dissolved in 40% (w/v) hydroxypropyl-β-cyclodextrin for subcutaneous administration or in 0.1 N hydrochloric acid/saline (1/9) for intravenous administration. Methamphetamine, phencyclidine, and SKF-82958 were dissolved in saline. The dosages of compounds were calculated in terms of the salt form of the compound. Recombinant human PDE1A, PDE1B, and PDE1C enzymes (SB Drug Discovery Ltd., Glasgow, UK), pig brain calmodulin (Enzo Life Sciences, Farmingdale, NY), [3H]-cGMP (Perkin Elmer, Waltham, MA), and cAMP-13C5 were purchased from commercial sources.
In Vitro Phosphodiesterase 1A, 1B, and 1C Assays.
Inhibition of PDE1A, 1B, and 1C activities was measured by scintillation proximity assays (Glickman et al., 2008). Recombinant human PDE1A, 1B, and 1C were used as enzymes, and [3H]-cGMP was used as the substrate. The concentration of each enzyme in the assay solution was 1.3 U/ml for PDE1A, 3.2 U/ml for PDE1B, and 1.1 U/ml for PDE1C. The substrate concentration in the assay solution was 100 nM, which is below the enzyme Km values for cGMP. The reaction was allowed to proceed for 30 minutes at 27°C. The reaction mixture contained 1 mM CaCl2 and 1 μg/ml calmodulin. Assays were independently performed three times on three different days to determine the mean and S.E. (S.E.M) of IC50 values for each PDE isoform.
In Vitro Assays of Other Phosphodiesterase Isoforms.
Other PDE isoform assays were conducted by SB Drug Discovery Ltd. Human recombinant PDE2A, PDE3 catalytic domain, PDE4 catalytic domain, PDE5 catalytic domain, PDE7A, PDE8A, PDE9A, PDE10A, and PDE11A were used for IMAP FP assays (Molecular Devices, Sunnyvale, CA). The IMAP FP assay was performed according to the standard protocol of the IMAP bulk assay (Molecular Devices). Fluorescein-labeled cAMP at 100 nM was used as the substrate for PDE2A, PDE3 catalytic domain, PDE4 catalytic domain, PDE7, PDE8, PDE10A, and PDE11A. Fluorescein-labeled cGMP at 100 nM was used as the substrate for PDE5 catalytic domain and PDE9A. Since DSR-141562 at 10,000 nM exhibited more than 50% of inhibition of PDE2A in our pilot study, the PDE2A inhibition assay was independently performed three times to determine the mean and S.E.M. of IC50 values of DSR-141562. The inhibitory effects of DSR-141562 on other PDE isoforms were measured at 10,000 nM, and data were analyzed by calculating the percentage of the inhibition values. Human recombinant PDE6A and PDE6B were used for the radiometric assay (Thompson and Appleman, 1971). [3H]-cGMP at 1000 nM was used as the substrate. The inhibitory effect of DSR-141562 on PDE6A/B was measured at 10,000 nM, and data were analyzed by calculating the percentage of the inhibition value.
In Vitro Off-Target Assays.
To evaluate the effects of DSR-141562 at 10,000 nM on the off-targets, Eurofins Pharma Discovery Services (Taipei, Taiwan) performed enzyme and radioligand-binding assays for 65 targets. The information about the assays is available on the Eurofins Discovery Services’ website (https://www.eurofinsdiscoveryservices.com/).
Pharmacokinetic Measurement in Animals.
For the pharmacokinetic study, DSR-141562 was diluted in 0.1 N hydrochloric acid/saline (1/9) for intravenous administration or suspended in 0.5% methylcellulose for oral administration and administered to male Sprague-Dawley rats or cynomolgus monkeys at an intravenous dose of 0.5 mg/kg or oral dose of 1 mg/kg. At 5 (intravenous only), 15, and 30 minutes, and 1, 2, 4, 6, and 24 hours after administration, venous blood was collected in sodium heparin. The blood was centrifuged for 15 minutes at 2500g at 4°C and the resulting plasma was frozen until analyzed.
For the plasma-brain exposure profile pharmacokinetic study, DSR-141562 was suspended in 0.5% methylcellulose solution and orally administered to male Sprague-Dawley rats at a dose of 30 mg/kg. At 0.5, 1, 2, and 6 hours after administration, rats were anesthetized with isoflurane and whole blood was collected in sodium heparin via the inferior vena cava. Plasma was obtained as described previously and frozen until analyzed. Following the blood collection, the brains were removed and stored at freezer temperature until analysis.
To estimate its plasma concentration for pharmacology studies, DSR-141562 was suspended in 0.5% methylcellulose solution and orally administered to ICR mice at doses of 1 and 30 mg/kg. Two hours after administration, venous blood was collected in sodium heparin. In Sprague-Dawley rats, DSR-141562 was dissolved in 40% hydroxypropyl-β-cyclodextrin and subcutaneously administered at a dose of 10 mg/kg. One hour after administration, venous blood was collected in sodium heparin. In common marmosets, DSR-141562 was suspended in 0.5% methylcellulose solution and orally administered at doses of 1 and 10 mg/kg. Two hours after administration, venous blood was collected in sodium heparin. Plasma was obtained as described previously and frozen until analyzed.
The plasma and brain homogenate samples were extracted by protein precipitation with methanol containing an internal standard (phenytoin) and analyzed by means of liquid chromatography–tandem mass spectrometry, using an API 4000 (SCIEX, Framingham, MA) mass spectrometer with electrospray ionization interface in positive ion mode and multiple reactions monitoring. An LC-20A Series HPLC system (Shimadzu, Kyoto, Japan) was used with a Unison UK-C18 column (2.0 × 20 mm, 3 μm; Imtakt Corporation, Kyoto, Japan) at a flow rate of 0.4 ml/min to achieve chromatographic separation. A gradient program was used with the mobile phase, combining solvent A [ammonium acetate buffer (10 mM, pH 4.0)] and solvent B (methanol) as follows: 10%−90% B for 0−2.5 minutes, 90% B for 2.5−3.7 minutes, and 10% B for 3.71−5.5 minutes. The column temperature was set at 40°C. The transitions (precursor to product) monitored for mass spectrometry were mass-to-charge ratios (m/z) 415.300–251.200 for DSR-141562 and 253.226–182.225 for phenytoin. Phoenix WinNonlin (Certara, Princeton, NJ) was used for all pharmacokinetic and noncompartmental analyses.
Brain Tissue Cyclic Nucleotide Measurements in Mice.
Brain tissue cGMP contents were measured as previously reported with some modifications (Schmidt et al., 2008). Male ICR mice were orally administered vehicle (0.5% methylcellulose) or DSR-141562 at 10 mg/kg. In the experiments evaluating the combined effects of DSR-141562 and a dopamine D1 receptor agonist, mice were subcutaneously administered saline or SKF-82958 (0.3 mg/kg) 1 hour after the administration of vehicle or DSR-141562. Two hours after vehicle or DSR-141562 treatment, mice were sacrificed by focused microwave irradiation of the brain. Brain regions of interest were isolated and stored at −80°C until use. Brain tissues were homogenized in 0.1 N hydrochloric acid followed by centrifugation. Supernatant concentration of cGMP or cAMP were measured using enzyme-linked immunosorbent assay kits (Direct cGMP EIA kit, ADI-900-014; Enzo Life Sciences) and homogeneous time-resolved fluorescence kits (HTRF cAMP Dynamic2; Cisbio Bioassays, Bedford, MA), respectively.
Cerebrospinal Fluid and Plasma Sampling in Monkeys.
