


Abstract
Blockade of interleukin (IL)-23 or IL-17 with biologics is clinically validated as a treatment of psoriasis. However, the clinical impact of targeting other nodes within the IL-23/IL-17 pathway, especially with small molecules, is less defined. We report on a novel small molecule inverse agonist of retinoid acid–related orphan receptor (ROR) γt and its efficacy in preclinical models of psoriasis and arthritis. 1-(2,4-Dichloro-3-((1,4-dimethyl-6-(trifluoromethyl)-1H-indol-2-yl)methyl)benzoyl)piperidine-4-carboxylic acid (A-9758) was optimized from material identified from a high-throughput screening campaign. A-9758 is selective for RORγt and exhibits robust potency against IL-17A release both in vitro and in vivo. In vivo, we also show that IL-23 is sufficient to drive the accumulation of RORγt+ cells, and inhibition of RORγt significantly attenuates IL-23–driven psoriasiform dermatitis. Therapeutic treatment with A-9758 (i.e., delivered during active disease) was also effective in blocking skin and joint inflammation. Finally, A-9758 exhibited efficacy in an ex vivo human whole blood assay, suggesting small molecule inverse agonists of RORγt could be efficacious in human IL-17–related diseases.
SIGNIFICANCE STATEMENT Using a novel small molecule inverse agonist, and preclinical assays, we show that RORγt is a viable target for the inhibition of RORγt/Th17–driven diseases such as psoriasis. Preclinical models of psoriasis show that inhibition of RORγt blocks both the accumulation and effector function of IL-17–producing T cells.
Introduction
Many therapeutic agents exist to treat a variety of inflammatory and autoimmune diseases. Currently, biologics dominate the group of agents that bring the most benefit to patients. Subsequently, there remains significant unmet medical need for efficacious small molecules.
The success of anti-interleukin (IL)-17 biologics (secukinumab/Cosentyx; ixekizumab/Taltz) or anti-IL-23/p40/p19 (ustekinumab/Stelara; guselkumab/Tremfya, risankizumab) in the treatment of psoriasis validates the IL-23/IL-17 pathway as an important target for therapy (Bartlett and Million, 2015). Therefore, alternative targets within this pathway have strong potential to become new efficacious therapies. In addition, emerging data suggest targeting the IL-23/IL-17 pathway may be efficacious in other diseases such as ankylosing spondylitis (Paine and Ritchlin, 2016; Sieper, 2016) and Crohn’s disease (Danese et al., 2017); highlighting the importance of supporting drug discovery efforts in this pathway. Retinoic acid–related orphan receptor (ROR)γt represents an attractive target in this area since it bridges the gap between IL-23 and IL-17 controlling processes such as T helper (Th)17 cell differentiation and effector function (Jetten, 2011). A number of companies have recently published data describing RORγt inverse agonist/antagonist tool compounds (Skepner et al., 2014; Wang et al., 2014; Xiao et al., 2014; Fauber et al., 2015; Scheepstra et al., 2015; Skepner et al., 2015; Banerjee et al., 2016; Guo et al., 2016; Hintermann et al., 2016; Smith et al., 2016; Xue et al., 2016; Guendisch et al., 2017; Takaishi et al., 2017). Recently, Vitae Pharmaceuticals reported a 4-week clinical proof-of-concept trial with the RORγt inhibitor VTP-43472 in psoriasis (Gege, 2016, 2017; Bronner et al., 2017).
The ROR isoforms RORα (NR1F1), RORβ (NR1F2), and RORγ (NR1F3) are members of the steroid nuclear hormone receptor superfamily (Jetten, 2009). RORs have been shown to play prominent roles in a variety of biologic processes including organ development, immunity, lipid homeostasis and metabolism, and circadian rhythm (Jetten, 2009). Mammalian RORγ exists in two distinct isoforms (RORγ and RORγt), which possess identical ligand binding domains (LBDs) and differ only in their N-terminal sequence (Medvedev et al., 1996). Expression of the RORγt isoform is restricted to lymphoid organs including the thymus, whereas RORγ is more broadly expressed (liver, muscle, and kidney) (Jetten, 2009). RORγt has been shown to be critical for the development of lymph nodes and Peyer’s patches and for the differentiation of thymocytes (Sun et al., 2000). Furthermore, RORγt is the obligatory transcription factor that controls the differentiation of naive CD4+ T cells into Th17 lineage, and regulates transcription of the effector cytokine IL-17 in Th17 cells and cells of the innate immune response in both rodents and humans (Ivanov et al., 2006).
Similar to other nuclear hormone receptors, the ROR family members are composed of both a LBD and a DNA-binding domain. Ligand binding causes a conformational change that modulates binding of coregulatory proteins. Agonists recruit coactivators and antagonists or inverse agonists disrupt the binding of coactivators or enhance the binding of corepressors, thereby repressing the transcription of target genes (Fauber and Magnuson, 2014). By inhibiting the recruitment of coactivators and promoting the recruitment of corepressors RORγ inhibitors (inverse agonists) may reduce RORγ transcriptional activity, Th17-cell differentiation, and IL-17 production. This impact on biology is expected to have a therapeutic effect on autoimmune diseases such as psoriasis. Indeed, small molecule inhibitors of RORγt have already been shown to be effective in the treatment of preclinical models of multiple sclerosis (Xiao et al., 2014; Guo et al., 2016), psoriasis (Skepner et al., 2015; Banerjee et al., 2016; Guo et al., 2016; Smith et al., 2016; Xue et al., 2016; Takaishi et al., 2017), and arthritis (Wang et al., 2014; Xue et al., 2016; Guendisch et al., 2017).
In this study, we describe the in vitro and in vivo characterization of a novel RORγt inverse agonist, 1-(2,4-dichloro-3-((1,4-dimethyl-6-(trifluoromethyl)-1H-indol-2-yl)methyl)benzoyl)piperidine-4-carboxylic acid (A-9758) with robust activity across animal species and therapeutic efficacy in skin and joint inflammatory models (Argiriadi et al., 2018).
Materials and Methods
RORγ Transactivation Assay.
Cos-7 cells were transiently transfected in 384-well plates with vector pSG5-GAL4-DBD/LBD-RORγ (Stratagene) (either human, dog, rat, or mouse RORγ) and pGAL4RE-pGL3, a reporter plasmid that contains five copies of the GAL4 response element (5′-TCGGAGGACAGTACTCC-3′) upstream of the thymidine kinase promoter (−105/+56) inserted in a pGL3-Basic vector (Promega). Twenty-four hours post-transfection, compounds were added for an additional 18 hours before luciferase activity was measured using an Envision Plate Reader (Perkin-Elmer). The relative light units at each concentration of compounds were normalized between 100% (high control) and 0% (low control using T0901317 at 10 µM) and plotted for IC50 determination.
AlphaScreen and Cofactor Recruitment Assay.
AlphaScreen Technology (i.e., amplified luminescent proximity homogeneous assay) was used to determine the compound-dependent interaction of the RORγt LBD [His-RORγ (amino acid T259-K518); Novalix] with the coactivator PGC1α (LXD1) peptide (N-biotin-QEAEEPSLLKKLLLAPANTQL-COOH; Bachem). The assay was developed in a 384-well format using Hepes 25 mM, NaCl 100 mM, and bovine serum albumin (BSA) 0.1%, pH 7.4, as the buffer. The buffer and PGC1α peptide at a concentration of 300 nM were distributed first, and then RORα, RORβ, or RORγ protein was added at concentrations of 3, 3, and 30 nM, respectively. The compound was added within a final concentration range of 10 µM to 0.3 nM and final DMSO concentration of 0.5%. The assay was incubated for 1 hour at room temperature and protected from light. Ni-chelate acceptor and streptavidin donor beads (Perkin-Elmer), both 20 µg/ml, were added and incubated for a further 2 hours. The fluorescence signal was then read on an Envision Plate Reader. The fluorescence signal at each concentration of compounds was normalized between 100% (high control) and 0% (low control without peptide) and plotted for IC50 determination.
