


Abstract
High-permeability–low-molecular-weight acids/zwitterions [i.e., extended clearance classification system class 1A (ECCS 1A) drugs] are considered to be cleared by metabolism with a minimal role of membrane transporters in their hepatic clearance. However, a marked disconnect in the in vitro-in vivo (IVIV) translation of hepatic clearance is often noted for these drugs. Metabolic rates measured using human liver microsomes and primary hepatocytes tend to underpredict. Here, we evaluated the role of organic anion transporter 2 (OAT2)–mediated hepatic uptake in the clearance of ECCS 1A drugs. For a set of 25 ECCS 1A drugs, in vitro transport activity was assessed using transporter-transfected cells and primary human hepatocytes. All but two drugs showed substrate affinity to OAT2, whereas four (bromfenac, entacapone, fluorescein, and nateglinide) also showed OATP1B1 activity in transfected cells. Most of these drugs (21 of 25) showed active uptake by plated human hepatocytes, with rifamycin SV (pan-transporter inhibitor) reducing the uptake by about 25%–95%. Metabolic turnover was estimated for 19 drugs after a few showed no measurable substrate depletion in liver microsomal incubations. IVIV extrapolation using in vitro data was evaluated to project human hepatic clearance of OAT2-alone substrates considering 1) uptake transport only, 2) metabolism only, and 3) transporter-enzyme interplay (extended clearance model). The transporter-enzyme interplay approach achieved improved prediction accuracy (average fold error = 1.9 and bias = 0.93) compared with the other two approaches. In conclusion, this study provides functional evidence for the role of OAT2-mediated hepatic uptake in determining the pharmacokinetics of several clinically important ECCS 1A drugs.
Introduction
We recently proposed a framework called extended clearance classification system (ECCS) to predict the rate-determining clearance mechanism of drugs or new molecular entities using simple molecular properties, including ionization state and molecular weight and in vitro membrane permeability (Varma et al., 2015; El-Kattan and Varma, 2018). According to this validated system, ECCS class 1A drugs (i.e., high-permeability–low-mol. wt. <400-Da acids/zwitterions) are thought to be cleared primarily by metabolism as the rate-determining step. However, despite the predominance of cytochrome 450 (P450)- and/or uridine 5′-diphospho-glucuronosyltransferase (UGT)-mediated metabolic pathways, many studies have indicated a marked disconnect in the in vitro–in vivo (IVIV) translation for this class of drugs. That is, metabolic rates measured by substrate depletion and/or metabolite formation in the incubations of human liver microsomes and human hepatocytes considerably underpredict human hepatic clearance of ECCS 1A drugs (Obach, 1999; Brown et al., 2007; Bowman and Benet, 2016; Wood et al., 2017). Several previous studies discussed plausible causes for IVIV disconnect in hepatic clearance as measured using hepatocytes and liver microsomes, which include permeation rate limitation, diffusion through unstirred water layer, suboptimal substrate concentration, reagent handling, and assay methods (Hallifax and Houston, 2012; Poulin et al., 2012; Bowman and Benet, 2016; Wood et al., 2017). Although the causes are thought to be multifactorial, empirical correction of prediction bias has been suggested as a pragmatic approach for pharmacokinetic predictions in drug discovery (Hallifax and Houston, 2012; Poulin et al., 2012; Wood et al., 2017).
Membrane transporters play a key role in the absorption, distribution, clearance, and elimination of drugs (Shitara et al., 2006; Giacomini et al., 2010; El-Kattan and Varma, 2018). Emphasis has been given to hepatic uptake clearance mediated by organic anion transporting polypetides (OATPs), where many high-molecular-weight acids/zwitterions (ECCS class 1B/3B) are shown to be substrates (Giacomini et al., 2010; Shitara et al., 2013; Varma et al., 2015, 2017b). OAT2, a member of solute carrier 22A (SLC22A7), is expressed in the liver and kidney and is known to transport several xenobiotics and endogenous compounds (e.g., creatinine and cGMP) (Lepist et al., 2014; Shen et al., 2017). Although protein abundance of OAT2 in the human liver is relatively similar to that of other major uptake transporters, such as OATP1B, Na+-taurocholate cotransporting polypeptide, and organic cation transporter (OCT)1 (Nakamura et al., 2016; Vildhede et al., 2018), little is known about its role in the clinical pharmacokinetics of drugs (Shen et al., 2017). We recently identified the role of OAT2-mediated hepatic uptake in the clearance of tolbutamide and R- and S-warfarin, which were previously assumed to be cleared by metabolism involving P450 2C9/19 (Bi et al., 2018a,b). Mechanistic in vitro studies and physiologically based pharmacokinetic (PBPK) modeling and simulations demonstrated the significance of OAT2-CYP2C interplay in their clinical pharmacokinetics (Bi et al., 2018,b). Given that these drugs are high-permeability–low-molecular-weight acids, we hypothesized that OAT2-mediated hepatic uptake contributes to the clearance of ECCS class 1A drugs.