The cerebrospinal fluid (CSF) sampling was conducted at Shin Nippon Biochemical Laboratories Ltd., as previously described with some modification (Kleiman et al., 2012). Adult cynomolgus monkeys were individually housed in cages (680 mm × 620 mm × 770 mm) kept in a temperature-controlled (23–29°C) and humidity-controlled (30%–70%) animal room. Under anesthesia with ketamine hydrochloride at 10 mg/kg, i.m. (Supriya Lifescience, Mumbai, India), and medetomidine hydrochloride at 0.08 mg/kg, i.m. (Nippon Zenyaku Kogyo, Tokyo, Japan), monkeys were fitted surgically with an indwelling cannula into the cisterna magna, and the cannula was connected to a subcutaneous access port to permit CSF sampling. The animals were administered ketoprofen at 2 m/kg, i.m. (Kissei, Nagano, Japan), for pain relief once a day on the day of surgery and for the next 3 days. To prevent infection, monkeys were administered benzylpenicillin procaine at 10,000 U/kg and dihydrostreptomycin sulfate at 12.5 mg/kg (Kyoritsu Seiyaku, Tokyo, Japan) once a day on the day of surgery and for the next 3 days. These antibiotics were administered again 1 day before each CSF sampling on four consecutive days. After the recovery period, DSR-141562 (30 and 100 mg/kg) or vehicle (10% hydroxy-β-cyclodextrin) was orally administered once a week in a crossover manner. The CSF samples were collected from the indwelling cannula before (baseline) or 0.5, 1, 2, 4, 6, 8, and 24 hours after the administration. The CSF samples were centrifuged at 600g for 10 minutes. The blood samples were collected from the femoral vein into the syringe containing heparin 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after the administration. The plasma samples were separated by centrifugation at 1710g for 15 minutes. The CSF and plasma samples were frozen and stored at −80°C for the analysis of cGMP, cAMP, and drug concentrations.
Measurement of Cyclic Nucleotides and DSR-141562 in Monkey CSF or Plasma.
The CSF samples mixed with internal standard (cAMP-13C5) solution were analyzed by means of liquid chromatography–tandem mass spectrometry, using a QTRAP 6500 (SCIEX) mass spectrometer with electrospray ionization interface in positive ion mode and multiple reactions monitoring. A Nexera ×2 series HPLC system (Shimadzu) was used with a Synergi Hydro RP column (2.0 × 50 mm, 2.5 μm; Phenomenex, Torrance, CA) at a flow rate of 0.4 ml/min to achieve chromatographic separation. A gradient program was used with the mobile phase, combining solvent A [water with 0.1% (v/v) formic acid] and solvent B [acetonitrile with 0.1% (v/v) formic acid] as follows: 1%−7% B for 0−2.5 minutes, 7%−95% B for 2.5−5.0 minutes, 95% B for 5.0−6.5 minutes, and 1% B for 6.6−9.0 minutes. The column temperature was set at 40°C. The transitions (precursor to product) monitored for mass spectrometry were m/z 330.1–136.0 for cAMP, 346.2–152.0 for cGMP, and 335.1–119.0 for cAMP-13C5. Relative change of the CSF cyclic nucleotide concentration from baseline in each animal was calculated by correcting with the baseline value obtained before administration with vehicle or DSR-141562 on each experimental day. The plasma concentrations of DSR-141562 were measured as described previously.
Brain Tissue c-fos Expression Measurement in Rats.
Brain tissue c-fos expression was measured according to a previous procedure with some modifications (Strick et al., 2010). Male Sprague-Dawley rats were subcutaneously administered with vehicle (40% hydroxypropyl-β-cyclodextrin) or DSR-141562 (3, 10, and 30 mg/kg). Sixty minutes later, the rats were sacrificed by decapitation. Frontal cortex, striatum, and cerebellum were dissected out and frozen at −80°C in RNA stabilization solution (RNAlater; Thermo Fisher Scientific Inc., Waltham, MA) until use. Total RNA of each sample was purified using an RNeasy Mini Kit (Qiagen N.V., Venlo, The Netherlands) and quantified with a RiboGreen RNA Quantification Kit (Thermo Fisher Scientific Inc.). Total RNA (1.0 μg) was converted into cDNA using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific Inc.) according to the manufacturer’s instructions. Real-time quantitative polymerase chain reaction was performed in 20 μl of reaction solution containing cDNA (50 ng), gene-specific primers for c-fos (Rn00487426-g1), 18S rRNA (T Eukaryotic 18S rRNA Endogenous Control), and TaqMan Gene Expression Master Mix (Thermo Fisher Scientific Inc.). The reactions were performed using an Applied Biosystems 7500 Fast Real-Time PCR System [Thermal cycling conditions: 50°C for 2 minutes, 95°C for 10 minutes, 95°C for 15 seconds (40 cycles), and 60°C for 1 minute]. Data were analyzed by using the 2−ΔΔC(T) method (Livak and Schmittgen, 2001) and normalized to the housekeeping gene, 18S rRNA. Relative change of c-fos expression was calculated by correcting with the average value of the vehicle group.
Locomotor Activity Measurement in Rats.
The locomotor activities were measured in male Sprague-Dawley rats as previously described with some modifications (Sumiyoshi et al., 2013). To evaluate the effects of DSR-141562 on spontaneous locomotor activities, rats were orally administered DSR-141562 (3, 10, and 30 mg/kg) or vehicle (0.5% methylcellulose) and returned to their home cages. Sixty minutes later, the rats were individually placed in other cages for locomotor measurement (220 mm × 380 mm × 205 mm). The locomotor activity was measured for 90 minutes with a Supermex activity measurement apparatus (Muromachi Kikai Co. Ltd., Tokyo, Japan). To evaluate the effect of DSR-141562 on methamphetamine-induced locomotor hyperactivity, rats were orally administered DSR-141562 (3, 10, and 30 mg/kg) or vehicle, and individually placed in other cages for locomotor measurement. After habituation to the new cage environment for 60 minutes, rats were intraperitoneally injected with methamphetamine (1 mg/kg). Locomotor activity after methamphetamine administration was measured for 90 minutes.
Catalepsy Test in Rats.
The catalepsy test was performed as previously described with some modifications (Ishibashi et al., 2010). Male Sprague-Dawley rats were orally treated with DSR-141562 (1, 10, and 100 mg/kg) or vehicle (0.5% methylcellulose). One hour later, rats’ forepaws were placed on a stainless-steel bar. The time that the animal maintained this position was measured three times, each with a maximum limit of 180 seconds. The maximum of the three measurements was regarded as the catalepsy time.
Social Interaction Test in Mice.
The social interaction test was performed as previously reported with some modification (Qiao et al., 2001). ICR mice were subcutaneously administered saline or phencyclidine (10 mg/kg) twice a day for 5 days (excluding weekends). One day after the last administration, mice were habituated to the experimental cages (400 mm × 250 mm × 300 mm) in a dark room (less than 10 lux) for 2 hours. Four mice from the same home cage were habituated together in the same experimental cage. On the next day, the mice were orally treated with vehicle (0.5% methylcellulose) or DSR-141562 (0.3, 1, and 3 mg/kg). One hour and 50 minutes later, the mice were individually placed in experimental cages for 10 minutes. After the habituation, two mice, which had received the same treatment in different home cages, were placed in the same experimental cage. Their behavior was recorded for 10 minutes with an infrared video camera. Social behavior duration was manually measured for each pair of mice. Social behaviors were defined as sniffing and grooming the partner, following, mounting, and crawling under or over the partner. The passive contact (sitting or lying with bodies in contact) was not included in the social interaction.
Novel Object Recognition Task in Rats.