To better profile the compound on a panel of coactivators and corepressors, similar conditions as described previously were used except that the final concentrations of RORγt and peptides were 0.1 µM. The following peptides were generated by Bachem: NCoA1 (LXD4) (N-biotin-CPSSHSSLTERHKILHRLLQEGSPS-COOH), EBIP37 (N-biotin-TGGGVSLLLHLLNTEQGES-COOH), NCoR1 (N-biotin-GHSFADPASNLGLEDIIRKALMGSF-COOH), and NCoR2 (N-biotin-EHASTNMGLEAIIRKALMGKY-COOH).
Radioligand Binding Assay.
One hundred nanograms of purified His-RORγ LBD was incubated for 2 hours, using Nunc 96-well polypropylene plates, with various concentrations of 25-[3H]hydroxycholesterol (Perkin-Elmer) in assay buffer [50 mM Hepes (pH 7.4), 150 mM NaCl, 5 mM MgCl2, 0.01% BSA, and 1 mM dithiothreitol]. Nonspecific binding was measured using an excess of nonradioactive 25-hydroxycholesterol, which was used for background removal in the calculation. The assay was terminated by rapid filtration through GFB Unifilter Plates (Perkin-Elmer) that were presoaked for 1 hour in 0.05% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate and then were washed four times with cold assay buffer. After filtration of the reaction mixture, filter plates were washed three times with ice-cold assay buffer and then dried for 1 hour at 50°C. Then, UltimaGold scintillation cocktail was added and the plates were read after 4 hours on a MicroBeta counter.
The radioligand binding results were analyzed using Prism Software (GraphPad Software, La Jolla, CA). The measured KD value of 25-[3H]hydroxycholesterol was 20 nM. For compound IC50 determination 20 nM of 25-[3H]hydroxycholesterol was used.
Human CD4+ T Cell IL-17A Release Assay.
Peripheral blood mononuclear cells were prepared from a buffy coat by Ficoll-Paque density grade centrifugation and CD4+ T cells were isolated by positive selection (Miltenyi Biotec). CD4+ T cells were frozen until further use. CD4+ T cells were thawed and cultured in a T75 flask with RPMI 1640 (Invitrogen) containing 10% inactivated FBS (Hyclone), 1% L-glutamine (Invitrogen), and 0.5% penicillin-streptomycin (Invitrogen). After overnight incubation, cells were transferred to anti-CD3 precoated 96-well plates (Becton Dickinson Biosciences). Cells were stimulated with anti-CD28 antibody (1 µg/ml) and test compound at the desired concentration (0.1% DMSO final concentration) in X-Vivo 15-cell culture media (Ozyme) containing 1% glutamine, 0.5% penicillin-streptomycin, and 3 mg/ml BSA (Invitrogen). After 72 hours, IL-17A was measured in cell culture supernatants by ELISA (Biolegend). Cell viability was evaluated by CellTiter-Glo (Promega). Optical density and luminescent parameters were measured using an Envision Multilabel Plate Reader (Perkin-Elmer). The IL-17A levels at each concentration of compound were normalized between 100% (high control) and 0% (low control using T0901317 at 10 µM) and plotted for IC50 determination.
Mouse Splenocyte IL-17A Release Assay.
A single cell suspension was generated from the spleens of C57BL/6J mice (Janvier Laboratories, France). Red blood cells were lysed using Pharmlyse (Beckton-Dickinson) and splenocytes were resuspended in culture media: RPMI 1640 (Invitrogen) containing 10% heat-inactivated FBS (Hyclone), 1% L-glutamine (Invitrogen), 0.5% penicillin-streptomycin (Invitrogen), 1% nonessential amino acids (Invitrogen), 1% sodium pyruvate (Invitrogen), and 10 mM Hepes. Cells were then plated in 96-well precoated plates with anti-CD3 (BD Pharmingen) and anti-CD28 (BD Pharmingen) antibodies. A Th17 polarizing cocktail of 5 ng/ml transforming growth factor-β (R&D), 50 ng/ml IL-6β (R&D), 10 ng/ml IL-1β (R&D), 10 µg/ml anti-IL-4 antibody (R&D), 10 µg/ml anti-interferon-γ (IFNγ) antibody (R&D), and 5 ng/ml IL-23 (Biolegend) was also added. Test compound was added at different concentrations (0.1% DMSO final concentration) and cells were incubated for 96 hours. Cell supernatant was then collected and IL-17A levels were measured by ELISA (Biolegend). Cell viability was also measured by CellTiter Glo assay (Promega). IL-17A levels at each concentration of compounds were normalized between 100% (high control) and 0% (low control using T0901317 at 10 µM) and plotted for IC50 determination.
Mouse Th17 Differentiation Assay.
Splenic CD4+ T cells were purified from C57BL/6 mice (6–8 weeks of age; Charles River Laboratories, MA) using the CD4 T Cell Isolation Kit (StemCell Technologies). CD4+ T cells were incubated for 3 days in the presence of anti-CD3 and anti-CD28 (10 μg/ml; BD Biosciences) precoated 96-well plates with or without Th17 polarization media [RPMI 1640 containing 10% FBS, 1X L-glutamine, 1X penicillin-streptomycin, 1X nonessential amino acids, 1 mM Na-pyruvate, and 10 mM Hepes (all from ThermoFisher Scientific, Waltham, MA) plus 5 ng/ml recombinant murine transforming growth factor-β1, 50 ng/ml recombinant murine IL-6, 10 ng/ml recombinant murine IL-1β, 10 μg/ml anti-mouse IL-4, and 10 μg/ml anti-mouse IFNγ (all from R&D Systems)] in the presence or absence of A-9758. Four hours prior to harvest, BD GolgiPlug/Brefeldin A (BD Biosciences) was added to the culture. Flow cytometry staining and analysis for intracellular IL-17A and RORγt measurements were then performed. A complete list of antibodies used for the flow-cytometric analysis is described in Supplemental Table 1. IL-17A levels in the supernatant were determined by ultrasensitive mouse IL-17A assay (Meso Scale Diagnostics, MD).
Human Whole Blood Assay.
Analogous to published methodology for the assessment of IL-17 production from human whole blood (Russell et al., 2018), blood was collected from healthy volunteers after informed consent and under an approved Institutional Review Board protocol (AbbVie Inc.). Whole blood was mixed 1:2.6 in RPMI 1640 (with Dutch modifications). Two hundred microliters of diluted blood was added to Nunc flat-bottom 96-well tissue culture plates that already contained vehicle (0.1% DMSO) or A-9758. CytoStim (Miltenyi Biotec) was added to a final concentration of 2.5 μl CytoStim per well. The final well volume was 250 μl. Plates were incubated for 48 hours at 37°C/5% CO2. Plates were then centrifuged at 1500 rpm for 5 minutes and plasma was collected for IL-17A analysis (Meso Scale Diagnostics).
Mice.