The main objective of this investigation was to evaluate the role of transporter-mediated hepatic uptake in the pharmacokinetics of high-permeability–low-molecular- weight acids and zwitterions. To this end, for a set of about 25 ECCS class 1A drugs (Varma et al., 2015), in vitro transport activity was characterized using transporter-transfected cells and primary human hepatocytes, and metabolic clearance was measured in human liver microsome (HLM) incubations supplemented with cofactors for oxidation and glucuronidation pathways. Moreover, IVIV extrapolation was evaluated to quantitatively predict human hepatic clearance considering the uptake and metabolic clearances separately and assuming transporter-enzyme interplay (extended clearance model).
Materials and Methods
Chemicals and Reagents.
Rosuvastatin was purchased from Carbosynth (Compton, UK). R- and S-warfarin and rifamycin SV were purchased from Sigma-Aldrich (St. Louis, MO). [3H]-cGMP was purchased from PerkinElmer Life Sciences (Boston, MA). All other test compounds were obtained from the Pfizer Chemical inventory system. InVitroGro-HT and CP hepatocyte media were purchased from BioreclamationIVT (Baltimore, MD). Collagen I–coated 24-well plates were obtained from Corning (Kennebunk, ME). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, nonessential amino acids, GlutaMAX-1, sodium pyruvate, penicillin, and streptomycin solution were obtained from Invitrogen (Life Technologies, Carlsbad, CA). Cryopreserved human plateable hepatocytes lot Hu8246 (female Caucasian, 37 years old) were obtained from Thermo Fisher Scientific (Carlsbad, CA). BCA protein assay kit was purchased from PierceBiotechnology (Rockford, IL). NP-40 protein lysis buffer was purchased from Thermo-Fisher (Franklin, MA). Human embryonic kidney (HEK)293 cells expressing human OATP1B1 were obtained from Absorption Systems (Exton, PA). HEK293 cells stably transfected with human OAT2(tv-1) were obtained from the laboratory of Ryan Pelis (Dalhousie University, Halifax, Canada).
In Vitro Transport Studies Using Transporter-Transfected Cells.
HEK293 cells singly transfected with OATP1B1 or OAT2 and wild-type cells were seeded at a density of 0.5–1.2 × 105 cells/well on BioCoat 48- or 96-well poly-d-lysine–coated plates (Corning Inc., Corning, NY), grown in DMEM containing 10% fetal bovine serum, 1% sodium pyruvate, 1% nonessential aminoacids, and 1% GlutaMAX for 48 hours at 37°C, 90% relative humidity, and 5% CO2. In addition, OAT2-HEK cells were supplemented with 1% gentamycin and hygromycin B (50 μg/ml) and OATP1B1-HEK cells were supplemented with 1% HEPES.
Methods adopted for uptake studies are similar to those previously reported by our group (Bi et al., 2017, 2018a; Mathialagan et al., 2017). Stock solutions of all compounds were made in DMSO. Cells were rinsed three times with warm uptake buffer (Hanks’ balanced salt solution (HBSS) with 20 mM HEPES, pH 7.4), followed by incubating test compounds at 1 µM (or 10 µM in some cases with analytical sensitivity issues) with a final concentration of 1% DMSO. At the end of incubation (2 minutes), the cellular uptake was terminated by washing the cells four times with ice-cold transport buffer, and then the cells were lysed with 0.2 ml of 1% NP-40 in water (radiolabeled compounds) or methanol containing internal standard (nonlabeled compounds). Transporter functionality was validated using in vitro probe substrates: 0.5 μM [3H]-cGMP (OAT2) or 0.5 μM rosuvastatin (OATP1B1) as described previously (Bi et al., 2017; Mathialagan et al., 2018). Intracellular accumulation was determined either by mixing the cell lysate with scintillation fluid followed by liquid scintillation analysis (PerkinElmer Life Sciences) for radiolabeled compounds or by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis for nonlabeled compounds. The total cellular protein content was determined using a Pierce BCA Protein Assay kit according to the manufacturer’s specifications. The uptake ratio was calculated as a ratio of accumulation in the transfected cells to the accumulation in wild-type cells.
Determination of Uptake Clearance Using Short-Term Cultured Plated Human Hepatocytes.