The novel object recognition task was performed as previously described with some modifications (Horiguchi et al., 2013). Female Long-Evans rats were intraperitoneally treated with saline or phencyclidine (3 mg/kg) twice a day for 10 days (excluding weekends). Three days after the last administration, eight or 10 rats were habituated for 1 hour to the experimental cage (500 mm × 500 mm × 350 mm) without any objects for three consecutive days. Six or 7 days later, the rats received the novel object recognition task. This task consists of a habituation phase, an acquisition trial, a 1-minute intertrial interval, and a retention trial. Rats were orally treated with vehicle (0.5% methylcellulose) or DSR-141562 (0.3, 1, and 3 mg/kg) 60 minutes before the acquisition trial. Rats were individually habituated to the experimental cage without any objects for 3 minutes, and then allowed to explore two identical objects (glass medium bottles, in which the trunk diameter was 65 mm and the height was 143 mm) for 3 minutes in the acquisition trial. After that, rats were returned to their home cages for 1 minute while one of the two objects was replaced with a novel object (a gold metal can, in which the trunk diameter was 65 mm and the height was 122 mm). Rats were allowed to explore the familiar object and the new object for 3 minutes in the retention trial. Object exploration was defined as animals’ sniffing and/or touching the object with the nose. The exploration time of each object in each trial was recorded manually using stopwatches. The discrimination index [(time spent exploring the novel object) − (time spent exploring the familiar object)/(total exploration time)] was calculated for the retention trials. The data from rats that took less than 10 seconds in total to explore during the acquisition trial or a retention trial were excluded from the data analysis.
Object Retrieval with Detour Task in Common Marmosets.
Object retrieval with detour task was performed using well-trained male and female marmosets to reach for the reward (a piece of kneaded cake) placed in a clear acrylic box (4 cm × 4 cm × 4 cm) open only on one side, as previously described (Kotani et al., 2016). Marmosets were tested twice a week: once on a drug-free day and once on a treatment day. The drug treatment tests were performed in a crossover manner with administrations separated by at least 1 week. DSR-141562 (0.3, 3, and 30 mg/kg) or vehicle (0.5% methylcellulose) was orally administered to the animals. Two hours later, the experimenter held a clear acryl box just outside the animal cage with the open side facing left, right, or toward the common marmoset. The positions of the reward in the box were outer edge, inner edge, or deep within the box. Each test session consisted of nine easy trials and eight difficult trials. In the easy trials, the reward was placed on the inner or outer edge of the box with the opening of the box facing left or right from the common marmoset, or inside the box with the opening facing the common marmoset. This enabled the animal to directly reach the reward. In the difficult trial, the reward was placed deep within the box with the opening facing the right or left side of the common marmoset. This arrangement required the animal to make a detour around the box to reach the reward. Reaching the reward without touching any wall of the box within 30 seconds was considered correct. When the common marmoset did not reach the reward within 30 seconds, it was defined as an omission. The performance of each animal in the drug-treated session was compared with that in the drug-free session of nine easy and eight difficult trials using the following equation: Change of success rate = (N drug treated) − (N drug free) × 100/9 or 8, where N drug treated represents the rate of correct responses in the drug-treated session, N drug free represents the rate of correct results in the drug-free session, and 9 or 8 represents the rate in the easy or difficult trial, respectively.
Statistical Analysis.
Data are expressed as mean ± S.D. or ± S.E.M. Data analyses were performed using Stat PreClinica (Takumi Information Technology Inc.) or GraphPad Prism7 (GraphPad Software Inc., La Jolla, CA). For mouse brain cyclic nucleotide measurement, mouse social-interaction test, and rat novel object recognition task, the statistical significance of the difference between two groups was determined by unpaired tests. For mouse brain cyclic nucleotide measurement, rat locomotor activity measurement, rat catalepsy test, mouse social-interaction test, and rat novel object recognition task, the statistical significance of the difference among multiple groups was analyzed by Dunnett’s or Tukey’s multiple comparisons tests. Brain tissue c-fos expression data were analyzed by two-way ANOVA followed by Dunnett’s multiple comparisons test. For monkey CSF cyclic nucleotide measurement, data were analyzed by two-way linear mixed models followed by Dunnett’s multiple comparisons test. For the marmoset object retrieval with detour task, data were analyzed by one-way linear mixed models followed by Dunnett’s multiple comparisons test.
Results
In Vitro Profiles of DSR-141562.
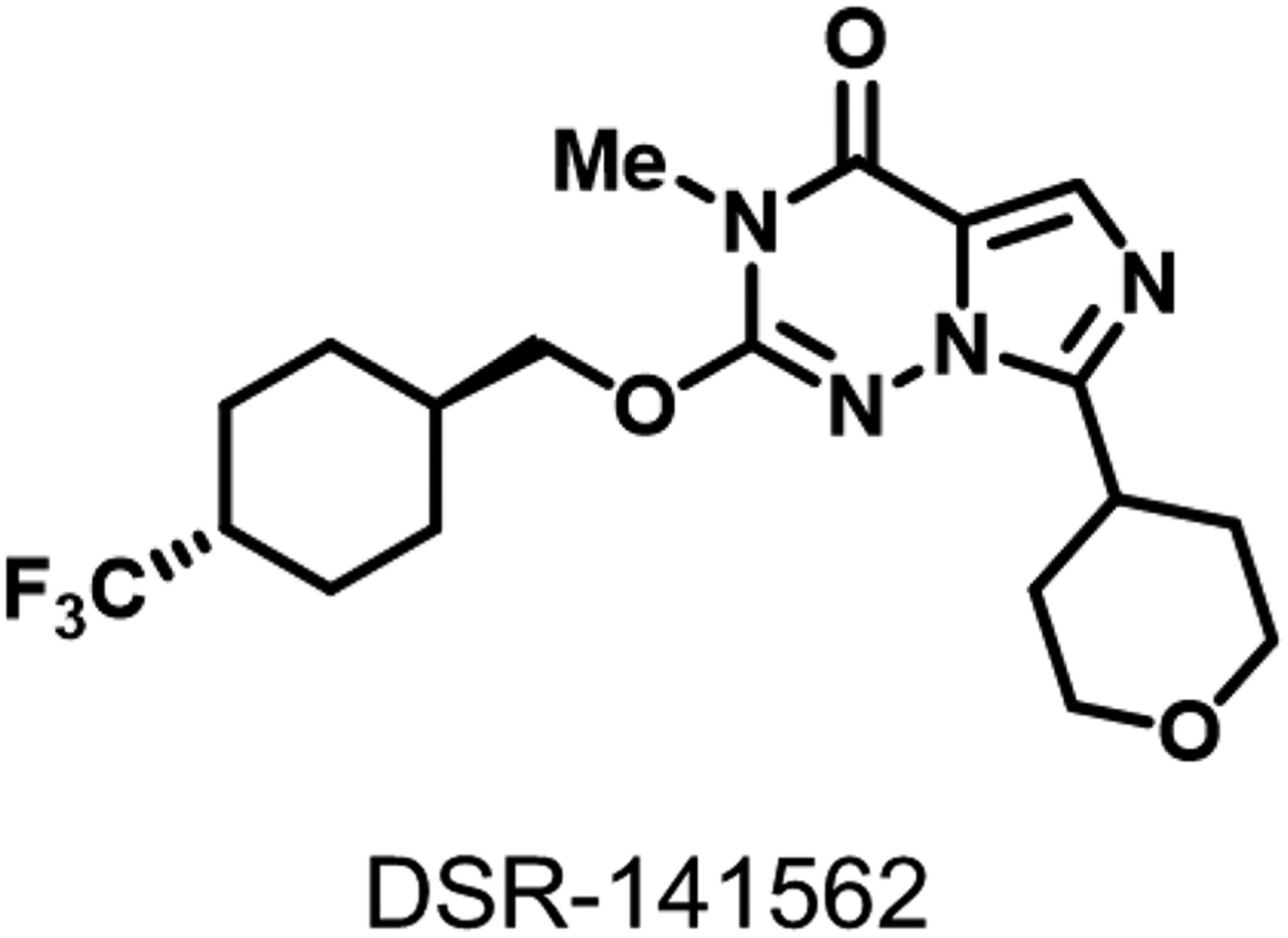
In our drug discovery program for PDE1B inhibitor, we identified a novel compound, DSR-141562 (Fig. 1). DSR-141562 inhibited human PDE1B in vitro with an IC50 value of 43.9 nM (Table 1). Since the IC50 values of DSR-141562 for human PDE1A and 1C were 97.6 and 431.8 nM, respectively, DSR-141562 had a 2.2- and 9.8-fold selectivity for PDE1B over PDE1A and PDE1C (Table 1). DSR-141562 had no effect or only weak inhibitory effects for other PDE families. The IC50 value of DSR-141562 for human PDE2A was 2480 nM, and DSR-141562 showed less than 50% inhibition for other PDE families at 10,000 nM (Table 1). The effects of DSR-141562 on 65 off-targets were evaluated in enzyme and radioligand-binding assays at Eurofins Pharma Discovery Services. Although DSR-141562 at 10,000 nM showed 64% and 51% inhibition of radioligand binding to melatonin MT1 and MT2 receptors (Supplemental Table 2), the inhibitory effects of DSR-141562 for all of the other 63 targets were less than 50% (Supplemental Tables 1 and 2).