All AbbVie-related animal studies were performed in accordance with approved guidelines and under approved Institutional Animal Care and Use Committee protocols for the Institution. Animal studies performed at Inventiva were conducted under European Union animal welfare regulations for animal use (European Directive 2010/63/EEC) and under a protocol approved by the Inventiva Ethical Committee (Comité de Reflexion Ethique en Expérimentation Animale, registered by the Ministère de l’Enseignement Supérieur et de la Recherche under No. 104). Animals were purchased from Charles River Laboratories, The Jackson Laboratory (Bar Harbor, ME), or Janvier Laboratories.
IL-23+/IL-1β Model.
Female C57BL/6 mice, aged 6–8 weeks, were dosed intraperitoneally 24 hours prior to challenge with 25 mg/kg mouse anti-p40 or by mouth 1 hour prior to challenge with A-9758 in 0.5% hydroxypropyl methylcellulose/0.02% Tween vehicle. In a model analogous to Fauber et al. (2015), mice were then challenged with a single intravenous injection of 300 ng IL-23+ and 1000 ng IL-1β. Three hours post challenge, mice were sacrificed by inhaled isofluorane and blood was collected via cardiac puncture. EDTA plasma was analyzed for cytokine levels via Meso Scale Discovery assay.
Dual Anti-CD3 Mouse Model.
Using a model similar to that described by Esplugues et al. (2011), female C57BL/6J mice (aged 8 weeks) were intraperitoneally injected with 10 µg/mouse of anti-CD3 (clone 2C11) 1 hour after compound treatment and again 48 hours later. Eleven mice per group received compound or vehicle twice a day via oral gavage. Blood samples from three mice were collected for evaluation of plasma compound exposure 0.5 hours after the last anti-CD3 injection. Blood samples from eight mice were collected for evaluation of serum IL-17A levels 4 hours after the last anti-CD3 injection. Plasma compound concentrations were measured by liquid chromatography–tandem mass spectrometry (mass spectrometer: API400 QTrap, Sciex; liquid chromatography system: UFLC-XR; Shimadzu) and serum IL-17A was quantified by the Milliplex Map Mouse Th17 Magnetic Bead Panel Kit (Millipore).
Myelin Oligodendrocyte Glycoprotein/Anti-CD3 Model.
A 35–55 peptide of myelin oligodendrocyte glycoprotein [(MOG)35–55; H2N-MEVGWYRSPFSRVVHLYRNGK-OH] was dissolved in PBS (5 mg/ml). Heat killed desiccated Mycobacterium tuberculosis H37Ra was suspended in incomplete Freunds adjuvant (10 mg/ml). Both reagents were emulsified in glass syringes in a one-to-one (vol:vol) ratio. Emulsion was kept on ice prior to final injection. Mice (C57BL/6) received 100 μl, s.c. (250 μg MOG35–55, 500 μg H37Ra), spread over three sites: one over each hip and one in the scruff of the neck. The mice were also given an intraperitoneal injection of 150 ng pertussis toxin in PBS on the day of immunization. After 5 days, mice were given an intravenous injection of 1 μg anti-CD3 antibody (2C11) in PBS/1% mouse serum. Following anti-CD3 challenge, mice were bled 2 hours to measure serum IL-17A. Vehicle or drug was dosed during the 5-day MOG/pertussis toxin priming and then 30 minutes prior to anti-CD3.
Intradermal IL-23 Injection Model.
Briefly, recombinant murine IL-23 (generated by AbbVie Inc.) or sterile PBS + 0.1% BSA was injected into a single ear of an anesthetized mouse using a 30 gauge needle every day for 4 days (days 0–3) as previously described (Gauld et al., 2018). Histologic, gene expression, and flow-cytometric profiling analysis of ear tissue was performed as previously described (Gauld et al., 2018).
IL-17A Protein Levels in Ear.
A 5-mm-diameter section was harvested from the distal ear and flash frozen, and ears were then homogenized in 500 μl of lysis buffer [5% 1 M Tris (pH 7.4), 3% 5 M NaCl, 1% Triton X-100 prepared in deionized water, including 1X protease inhibitor (Sigma, St. Louis, MO)] with 1.4 mm ceramic beads in a 2 ml tube using Bead Ruptor 24 with CryoCool (OMNI, Kennesaw, GA). Tubes containing ear tissue lysates were centrifuged at 12,000g for 10 minutes at 4°C. The lysates were transferred and stored at −80°C until analysis. IL-17A was measured by Luminex Multiplex (ThermoFisher Scientific) following the manufacturer’s instructions. Ear protein levels were normalized to total protein (Pierce Protein Assay; ThermoFisher Scientific) and expressed as picograms per milligram total protein.
Glucose-6-Phosphate Isomerase/Arthritis Model.
Similar to Schubert et al. (2004), male DBA/J mice (Jackson Laboratories) were immunized intradermally at the base of the tail with 100 µl of 1:1 (v/v) emulsion containing 300 µg of glucose-6-phosphate isomerase (GPI) and 200 µg of heat-inactivated M. tuberculosis H37Ra (Complete Freund’s Adjuvant; Difco, Laurence, KS). Mice were dosed orally two times daily with 100 mg/kg A-9758 in 0.5% hydroxypropyl methylcellulose/0.02% Tween 80. For prophylactic treatment mice were dosed two times daily with 100 mg/kg A-9758 starting on day 0 prior to immunization. For late prophylactic treatment mice were dosed two times daily with 100 mg/kg A-9758 starting on day 7 after immunization. Paw swelling in rear paws was measured using Dyer spring calipers, with baseline paw thickness assessed on day 7 after immunization and additional measurements assessed between days 10 and 17.
Statistical Analysis.
All data are expressed as mean ± S.E.M. unless otherwise noted. Statistical significance was calculated by one-way ANOVA, followed by Bonferroni’s or Dunnett’s multiple comparisons post hoc test using GraphPad Prism 5.0. Differences were considered significant at *P ≤ 0.05, **P ≤ 0.01, and *** P ≤ 0.001 and not significant at P > 0.05.
Results
A-9758 Is a RORγ Ligand Displaying Inverse Agonist Properties.
To identify potential RORγt inverse agonists, a high-throughput screening campaign was performed on a library of proprietary compounds using a Gal4-hourRORγ LBD transactivation assay. Resultant from this screen was the identification of a benzoxyquinoline chemical series with RORγt inverse agonist properties (Amaudrut et al., 2019). Further optimization of this initial chemical matter was performed and led to the identification of an indole chemical series and A-9758.
In Fig. 1A we show that A-9758 inhibits, in a concentration-dependent manner, human RORγ transactivation (but not the related human pregnane receptor) with an IC50 value of 38 nM. In addition, A-9758 also inhibits mouse RORγ transactivation with similar activity (20 nM) (Fig. 1B) and is equally active on dog and rat RORγ (25 and 64 nM, respectively; data not shown). To further demonstrate the human RORγ ligand properties of the compound, A-9758 was assayed in a radioligand competition binding assay with 25-hydroxycholesterol and displayed an IC50 value of 27 nM (Fig. 1C).

Biochemical and in vitro activities of A-9758. (A) Transactivation assay. Cos-7 cells transfected with human RORγt (hRORγt) or human pregnane X receptor (hPXR) Gal4 DNA-binding domain (DBD)/LBD and pGL3 reporter plasmid in the presence of titrated A-9758. (B) Transactivation assay. Cos-7 cells transfected with mouse RORγt (mRORγt) Gal4 DBD/LBD and pGL3 reporter plasmid in the presence of titrated A-9758. (C) Radioligand binding. His-RORγ LBD incubated with 25-[3H]hydroxycholesterol in the presence of titrated A-9758. (D) AlphaScreen assay. Biotinylated PGC-1 and human His-tagged RORα, RORβ, or RORγ LBD was incubated with acceptor and donor beads in the presence of titrated A-9758. (E) Cofactor recruitment assay. AlphaScreen assay using RORγt LBD with specific coactivator or corepressor peptides along with acceptor and donor beads in the presence of titrated A-9758. (F) Human or mouse IL-17A release assays. Human CD4+ T cells or Th17 polarized mouse splenocytes were stimulated with anti-CD3/28 antibodies in the presence of titrated A-9758. Studies are representative of at least two independent studies, plotted by percentage of activity or binding of DMSO control vs. compound concentration and are presented as mean ± S.E.M.
To determine the specificity of the compound within a broader subset of nuclear receptors, we established NR/Gal4 cell-based assays in Cos7 cells for 25 nonorphan nuclear receptors. Supplemental Fig. 1 shows that A-9758 is highly selective, with maximum effect values at the highest dose tested (30 µM) below 20% (significant threshold) for all members except human RORγ. To assess the selectivity of A-9758 within the ROR family, an AlphaScreen assay was performed using PGC1α as the coactivator (Fig. 1D). A-9758 was shown to be approximately 14-fold specific for RORγt (5 nM) over RORα (73 nM) and nearly 270-fold specific over RORβ (1370 nM).
Like other nuclear receptors, RORγt interacts with coactivators or corepressors to regulate gene transcription (Collingwood et al., 1999). Therefore, we evaluated the effect of A-9758 on a wider selection of coactivator and corepressors that have been identified as binders of RORγ. Our data showed that A-9758 inhibits binding of the coactivators PGC1α and NCoA1 and conversely increases binding of the corepressors NCoR1 and NCoR2. In contrast, EBIP37, which has been shown to antagonize RORγ-mediated transactivation, was weakly inhibited (Fig. 1E).
A-9758 Inhibits IL-17A Produced by Stimulated Human CD4+ and Mouse Splenocytes.
RORγt is the master transcription factor for Th17 cell differentiation and the production of IL-17 by T cells. Therefore, an inverse agonist compound should inhibit IL-17 secretion. Using either human CD4+ T cells or in vitro differentiated mouse Th17 cells, A-9758 inhibited T cell receptor (TCR)–mediated IL-17A secretion with IC50 values of 100 and 38 nM, respectively (Fig. 1F).
To assess the impact of A-9758 in more detail, flow cytometry was used to define the relationship between RORγt and IL-17A under Th17 differentiation conditions. Studies confirmed the expected increase in frequency of RORγt positive cells under Th17 differentiation conditions and IL-17A production was limited to RORγt-expressing cells (Fig. 2A). A-9758 was shown to be effective in reducing the frequency of RORγt positive cells under Th17 polarizing conditions (Fig. 2, A and B), and ultimately reducing IL-17A secretion (Fig. 2, A and C). Collectively, these studies confirm the ability of A-9758 to attenuate the differentiation of RORγt-expressing Th17 cells and/or their effector function.

Impact of A-9758 on Th17 differentiation and effector function. (A) Expression of RORγt and IL-17A under Th0 and Th17 differentiation conditions and in the presence of A-9758. (B) Enumeration of the frequency of RORγt positive cells within CD4+ parent population under Th0 or Th17 differentiation conditions and in the presence of A-9758. (C) IL-17A levels secreted from CD4+ T cells cultured under Th0 or Th17 differentiation conditions and in the presence of A-9758. Data generated from a single representative study with A-9758. Graphs are presented as mean ± S.E.M.; ns, not significant, P < 0.05 (*) vs. challenge/vehicle group (black bars).
A-9758 Inhibits Circulating IL-17A in Multiple Acute Models.
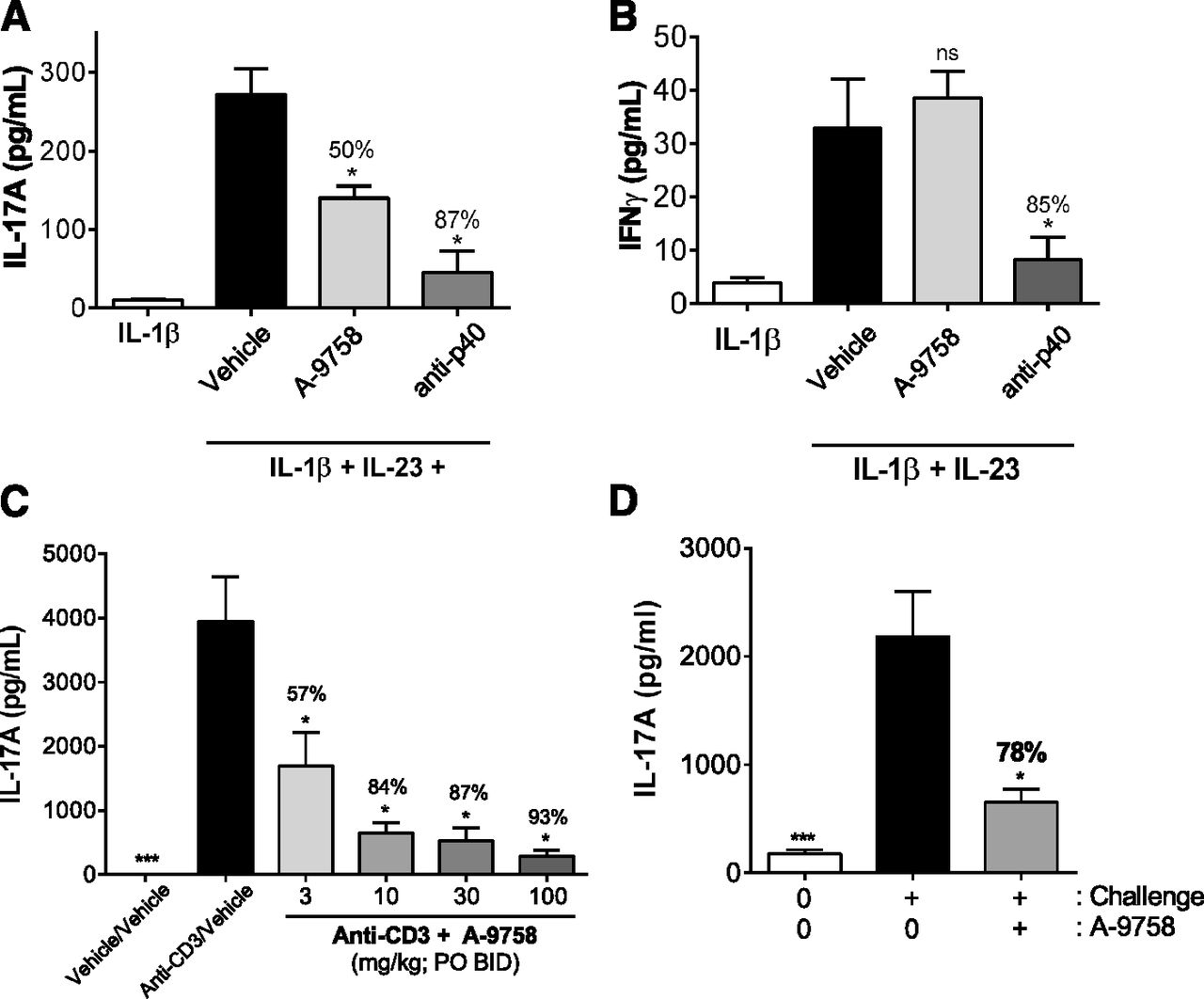
In vivo, IL-17A can be rapidly induced by the coinjection of IL-1β and IL-23 (Fauber et al., 2015). Utilizing similar conditions, we observed circulating IL-17A levels to increase from a background level of 10 pg/ml to almost 275 pg/ml (Fig. 3A). A single oral dose of A-9758 (100 mg/kg), delivered 1 hour prior to IL-1β/IL-23 challenge, was shown to block approximately 50% ± 6% of the systemic IL-17A signal driven by this stimuli (Fig. 3A). Systemic exposure of A-9758 (total drug) was approximately 10 μg/ml at the time when cytokine levels were assessed (data not shown). For a positive control, anti-p40 (25 mg/kg) was delivered to an additional cohort of animals and was shown to significantly block IL-17A induction (87% ± 11%) mediated by IL-1β/IL-23 challenge (Fig. 3A). A-9758 did not block the IFNγ response initiated by IL-1β/IL-23 challenge, highlighting the specificity of A-9758 to RORγt biology (Fig. 3B).