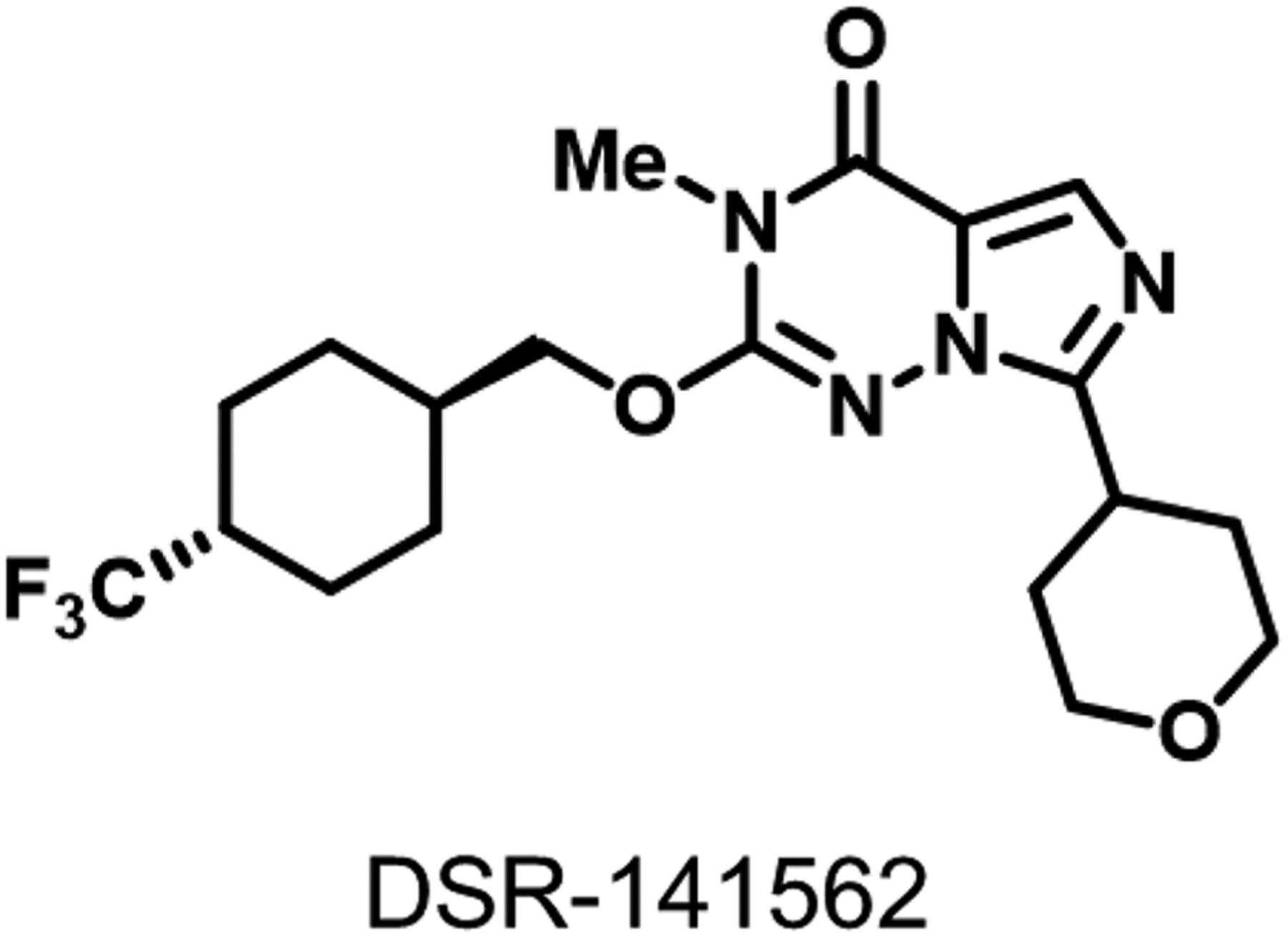
The structure of DSR-141562.
IC50 or inhibitory values of DSR-141562 for human PDEs in vitro
Mean ± S.E.M. (n = 3).
Pharmacokinetic Profiles of DSR-141562.
Plasma concentrations of DSR-141562 were measured after administration of DSR-141562 at 1 mg/kg p.o. and 0.5 mg/kg i.v. in rats and cynomolgus monkeys (Fig. 2, A and B). As summarized in Supplemental Table 3, the oral and intravenous pharmacokinetic profiles of DSR-141562 indicated that DSR-141562 was orally bioavailable. To evaluate the brain permeability of DSR-141562, plasma and brain exposures were measured 0.5, 1, 2, and 3 hours after the oral administration of DSR-141562 at 30 mg/kg in rats. DSR-141562 exhibited good brain uptake, with the brain-to-blood concentration ratio of unbound drug being 0.99 in rats (Fig. 2C). The plasma drug concentrations of this compound for estimating the exposure in pharmacology studies in mice (p.o.), rats (s.c.), and common marmosets (p.o.) are shown in Supplemental Table 4.

Plasma concentrations of DSR-141562 in rats and cynomolgus monkeys. DSR-141562 was diluted in 0.1 N hydrochloric acid/saline (1/9) for intravenous administration or suspended in 0.5% methylcellulose for oral administration. Rats (A) and cynomolgus monkeys (B) were treated with DSR-141562 at an oral dose of 1 mg/kg or intravenous dose of 0.5 mg/kg. Plasma samples were collected 0.083, 0.25, 0.5, 1, 2, 4, 6, and 24 hours after administration. For the plasma-brain exposure profile pharmacokinetic study in rats (C), rats were orally treated with DSR-141562 at 30 mg/kg. Plasma and brain samples were collected 0.5, 1, 2, and 6 hours after administration. Unbound drug concentration was measured in the samples. Data are expressed as mean ± S.D. (n = 3 for oral administration and n = 2 for intravenous administration).
Effects of DSR-141562 on Cyclic Nucleotide Contents In Vivo.
To evaluate the effects of DSR-141562 on cyclic nucleotide levels, the cyclic nucleotide concentrations were measured in the mouse striatum and frontal cortex 2 hours after oral administration of DSR-141562. Compared with vehicle, DSR-141562 at 10 mg/kg p.o. slightly but significantly increased cGMP contents in the frontal cortex (P < 0.05) (Fig. 3A) and striatum (P < 0.01) (Fig. 3B). On the other hand, DSR-141562 did not affect the cAMP content in both brain regions (Supplemental Fig. 1, A and B). To evaluate the effect of DSR-141562 on dopamine D1 receptor signaling, mice were administered DSR-141562 and/or the dopamine D1 receptor agonist SKF-82958. Cotreatment with DSR-141562 and SKF-82958 significantly increased the striatal cGMP level compared with treatment with DSR-141562 and saline (P < 0.01) (Fig. 3C) or vehicle and SKF-82958 (P < 0.01) (Fig. 3C). The treatment with DSR-141562 and/or SKF-82958 did not affect the striatal cAMP level (Supplemental Fig. 1C).
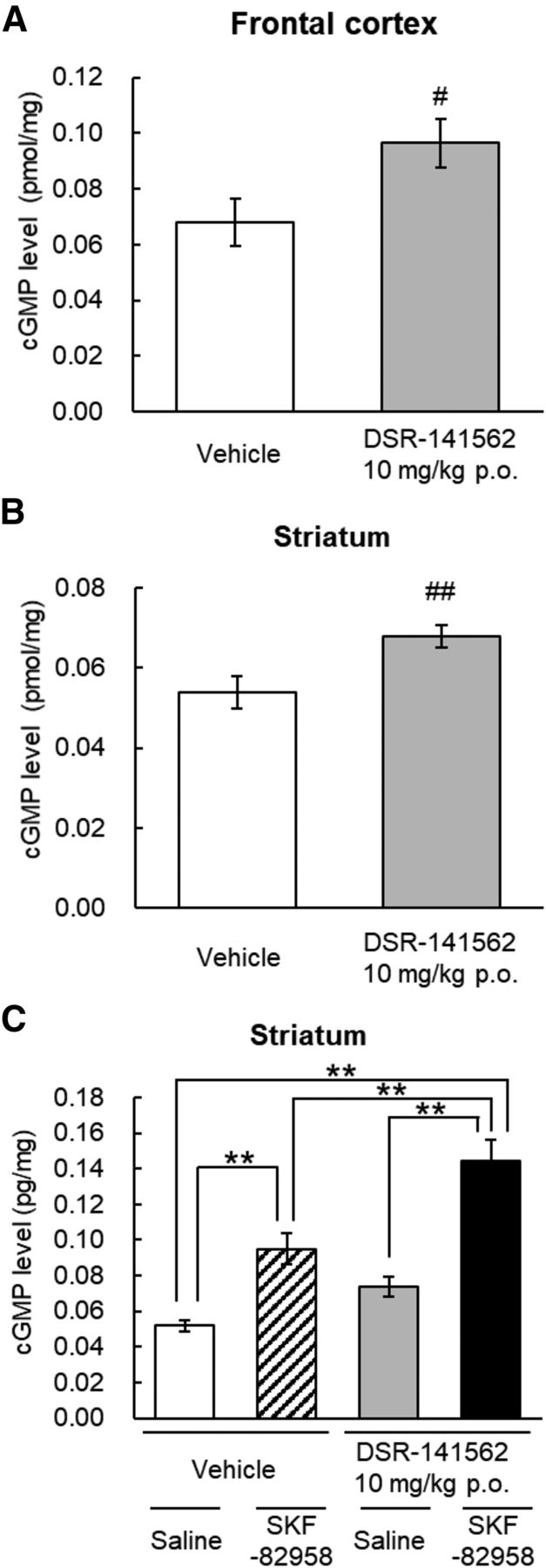
Effects of DSR-141562 on cGMP concentration in mouse brain tissues. Mice were orally administered vehicle (0.5% methylcellulose) or DSR-141562 (10 mg/kg) 120 minutes before brain tissue sampling. In the experiments for the combined effects of DSR-141562 and a dopamine D1 agonist (C), mice were subcutaneously administered saline or SKF-82958 (0.3 mg/kg) 1 hour after the administration of vehicle or DSR-141562. cGMP levels in the striatum (A and C) and frontal cortex (B) were measured by the enzyme-linked immunosorbent assay. Data are expressed as mean ± S.E.M. The number of animals per group was 15 (A and B). The number of animals in the vehicle/saline–, vehicle/SKF-82958 –, DSR-141562/saline–, and DSR-141562/SKF-82958–treated groups was 12, 10, 11, and 11, respectively (C). #P < 0.05; ##P < 0.01 vs. vehicle-treated group (unpaired t test). **P < 0.01 (Tukey’s multiple comparisons test).
Next, we evaluated the possibility that the cGMP level in the CSF is a translational biomarker. When cynomolgus monkeys were orally treated with DSR-141562 at 30 and 100 mg/kg, the plasma concentrations of unbound DSR-141562 were above the IC50 values of DSR-141562 for PDE1B in vitro (43.9 nM) (Fig. 4A). DSR-141562 at 30 mg/kg and 100 mg/kg (compared with vehicle) caused a significant increase in cGMP concentration in monkey CSF (P < 0.01) (Fig. 4B). On the other hand, DSR-141562 (compared with vehicle) induced only a marginal increase in cAMP in monkey CSF (P < 0.01 at 30 mg/kg and P = 0.078 at 100 mg/kg) (Fig. 4C).

Effects of DSR-141562 on cyclic nucleotide concentrations in monkey CSF. Cynomolgus monkeys were orally treated with vehicle (10% hydroxy-β-cyclodextrin) or DSR-141562 (30 and 100 mg/kg) once a week in a crossover manner. The CSF samples were collected before (baseline) or 0.5, 1, 2, 4, 6, 8, and 24 hours after the administration. The blood samples were collected 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after the administration. The unbound drug concentrations in plasma (A) and the cGMP (B) and cAMP (C) concentrations in CSF were measured. The plasma drug concentrations are expressed as mean ± S.D. Change in CSF cyclic nucleotide concentration from baseline is expressed as mean ± S.E.M. The number of animals per group was four. **P < 0.01 vs. vehicle-treated group (two-way linear mixed models followed by Dunnett’s multiple comparisons test).
Effects of DSR-141562 on c-fos Expression in Rat Brains.
To evaluate the effects of DSR-141562 on neuronal activity, we measured the early immediate gene c-fos mRNA expression level in rat brain. When rats were treated with vehicle or DSR-141562 (3, 10, and 30 mg/kg s.c.), DSR-141562 at 10 and 30 mg/kg (compared with vehicle) significantly increased the c-fos mRNA expression level in the striatum (P < 0.01) (Fig. 5), but not in the frontal cortex and cerebellum (Fig. 5).

Effects of DSR-141562 on c-fos mRNA expression in rats. Rats were subcutaneously administered vehicle (40% hydroxy-β-cyclodextrin) or DSR-141562 (3, 10, and 30 mg/kg) 60 minutes before brain tissue sampling. The c-fos mRNA expression was measured by the real-time polymerase chain reaction method in the frontal cortex, striatum, and cerebellum. The c-fos mRNA expression level relative to the control level after vehicle treatment in each brain region is expressed as mean ± S.E.M. The number of animals per group was five. Veh, vehicle. **P < 0.01 vs. vehicle-treated group (two-way ANOVA followed by Dunnett’s multiple comparisons test).
Behavioral Effects of DSR-141562.
We evaluated the effects of DSR-141562 on methamphetamine-induced locomotor hyperactivity in rats using a behavioral assay of antipsychotic efficacy (Jones et al., 2008; Ishibashi et al., 2010). DSR-141562 significantly reversed methamphetamine-induced locomotor hyperactivity (P < 0.05 at 3 mg/kg, and P < 0.01 at 10 and 30 mg/kg p.o.) (Fig. 6A), had no effect on spontaneous locomotor activity at 3 and 10 mg/kg (Fig. 6B), and showed a nonsignificant decrease of spontaneous locomotor activity only at the high dose (30 mg/kg p.o.; P = 0.08) (Fig. 6B).