Modulation of IL-17A responses by A-9758 in multiple acute in vivo models. (A). Inhibition of intravenous IL-23/IL-1β induced IL-17A by A-9758 (100 mg/kg) or anti-p40 (25 mg/kg). (B) Inhibition of intravenous IL-23/IL-1β induced IFNγ by anti-p40 (25 mg/kg) but not A-9758 (100 mg/kg). (C) Inhibition of dual anti-CD3–mediated IL-17A release by A-9758. (D) Inhibition of MOG33–55+ anti-CD3–mediated serum IL-17A levels by 60 mg/kg A-9758. Studies are representative of at least two independent studies, plotted mean ± S.E.M.; ns, not significant, P < 0.05 (*) vs. challenge/vehicle group (black bars).
Other acute mouse models have been used to assess IL-17A production in vivo. One model includes two sequential injections of anti-CD3 that is shown to induce Th17 differentiation and increase systemic IL-17A levels (Mele et al., 2013). Our data showed that dual anti-CD3 injection induced a high circulating level of IL-17A (4000 pg/ml) compared with undetectable levels in control animals (Fig. 3C). Oral (two times daily) administration of A-9758 reduced IL-17A in a dose-dependent manner, reaching 93% ± 2.5% reduction at the highest dose group (Fig. 3C). Under the same conditions, IL-17F plasma levels were equally reduced (data not shown). Systemic exposure of A-9758 (total drug) was approximately 1.1–20.3 μg/ml, depending on dose group, taken 1.5 hours after final dose (data not shown).
Inhibition of IL-17A production driven by MOG35–55 peptide/pertussis toxin immunization and anti-CD3 challenge was also assessed. A-9758 (60 mg/kg, by mouth two times daily) resulted in a 78% ± 5% reduction in systemic IL-17A levels driven by MOG/anti-CD3 challenge (Fig. 3D). Similar to our IL-1β/IL-23 model findings, A-9758 had no effect on IFNγ (Table 1). To determine the broader impact of A-9758 on other T cell–derived cytokines, levels of IL-17F, IL-22, IL-2, IL-6, IL-10, and tumor necrosis factor α (TNFα) were also assessed. A-9758 was effective in robustly blocking the production of IL-17F, and to a lesser extent IL-22. No impact on IL-2, IL-6, IL-10, and TNFα was observed with A-9758 (Table 1). A-9758 exposures were approximately 23.9 ± 5.2 μg/ml taken 1 hour after the last dose of compound. Collectively, these studies highlight the specificity of A-9758 and its ability to suppress cytokines associated with Th17 effector function (IL-17 and IL-22) as opposed to those more broadly associated with other T helper lineages (IL-10, IL-2, IL-6, and TNFα).
Inhibition of MOG33–55+ anti-CD3–mediated serum IL-17A levels of A-9758
Levels of serum cytokines (picograms per milliliter) in animals treated with vehicle or A-9758 (60 mg/kg, by mouth two times daily); data are shown as mean ± S.E.M.
Inhibition of IL-23–Mediated Psoriasiform Dermatitis by A-9758.
We, and others, have done work in which it was shown that intradermal injections of IL-23 over a 5-day period are sufficient to drive inflammatory skin pathology akin to human psoriasis with a robust IL-17 signature (Suárez-Fariñas et al., 2013; Gauld et al., 2018). While both IL-23 and IL-17 are clinically validated for the treatment of psoriasis, a role for RORγt (although expected) has yet to be clinically validated.
Our initial studies confirmed the presence and time-dependent accumulation of RORγt-expressing cells following IL-23 treatment (Supplemental Fig. 2A). RORγt cells were found to be distributed across both the dermis and epidermis of affected skin (Supplemental Fig. 2B). Using flow cytometry, it was confirmed that RORγt-expressing cells were of hematopoietic origin (CD45+) and were increased significantly in number after 4 days of IL-23 treatment (Supplemental Fig. 2D).
Having confirmed the accumulation of RORγt-expressing cells by IL-23, we next assessed whether inhibition of RORγt would impact IL-23–mediated psoriasiform dermatitis. Mice were treated with A-9758 (1, 10, or 100 mg/kg, by mouth two times daily) for 4 days alongside intradermal injections of IL-23. A-9758 was effective in reducing ear inflammation driven by IL-23 in a dose-dependent manner (Fig. 4, A and B), with a strong exposure-efficacy relationship (Fig. 4C). Additional analysis confirmed that epidermal thickening (acanthosis) was significantly attenuated in the presence of A-9758 (Fig. 4D). Finally, A-9758 was shown to be effective in suppressing the gene signature associated with IL-23 exposure (Table 2). This included a significant decrease in Il17a, Il17f, Il23r, Ccr6, Defb4, and s100a7a. Collectively, these genes reflect known targets of RORγt biology and are upregulated in skin biopsies from psoriasis patients. Inhibition of this signature confirms the effectiveness of A-9758 as a modulator of RORγt biology in vivo.

Effect of RORγt inhibition on IL-23–mediated psoriasiform dermatitis. (A) Representative hematoxylin and eosin images of mouse skin (ear) treated with vehicle, IL-23, or IL-23+ A-9758 for 5 days. (B) Quantification of ear thickness [area under the curve (AUC); days 0–4] mediated by vehicle, IL-23, or IL-23+ A-9758 (1, 10, and 100 mg/kg). (C) Relationship between percentage of inhibition of ear thickness (AUC) vs. systemic exposures of A-9758 (each closed circle is the mean of n = 6 animals). (D) Quantification of changes to epidermal thickness mediated by vehicle, IL-23, or IL-23+ A-9758 (1, 10, and 100 mg/kg). Studies are representative of at least two independent studies, plotted mean ± S.E.M.; ns, not significant, P < 0.05 (*) vs. challenge/vehicle group (black bars).
Inhibition of IL-23–treated skin gene signature by A-9758
Percentage of change of gene expression comparing IL-23/vehicle to IL-23/A-9758 groups. All genes were decreased in the A-9758-treated group besides keratin16.
Together, these studies confirm that IL-23 exposure is sufficient to drive the accumulation of RORγt-expressing cells and that inhibition of RORγt by a small molecule approach is sufficient to attenuate skin pathology mediated by IL-23; the latter being in agreement with previously published findings (Xue et al., 2016).
Inhibition of RORγt by A-9758 Blocks Both the Accumulation and Effector Function of RORγt+ Cells.
Our data highlighted that small molecule inhibition of RORγt can attenuate IL-23–mediated skin inflammation and associated gene signature (Fig. 4). To assess its direct impact on RORγt-expressing cells we investigated the number, lineage commitment, and effector function of RORγt-expressing cells in IL-23–treated mice with or without A-9758 treatment. IL-23 exposure supported an increase in CD45+RORγt+ (Fig. 5A; Supplemental Fig. 2, C and D). The majority of CD45+RORγt+ cells were T cells, none were B220+ cells, and only a small number were TCR–B220− cells (data not shown) (Fig. 5A). In vehicle-treated ears, RORγt+ T cells were equally distributed across αβ or γδ T cells. However, upon IL-23 treatment, the number of αβTCR+RORγt+ cells significantly increased, while those expressing γδ TCRs remained constant (Fig. 5A). Treatment with A-9758 reduced the number of CD45+RORγt+ cells and αβTCR+ cells normally observed after IL-23 exposure (Fig. 5A). A-9758 had no effect on the number of γδTCR+RORγt+ cells.

Flow-cytometric profiling of RORγt-expressing cells in the presence of IL-23 and the RORγt inverse agonist A-9758. (A) Quantification of RORγt-expressing cells within various immune cell subsets (total CD45+, CD45+αβTCR+, CD45+γδTCR+, and CD45+TCR−B220−) in the presence or absence of IL-23 and A-9758 (100 mg/kg, by mouth two times daily). (B) Contour plots of CD45+αβTCR+ cells examining expression of RORγt and IL-17A. (C) Quantification of the frequency of RORγt+IL-17A+ cells from within the CD45+αβTCR+ population. Data generated from a single study with A-9758. Similar data were generated with other analogs from the same chemical series. Graphs are presented as mean ± S.E.M.; ns, not significant, P < 0.05 (*) vs. challenge/vehicle group (black bars).
Effector function of RORγt+ cells was determined by the expression of IL-17A. We focused on αβTCR+ cells due to their increase in number upon IL-23 treatment. IL-17A production was only observed in αβTCR+ cells that coexpressed RORγt and these cells represented a small fraction of all αβTCR+ cells. Inhibition of RORγt with A-9758 significantly reduced the frequency of αβTCR+RORγt+IL-17A+ cells (Fig. 5, B and C). Collectively, these data suggest that IL-23 exposure supports the expansion of IL-17A–producing αβTCR+RORγt+ cells and that inhibition of RORγt blocks the accumulation and effector function of these cells.
Inhibition of RORγt Is Sufficient to Block Preexisting Disease in IL-23–Treated Animals.
In our previous studies, we have shown that prophylactic treatment with an RORγt inverse agonist will attenuate IL-23–mediated psoriasiform dermatitis. Knowing that the expansion of RORγt-expressing cells (in response to IL-23) occurs predominately around day 2, we delayed treatment of animals with A-9758 until day 2 (herein referred to as therapeutic dosing). Our studies showed that therapeutic dosing with A-9758 was sufficient to attenuate (85% ± 9%) further increases in IL-23–driven ear inflammation (Fig. 6, A and B). Therapeutic dosing also reduced the expression of genes known to be key drivers of psoriasis, including IL17A and IL17F and the antimicrobials s100a7a and beta-defensin (Fig. 6C). IL-17A protein levels were also significantly reduced with therapeutic dosing of A-9758 (Fig. 6D). Together, these data suggest that inhibition of RORγt may be an effective therapeutic option to treat/manage ongoing inflammation.

Inhibition of RORγt during active IL-23–mediated inflammation is sufficient to block further disease progression. (A) Quantification of ear thickness (daily ear thickness measurements) mediated by vehicle, IL-23, or IL-23+ A-9758 (100 mg/kg). (B) Ear thickness data for days 2–4 represented as area under the curve (AUC) for vehicle, IL-23, or IL-23+ A-9758 (100 mg/kg) groups. (C) Changes in expression of key psoriasis-related genes taken from ear samples on day 4. (D) Levels of IL-17A protein determined by ELISA from whole ear lysates of the vehicle, IL-23, or IL-23+ A-9758 (100 mg/kg) groups. Data generated from a single study with A-9758. Similar data were generated with other analogs from the same chemical series. Graphs are presented as mean ± S.E.M.; ns, not significant, P < 0.05 (*) vs. challenge/vehicle group (black bars).
Inhibition of RORγt Is Sufficient to Block GPI-Mediated Arthritis.
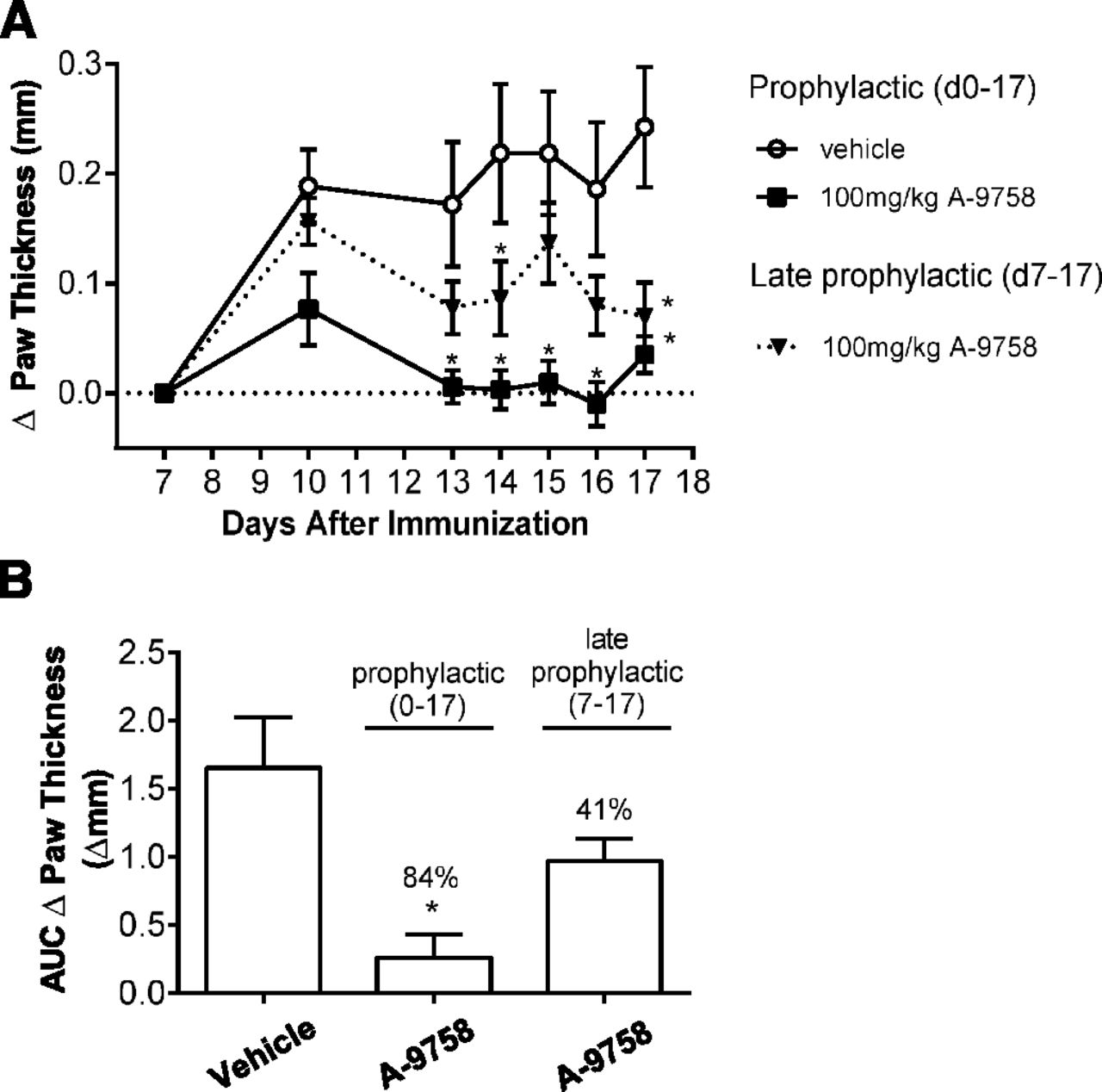
Clinical studies have shown that biologics against IL-23 are relatively ineffective against rheumatoid arthritis (Smolen et al., 2017), while those targeting IL-17 may show promise (Blanco et al., 2017). To assess the impact of RORγt inhibition in rheumatoid arthritis, at the preclinical level, we used the GPI arthritis model. We showed that A-9758 was capable of reducing GPI-induced paw swelling when dosed either prophylactically or late prophylactically (Fig. 7A). Quantitatively, when treated prophylactically, A-9758 reduced paw swelling (area under the curve paw thickness, Δmm) by 84% ± 10%, and when treated late prophylactically paw swelling was reduced by 41% ± 10% (Fig. 7B). These values compare favorably to anti-TNFα therapy in the same model, which when dosed prophylactically or late prophylactically reduced paw swelling (area under the curve) by ∼100% and ∼40%, respectively (data not shown). In both dosing regimens, terminal exposures of A-9758 reached approximately 13.75 μg/ml (serum; total drug). These results are in agreement with previous work by Xue et al. (2016), who showed efficacy of a RORγt small molecule in the mouse collagen-induced arthritis model. Together, they highlight that inhibition of RORγt may be an effective therapy for the treatment of early, or developed, rheumatoid arthritis.