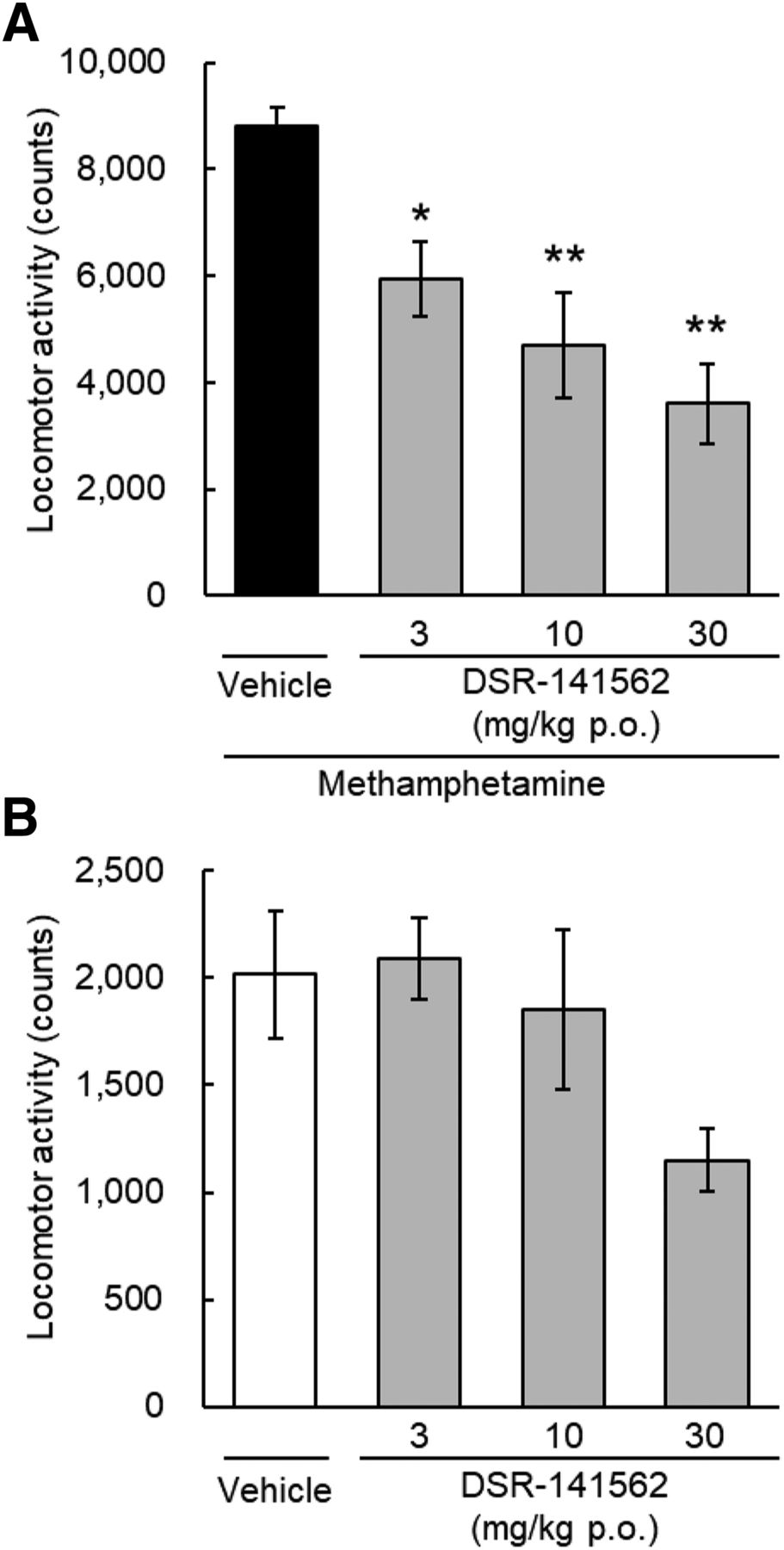
Effects of DSR-141562 on locomotor activity in rats. Rats were orally treated with vehicle (0.5% methylcellulose) or DSR-141562 (3, 10, and 30 mg/kg). Sixty minutes later, methamphetamine (1 mg/kg, i.p.)-induced locomotor hyperactivity (A) or spontaneous locomotor activity (B) was measured for 90 minutes. Data are expressed as mean ± S.E.M. (n = 6). *P < 0.05; **P < 0.01 vs. vehicle/methamphetamine-treated group (Dunnett’s multiple comparisons test).
Next, the potential of DSR-141562 on extrapyramidal side effects was evaluated using the rat catalepsy test. DSR-141562 at 1, 10, and 100 mg/kg did not cause catalepsy. The catalepsy times in the vehicle-treated and DSR-141562-treated groups at 1, 10, and 100 mg/kg were 1.8 ± 0.6, 2.0 ± 0.9, 4.9 ± 2.2, and 2.6 ± 0.8 seconds, respectively (mean ± S.E.M., n = 6).
We evaluated the effects of DSR-141562 on the decrease of social interaction time in mice induced by repeated treatment with the N-methyl-D-aspartate receptor antagonist, phencyclidine, which has been used as a preclinical model for negative symptoms in schizophrenia (Sams-Dodd, 1999; Qiao et al., 2001; Enomoto et al., 2007). Repeated treatment with phencyclidine (10 mg/kg s.c., twice a day for 5 days) significantly decreased the social interaction time compared with repeated treatment with saline (P < 0.01) (Fig. 7). Compared with vehicle, DSR-141562 significantly reversed the phencyclidine-induced decrease of social interaction time in mice (P < 0.05 at 0.3 mg/kg, and P < 0.01 at 1 and 3 mg/kgp.o.) (Fig. 7).

Effects of DSR-141562 on phencyclidine-induced decrease of social interaction time in mice. Mice were repeatedly treated with saline or phencyclidine (10 mg/kg s.c., twice a day for 5 days). Two days after the last administration of saline or phencyclidine, mice were acutely treated with vehicle (0.5% methylcellulose) or DSR-141562 (0.3, 1, and 3 mg/kg p.o.). Two hours later, social interaction time between two mice, which received the same treatment in different home cages, was measured for 10 minutes. Data are expressed as mean ± S.E.M. (n = 16). ##P < 0.01 vs. vehicle/saline–treated group (unpaired t test). *P < 0.05; **P < 0.01 vs. vehicle/phencyclidine–treated group (Dunnett’s multiple comparisons test).
To evaluate the effects of DSR-141562 on cognitive function, we performed the novel object recognition task in subchronic phencyclidine-treated rats and the object retrieval with detour task in common marmosets. Repeated treatment with phencyclidine (3 mg/kg i.p., twice a day for 10 days) decreased the discrimination index compared with repeated treatment with saline (P < 0.01) (Fig. 8). DSR-141562 at 0.3, 1, and 3 mg/kg p.o. reversed the decrease of the discrimination index induced by repeated phencyclidine (P < 0.01) (Fig. 8). In the object retrieval with detour task, DSR-141562 (0.3, 3, and 30 mg/kg p.o.) did not affect the success rate in the easy trials (data not shown). On the other hand, DSR-141562 at 3 and 30 mg/kg significantly improved the success rate compared with vehicle in the difficult trials (P < 0.01) (Fig. 9).