Prophylactic treatment with A-9758 inhibits paw swelling in GPI arthritis. DBA/J mice (n = 15) were immunized with GPI on day 0 and paw swelling was assessed. Mice we dosed orally two times daily with 100 mg/kg A-9758 starting on day 0 (prophylactic) or day 7 (late prophylactic treatment). (A) Change in paw thickness over time with baseline measurement assessed on day 7. (B) Area under the curve (AUC) for change in paw thickness from day 7 to 17 calculated for all groups. Data generated from a single study with A-9758. Graphs are presented as mean ± S.E.M.; P < 0.05 (*) vs. prophylactic vehicle group.
Suppression of T Cell–Derived IL-17A in Whole Blood by Inhibition of RORγt.
A recent study by Russell et al. (2018) described the use of plate-based ex vivo human whole blood stimulation assays to assess IL-17 levels. Using a similar method, we assessed the potency and efficacy of A-9758 in the inhibition of IL-17A release from whole blood. CytoStim was shown to induce a significant increase in IL-17A from whole blood, increasing from ∼0.5 pg/ml at rest to ∼800 pg/ml after stimulation for 48 hours (Fig. 8A). We show that A-9758 is capable of blocking approximately 80% of the IL-17A induced by CytoStim at the top concentration used with an IC50 value of approximately 1 μM total drug concentration (approximately 0.5 μg/ml) (Fig. 8B). Adjusting for plasma protein binding (fraction unbound, 0.003), we observe an IC50 value of approximately 3 nM, which is well aligned with our in vitro data previously described (Fig. 1).

Inhibition of CytoStim induced IL-17A from human whole blood by A-9758. (A) Whole blood from healthy volunteers (n = 2) was stimulated with CytoStim to induce IL-17A release (representative of >6 independent studies, data shown as mean ± S.E.M., student t test, *P = 0.0469). (B) Whole blood stimulated with CytoStim for 48 hours in the presence of increasing concentrations of A-9758 (representative of two independent studies, data shown as mean ± S.E.M.). Data are representative of data from two independent studies generated with A-9758. Similar data have also been generated with other analogs from the same chemical series. Graphs are presented as mean ± S.E.M. P < 0.05 (*).
Discussion
RORγt has emerged as an important drug target due to its role as an intermediate in the IL-23/IL-17 pathway (Skepner et al., 2014; Wang et al., 2014; Xiao et al., 2014; Fauber et al., 2015; Scheepstra et al., 2015; Skepner et al., 2015; Banerjee et al., 2016; Guo et al., 2016; Hintermann et al., 2016; Smith et al., 2016; Xue et al., 2016; Guendisch et al., 2017; Takaishi et al., 2017). Here, we describe the identification and characterization of a novel small molecule inverse agonist (A-9758) against RORγt. Similar to other reports, A-9758 is effective in the suppression of IL-17A both across assays and species. We expand on previously published studies in three key areas. First, we provide a comprehensive analysis of RORγt-expressing cells under conditions in which IL-23 drives psoriasiform dermatitis. This is important since the transcriptomic profile of the inflamed skin from this preclinical mouse model is closely aligned to the transcriptomic profile of human psoriasis (Suárez-Fariñas et al., 2013; Gauld et al., 2018). Second, RORγt activity is regulated by the recruitment of endogenous coactivators or corepressors (Fauber and Magnuson, 2014). We show that A-9758 is effective in recruiting corepressors of RORγt activity while derecruiting coactivators. Finally, our data in both IL-23–mediated psoriasiform dermatitis and GPI-mediated arthritis identify RORγt as a therapeutic target due to the ability of A-9758 to suppress ongoing disease. These data are also in agreement with previously published studies (Xue et al., 2016). Our studies also show that A-9758 is effective in suppressing both Th17 differentiation and Th17 effector function (Fig. 2). This has important implications for human disease and differentiates a small molecule approach against RORγt from biologics against IL-17 or IL-17R, which work predominately by blocking the effector function of IL-17–producing cells.
While the assessment of RORγt-expressing cells in humans has been hindered by the absence of robust antibodies for flow/mass cytometry, this is not the case for mice. We performed an extensive assessment of RORγt+ cells in naive and IL-23–treated tissue. Few RORγt+ cells were found in naive/sham–treated ears; however, those present were predominately dermal in locale and appeared to be αβ or γδ T cells (Fig. 5). This contrasts with IL-23–treated skin where RORγt+ cells accumulate, are mainly αβ T cells, and are distributed across both dermis and epidermis (Fig. 5). Whether the accumulation of the described RORγt+ T cells is supported by de novo generation in the skin or influx from other lymphoid tissue remains to be determined.
In the absence of ligand, RORγt is in an active conformation, capable of recruiting coactivators. From a structural point of view, His479, Tyr 502, and Phe506 triplets have been identified as the primary structural elements responsible for stabilizing H12 and anchoring RORγt in the active conformation, allowing coactivator interactions (Williams et al., 2003; Carcache et al., 2018; Schnute et al., 2018). Agonist compounds stabilize the active conformation either directly or indirectly and it has been shown that a putative natural ligand like 25-hydroxy-cholesterol stabilizes H12 indirectly through a water-mediated hydrogen bond between His479 and Tyr502 (Li et al., 2017; Noguchi et al., 2017; Jetten et al., 2018). A number of studies have described inverse agonist compounds that induce conformational changes in the LBD, resulting in corepressor recruitment and subsequent inhibition of RORγt transcriptional activities (Xue et al., 2016). Several structural changes have been observed with different classes of inverse agonists. The first class of compounds induces a steric clash that disrupts the His-Tyr lock. A second class acts by nonsteric clash and can include water trapping or close contact with His479. In this class, H12 could be maintained in an agonist or inverse agonist position (Kallen et al., 2017; Jetten et al., 2018). For example, the GlaxoSmithKline/Tempero group demonstrated that one inverse agonist, TMP920, interacts with the ligand-binding pocket to disrupt RORγt binding to DNA, whereas two others, TMP778 and GSK805, do not (Xiao et al., 2014). The same GlaxoSmithKline group has recently described that small changes in chemical structures starting from an agonist compound could lead to two inverse agonists: one recruiting the corepressor NCoR2 and derecruiting the coactivator NCoA1, whereas the second one derecruits both cofactors (Wang et al., 2018). A third class of compounds has been shown to bind to an allosteric rather than the canonical orthosteric binding site (Scheepstra et al., 2015; Jetten et al., 2018). It is conceivable that such differences in inverse agonist mechanisms could affect binding of RORγt to Il17a and Il23r promoter regions by modifying the interaction with cofactors, leading to different acetylation and methylation patterns that control chromatin structure and finally target gene expression and Th17 cell function (Jetten et al., 2018; Tanaka et al., 2018).
To assess how A-9758 modulates cofactor recruitment, or derecruitment, a selection was made of two coactivators (NCoA1 and PGC1α) and two corepressors (NCoR1 and NCoR2). This selection was based on good AlphaScreen signal windows, which denote a good interaction level between RORγt and the cofactor peptides. EBIP37 has been described to inhibit the transcriptional activity of RORγ, meaning that it could be considered as a corepressor (Kurebayashi et al., 2004). We showed that A-9758 displayed a cofactor profile in recruiting corepressors (NCoR1: EC50 = 60 nM; NCoR2: EC50 = 43 nM) and derecruiting coactivators (NCoA1: IC50 = 110 nM; PGC1α: IC50 = 49 nM). A-9758 showed only a weak effect on derecruiting EBIP37.
This favorable profile is in good alignment with the biologic mechanisms involved in nuclear receptor RORγt functions. Together with the activities seen in in vitro and in vivo in Th17-driven models, these data confirm that modulating RORγt with an inverse agonist compound translates into beneficial effects. Further crystallographic experiments are needed to decipher the exact mode of binding of A-9758.
Previous studies have shown that inhibition of RORγt attenuates development of disease models of psoriasis (imiquimod and IL-23) (Skepner et al., 2014; Banerjee et al., 2016; Guo et al., 2016; Smith et al., 2016; Xue et al., 2016; Takaishi et al., 2017), arthritis (collagen-induced arthritis and adjuvent-induced arthritis) (Xue et al., 2016; Guendisch et al., 2017), and multiple sclerosis (experimental autoimmune encephalomyelitis) (Wang et al., 2014; Xiao et al., 2014; Guo et al., 2016). Our studies are well aligned and build upon these earlier reports. We show that our RORγt inverse agonist promotes a significant decrease in skin inflammation (Fig. 4) and does so by influencing RORγt in a way consistent with our in vitro Th17 differentiation assay data (Fig. 2). First, A-9758 prevents the accumulation of RORγt+ cells (predominately αβT cells) in the ear following IL-23 exposure, and second it blocks the effector function of remaining RORγt+ cells. Mechanistically, we propose that A-9758 modulates the interaction between coactivators and the LBD of RORγt. This prevents the ability of RORγt to induce productive changes in gene transcription at related gene loci/promoter regions. This helps explain the profound effect of A-9758 on the effector function of RORγt-expressing cells (i.e., IL-17 production). The mechanism of action for the reduced accumulation of RORγt-expressing T cells (Th17 cells) after IL-23 exposure is less clear. It is possible that A-9758 prevents the differentiation of Th17 cells by modulating the stability of RORγt induced by factors such as IL-6, transforming growth factor-β, and IL-23, or it may limit the survival of existing Th17 cells due to the lack of RORγt transcriptional activity.
To assess the role for A-9758 as a novel therapeutic, we investigated the impact of therapeutic dosing in the IL-23 model of psoriasiform dermatitis and GPI model of arthritis (Figs. 6 and 7). In both models, A-9758 was effective in attenuating further disease progression at the tissue (ear or paw thickness) or mechanistic level (decreased IL-17A production). Together, our preclinical studies highlight the potential for RORγt inverse agonists to be an effective therapy in humans with active disease. However, the efficacy of RORγt inverse agonists in human disease states remains to be fully explored. To date, only one set of data has been released that provides support for this approach. Vitae Pharmaceuticals released data in 2016 showing 24% and 30% placebo-adjusted improvements in the psoriasis area and severity index in a 4-week trial of moderate-to-severe psoriasis patients (Gege, 2016, 2017). While this limited clinical data set is supportive of a therapeutic role for the inhibition of RORγt in psoriasis, it is not conclusive, nor does it suggest efficacy will be aligned with current anti-IL-23/IL-17 biologics therapies. Furthermore, potential toxicology liabilities resulting from the inhibition of RORγt, if any, remain unclear.
Overall, our studies confirm that RORγt represents a viable target for small molecule intervention with multiple potent and selective small molecule inverse agonists reported. Our data show the impact of RORγt inhibition on cell differentiation and effector function (beyond IL-17A production) and that it can be used to successfully suppress ongoing disease in multiple preclinical models of human disease.
Acknowledgments
We thank the Comparative Medicine Group at AbbVie, Lake County, IL, and especially Paige Ebert and Donna Strasburg.
Authorship Contributions
Participated in research design: Gauld, Jacquet, Wallace, McCarthy, Goess, McGaraughty, Luccarini, Breinlinger, Cusack, Potin, Kort, Masson.
Conducted experiments: Gauvin, Wallace, Wang, McCarthy, Goess, Leys, Huang, Su, Wetter, Salte.
Performed data analysis: Gauld, Jacquet, Wallace, Wang, McCarthy, Goess, Su, Edelmayer, Argiriadi, Bressac, Desino, Cusack, Masson.
Wrote or contributed to the writing of the manuscript: Gauld, Cusack, Masson, Honore, Kort.
Footnotes
- Received March 25, 2019.
- Accepted July 18, 2019.
All authors are current employees of AbbVie or Inventiva or were employees of AbbVie or Inventiva at the time of the study. (D.G. and R.E. are former AbbVie Inc. employees. S.J. is a former Inventiva employee.) The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie and Inventiva participated in the interpretation of data, review, and approval of the publication. The authors declare no competing financial interests.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- A-9758
- 1-(2,4-dichloro-3-((1,4-dimethyl-6-(trifluoromethyl)-1H-indol-2-yl)methyl)benzoyl)piperidine-4-carboxylic acid
- BSA
- bovine serum albumin
- GPI
- glucose-6-phosphate isomerase
- IFNγ
- interferon γ
- IL
- interleukin
- LBD
- ligand-binding domain
- MOG
- myelin oligodendrocyte glycoprotein
- ROR
- retinoic acid–related orphan receptor
- TCR
- T cell receptor
- Th
- T helper
- TNFα
- tumor necrosis factor α
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}