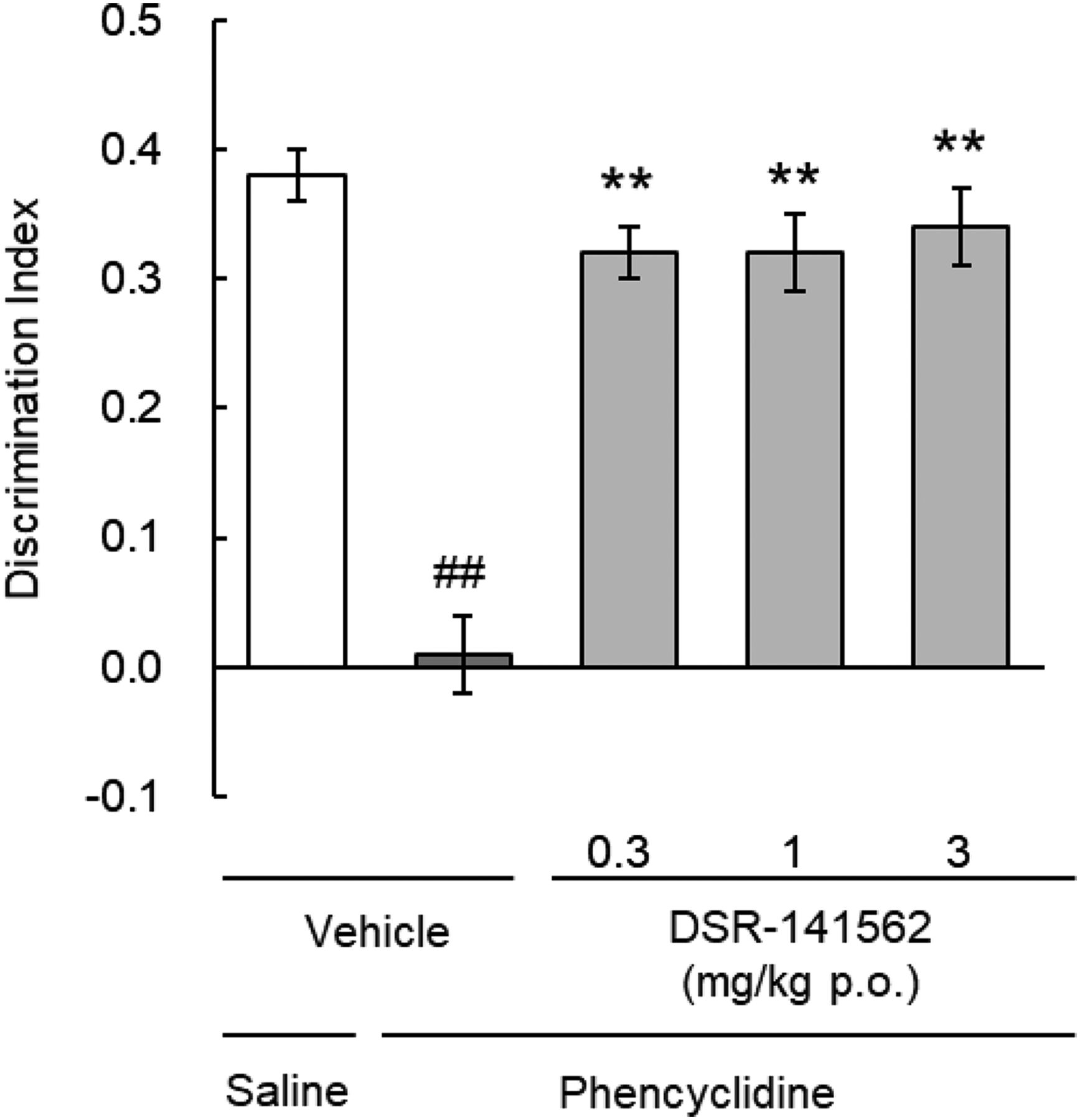
Effects of DSR-141562 on phencyclidine-induced learning and memory impairments in the rat novel object recognition test. Rats were repeatedly treated with saline or phencyclidine (3 mg/kg i.p., twice a day for 10 days). Eleven or 12 days after the last administration with saline or phencyclidine, rats were acutely treated with vehicle (0.5% methylcellulose) or DSR-141562 (0.3, 1, and 3 mg/kg p.o.). One hour later, the novel object recognition task was performed. The discrimination index [(time spent exploring the novel object) − (time spent exploring the familiar object)/(total exploration time)] was measured. Data are expressed as mean ± S.E.M. (n = 14 for vehicle/phencyclidine–treated and DSR-141562 at 0.3 mg/kg/phencyclidine-treated groups, n = 15 for other groups). ##P < 0.01 vs. vehicle/saline–treated group (unpaired t test). **P < 0.01 vs. vehicle/phencyclidine–treated group (Dunnett’s multiple comparisons test).

Effects of DSR-141562 on executive function in common marmosets. Marmosets were orally treated with vehicle (0.5% methylcellulose) or DSR-141562 (0.3, 3, and 30 mg/kg) 2 hours before the object retrieval with detour task. The test was performed in a crossover manner with administrations separated by at least 1 week. The change in success rate from the baseline performance was measured in the difficult trials. Data are expressed as mean ± S.E.M. (n = 10). **P < 0.01 vs. vehicle-treated group (linear mixed model followed by Dunnett’s multiple comparisons test).
Discussion
Here, we report that the PDE1 inhibitor DSR-141562 has efficacy in animal models for all three major symptoms in schizophrenia. As far as we know, this is the first paper to show that the PDE1 inhibitor is efficacious in animal models of schizophrenia-associated positive and negative symptoms. Furthermore, it provides the first evidence that the PDE1 inhibitor improves cognitive function in nonhuman primates.
Although PDE1, especially PDE1B, is believed to be an attractive treatment target for neuropsychiatric disorders, the research has been hampered by the shortage of brain-penetrant compounds with PDE1B selectivity. DSR-141562 has remarkable brain permeability with a brain-to-blood unbound drug concentration ratio of 0.99. While the IC50 value of DSR-141562 for PDE1B is 43.9 nM, it acts on other PDE families and other tested targets only in the micromolar range or above. Furthermore, this compound has approximately 2–10 times selectivity for the PDE1B isoform over other PDE1 family isoforms.
DSR-141562 elevated the tissue level of cGMP in mouse striatum and frontal cortex. Furthermore, the cGMP level in the monkey CSF was also elevated after administration of DSR-141562. Cyclic nucleotide concentration has been used as a biomarker in preclinical and clinical studies with PDE9A inhibitors (Kleiman et al., 2012; Boland et al., 2017). Taken together, the data suggest that cGMP in the CSF might be a valuable translational biomarker to detect PDE1 inhibition in the brain. On the other hand, this compound did not elevate the cAMP level in mouse brain tissues, and only marginally increased the cAMP level in monkey CSF. This suggests that cAMP in CSF might not be the most reliable biomarker for PDE1 inhibition. The efficacy of PDE1 inhibition for increasing cAMP levels in brain homogenates might be masked by the high cAMP baseline (Chen et al., 2019). In addition, it might be related to the fact that PDE1B and PDE1A enzymes prefer cGMP (Km = 1.2–5.9 μM for PDE1B; Km = 2.6–3.5 μM for PDE1A) to cAMP (Km = 10–24 μM for PDE1B; Km = 72.7–124 μM for PDE1A) (Bender and Beavo, 2006). However, it is worth mentioning that the brain’s cyclic nucleotide content is not a highly sensitive indicator, especially considering the on-demand nature of PDE1 enzyme activation by phasic Ca2+ influx (Grauer et al., 2009; Wennogle et al., 2017; Pekcec et al., 2018; Betolngar et al., 2019). Strick et al. (2010) have reported that immediate early gene c-fos expression is regulated by the cAMP pathway but not the cGMP pathways after administration of an inhibitor of PDE10A, which is another striatum-enriched dual-substrate PDE isoform. When we assessed neural activity by measuring c-fos mRNA expression, DSR-141562 elevated the c-fos expression level in the rat striatum but not the frontal cortex and cerebellum. PDE1B is one of the most robustly expressed enzymes in the striatum among PDEs, and its striatal expression is relatively high compared with the expression of PDE1 isoforms in other brain regions (Lakics et al., 2010; Kelly et al., 2014). Thus, DSR-141562 might affect the cAMP downstream signaling pathway at least in the striatum where PDE1B is predominantly expressed.
In the present study, we found that DSR-141562 at 3–30 mg/kg inhibits methamphetamine-induced locomotor hyperactivity in rats, which has been widely used as a preclinical behavioral assay system for antipsychotic activity (Jones et al., 2008; Ishibashi et al., 2010). Importantly, the effective dose of DSR-141562 in this antipsychotic test has only minimal effects on spontaneous locomotor activity, which indicates that the effects of DSR-145162 cannot be attributed to general sedation or motor deficits. These findings are somewhat surprising since previous studies have demonstrated that PDE1B-deficient mice exhibit increased spontaneous locomotor activity and exaggerated methamphetamine- or amphetamine-induced locomotor hyperactivity (Reed et al., 2002; Siuciak et al., 2007). We recognize that current experimental results cannot exclude the possibility that PDE1A, PDE1C, or other untested pharmacological targets of DSR-141562 or its metabolites might have antipsychotic effects, although DSR-141562 has a high selectivity for PDE1 family isoforms among our tested targets, which include potential antipsychotic targets (Nishi and Snyder, 2010; Girgis et al., 2019). However, sometimes the discrepancies in the behavioral phenotype of genetically manipulated mice and in the pharmacological effects of compounds are due to compensatory changes during development (Hufgard et al., 2017a). For example, PDE1B-deletion mice manifest a compensatory increase in PDE10A in the striatum (Hufgard et al., 2017a). In addition, the immobility-resistant phenotype of PDE1B-deficient mice is detected when the PDE1B gene deletion occurs during postnatal days 0 or 32 but not during adulthood (Hufgard et al., 2017b). Furthermore, PDE1B-deficient mice display spatial learning and memory impairments in the Morris water maze task (Reed et al., 2002), while PDE1 inhibitors improve learning and memory function in several behavioral tasks in mice and rats (Li et al., 2016a; Snyder et al., 2016; Pekcec et al., 2018; McQuown et al., 2019). It is worth mentioning that PDE1B is expressed not only in D1 receptor–expressing but also D2 receptor–expressing medium spiny neurons, which are targets of current antipsychotics (Polli and Kincaid, 1994; Yan et al., 1994; Nishi and Snyder, 2010). The regulation of D2 receptor downstream signaling by DSR-141562 might be involved in its antipsychotic efficacy. Indeed, Betolngar et al. (2019) have reported that PDE1 inhibitor suppresses the N-methyl-D-aspartate–induced decrease in cAMP not only in D1 receptor–expressing but also D2 receptor–expressing striatal neurons in vitro. Thus, the antipsychotic mechanism of DSR-141562, especially the contribution of the D2 receptor–expressing indirect pathway, remains to be evaluated.
Most current antipsychotics induce catalepsy at near effective doses in antipsychotic behavioral assays (Ishibashi et al., 2010). DSR-141562 does not induce any sign of catalepsy at even 100 mg/kg, which is a 33-times higher dose than the effective dose on methamphetamine-induced locomotor hyperactivity. This is quite in contrast to the concept that antipsychotic candidate PDE10A inhibitors typically operate within a narrow dose range because of extrapyramidal side effects in mice, rats, monkeys, and humans (Schmidt et al., 2008; Grauer et al., 2009; Uthayathas et al., 2014; Li et al., 2016b; Goldsmith et al., 2017). Since the extrapyramidal side effects were dose limiting in clinical studies with D2 receptor antagonists and PDE10A inhibitors, the wide safety margin of the DSR-141562 dose for catalepsy would be of great benefit in schizophrenia treatment.
Since phencyclidine is known to mimic schizophrenia-like symptoms in humans including positive, negative, and cognitive symptoms (Javitt and Zukin, 1991), it has been widely used in animals to model schizophrenia, especially its negative and cognitive symptoms (Jentsch and Roth, 1999; Sams-Dodd, 1999; Enomoto et al., 2007). The present study demonstrated that DSR-141562 reversed the decrease of social interaction time in mice repeatedly injected with phencyclidine, which is an animal model for negative symptoms. In addition, we found that DSR-141562 reversed learning and memory impairments in rats repeatedly injected with phencyclidine. Furthermore, we assessed the effects of DSR-141562 in common marmosets, which have a well-developed prefrontal cortex (Lewis and González-Burgos, 2008), since the pathophysiology of schizophrenia has been thought to involve the prefrontal cortex (Marvanová et al., 2003; Kaiser and Feng, 2015). The object retrieval with detour task is a behavioral test for the attentional and inhibitory aspects of executive function, which is dependent on the prefrontal cortex (Diamond et al., 1989; Wilkinson et al., 1997; Wallis et al., 2001). We have previously reported that most antipsychotics have detrimental effects or no beneficial effects on executive function in this task (Murai et al., 2013; Baba et al., 2015). On the other hand, DSR-141562 potently improved executive function in marmosets in the present study, which suggests that this compound has great potential as a cognitive enhancer.
The dopamine D1 receptor is expected to be an attractive target for the development of agonists that are effective against schizophrenia-associated negative and cognitive symptoms (Goldman-Rakic et al., 2004; Remington et al., 2011). However, dopamine D1 receptor agonism has peripheral side effects such as hypotension, which has hampered the clinical development of agonists for these indications (Arnsten et al., 2017). PDE1B enzymes are colocalized with D1 receptors in the striatum and prefrontal cortex (Polli and Kincaid, 1994; Yan et al., 1994; Pekcec et al., 2018), and mainly based on research with genetically manipulated animals it has been hypothesized that PDE1B inhibition augments downstream D1 receptor signaling (Reed et al., 2002; Nishi and Snyder, 2010). We found that DSR-141562 enhanced the increase of cGMP induced by a dopamine D1 receptor agonist. Although the physiological roles of the D1 receptor agonist–induced cGMP pathway are relatively unknown compared with the traditional cAMP pathway (Altar et al., 1990; Snyder and Vanover, 2014), cGMP signaling has recently attracted a great deal of attention as a therapeutic target for schizophrenia-associated negative and cognitive symptoms (Reneerkens et al., 2009; Shim et al., 2016). DSR-141562–induced augmentation of D1 receptor signaling might be related to its efficacy for negative and cognitive symptoms.
In summary, we identified the orally-available and brain-penetrating PDE1 inhibitor DSR-141562. Since DSR-141562 exhibits potent efficacy in animal models for positive, negative, and cognitive symptoms associated with schizophrenia, this compound could have great potential as a novel therapeutic agent for schizophrenia.
Acknowledgments
We express our appreciation to Akemi Nishihara, Hiroko Ikeda, Yuji Yasuda, Yuji Ogi, Tatsuo Nakayama, and Tomokazu Nakako for technical expertise, and Nobuyuki Yamaguchi, Tatsuya Ishikawa, Junji Ichihara, Kumiko Ishida, and Hiroyuki Nishikawa for helpful discussions.
Authorship Contributions
Participated in research design: Enomoto, Nishizato, Nishigori, Kitamura, Hashimoto.
Conducted experiments: Enomoto, Tatara, Goda, Nishizato, Nishigori, Kamada, Taga, Ikeda.
Contributed new reagents or analytic tools: Fujii.
Performed data analysis: Enomoto, Nishizato, Nishigori, Kitamura.
Wrote or contributed to the writing of the manuscript: Enomoto, Nishigori, Kitamura, Kamada.
Footnotes
- Received June 17, 2019.
- Accepted September 18, 2019.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CSF
- cerebrospinal fluid
- DSR-141562
- 3-methyl-7-(tetrahydro-2H-pyran-4-yl)-2-{[trans-4-(trifluoromethyl)cyclohexyl]-methoxy}imidazo[5,1-f][1,2,4]triazin-4(3H)-one
- ICR
- Institute of Cancer Research
- ITI-214
- (6aR,9aS)-2-[4-(6-Fluoropyridin-2-yl)benzyl]-5-methyl-3-(phenylamino)-5,6a,7,8,9,9a-hexahydrocyclopenta-[4,5]imidazo[1,2-a]pyrazolo[4,3-e]pyrimidin-4-(2H)-one phosphate
- m/z
- mass-to-charge ratios
- PDE
- phosphodiesterase
- s.c.
- subcutaneous administration
- SKF-82958 hydrobromide
- 3-Allyl-6-chloro-1-phenyl-2,3,4,5-tetrahydro-1H-benzo[d]azepine-7,8-diol hydrobromide
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}