The hepatic uptake assay was performed using short-term cultured, cryopreserved human hepatocytes as described previously (Bi et al., 2017, 2018a). Briefly, plateable cryopreserved human hepatocytes (Hu8246 lot; Thermo Fisher Scientific) were thawed in a water bath at 37°C and placed on ice. The cells were then poured into In VitroGro-HT medium at 37°C at a ratio of one vial/50 ml in a conical tube. The cells were centrifuged at 50g for 3 minutes and resuspended at 0.75 × 106 cells/ml in In VitroGro-CP medium. Cell viability was determined by trypan-blue exclusion and exceeded 85%. Hepatocyte suspensions were plated in collagen-coated 24-well plates at a density of 0.375 × 106 cells/well in a volume of 0.5 ml/well and incubated overnight (∼18 hours). Cells were first rinsed twice with HBSS or ice-cold HBSS and preincubated with HBSS in the presence or absence of 1 mM rifamycin SV or ice-cold HBSS for 10 minutes at 37°C or 4°C. After aspirating the preincubation buffer, 0.5 ml of HBSS or ice-cold HBSS containing a substrate (1 or 10 µM) was added in the presence or absence of 1 mM rifamycin SV, a pan-SLC inhibitor (Bi et al., 2017, 2018a; Mathialagan et al., 2018). The uptake was terminated at designated times (0.25, 0.5, and 1 or 0.5, 1, and 2 minutes) by adding 0.5 ml of ice-cold standard HBSS after removal of the incubation buffer. Cells were then washed three times with 0.5 ml of ice-cold HBSS. Hepatocytes were lysed with methanol containing the internal standard for LC-MS/MS quantification.
A two-compartment mathematical model (compartments representing the media and cell) was developed to estimate the intrinsic passive clearance (PSpassive) and total intrinsic active uptake clearance (PSactive) by simultaneously fitting the cell accumulation (Acell) data in plated human hepatocytes with and without rifamycin SV (1 mM), a pan-inhibitor of active uptake transporters. This model is analogous to the method described previously to analyze transport data in other cell systems (Poirier et al., 2008). Equations 1–6 are used in this modeling process: (1)
(1) (2)
(2) (3)
(3) (4)
(4) (5)
(5) (6)where, Cew, Ciw, Aew, Aiw, Vew (0.5 ml), Viw, fu,iw, and fu,ew represent the concentration (C), amount (A), volume (V), and unbound fraction (fu) of the extracellular (ew) and intracellular (iw) compartments. PR is the measured protein concentration per well, CpPR is the number of cells per measured protein (1.5 million cells/mg) (Sohlenius-Sternbeck, 2006), and VpC is cell volume (2.6 µl/million cells, internal data) measured assuming a spherical structure (17.2 µm diameter, internal data).
(6)where, Cew, Ciw, Aew, Aiw, Vew (0.5 ml), Viw, fu,iw, and fu,ew represent the concentration (C), amount (A), volume (V), and unbound fraction (fu) of the extracellular (ew) and intracellular (iw) compartments. PR is the measured protein concentration per well, CpPR is the number of cells per measured protein (1.5 million cells/mg) (Sohlenius-Sternbeck, 2006), and VpC is cell volume (2.6 µl/million cells, internal data) measured assuming a spherical structure (17.2 µm diameter, internal data).
In Vitro Metabolism in Human Liver Microsomes Incubations.
Pooled HLMs (HLM-103 lot prepared from a mixed gender pool of 50 donors; Sekisui XenoTech, LLC, Kansas City, KS; final protein concentration, 1 mg/ml) were diluted in 0.1 M potassium phosphate buffer (pH 7.4) containing 5 mM magnesium chloride. The microsomes were then activated by adding alamethicin at a final concentration at 10 µg/ml and allowed to remain on ice for 15 minutes (Walsky et al., 2012). The stock solutions were first prepared in DMSO (10 mM), and then subsequent substock 100× solutions (0.1 mM) were prepared in 50% acetonitrile:water. The final concentration of acetonitrile in the incubation was 0.5% (v/v). The total incubation volume in the experiment was 0.5 ml. The microsome and buffer mixture was then prewarmed at 37°C for 5 minutes on a heat block before the addition of both cofactors NADPH (1.3 mM) and UDPGA (5 mM). The reaction was initiated immediately, after the addition of cofactors, with compound solutions at a final concentration of 1 µM (Kilford et al., 2009). The incubations were terminated by removing (50-μl) aliquots of the reaction mixture at 0-, 5-, 10-, 15-, 30-, 45-, 60-, 90-, 120-, and 180-minute time points (n = 2 at each time point) and were added to acetonitrile (200 μl) containing a cocktail of internal standards. The samples were then vortexed for 1 minute and centrifuged (Allegra X-12R; Beckman Coulter, Fullerton, CA) at 3000 rpm for 5 minutes. The supernatant (150 μl) was transferred to a 96-deep-well injection block containing (150 μl) of high-performance liquid chromatography water containing 0.2% formic acid and mixed before LC-MS/MS analysis for the disappearance of compound. The apparent metabolic intrinsic clearance (CLint,met,app) was determined based on the substrate-depletion half-life estimated from the ratio of the peak area response of each compound to that of the internal standard, as described earlier (Di et al., 2012).
In Vitro Substrate-Depletion Assay Using Human Hepatocytes.
The high-throughput human hepatocyte substrate-depletion assay was performed in a 384-well formatted as described previously (Di et al., 2012). Briefly, the cryopreserved human hepatocytes (DCM lot, 10 donor pool; Bioreclamation IVT, Westbury, NY) were thawed and resuspended in Williams’ E medium supplemented with HEPES and Na2CO3. The cells were counted using the trypan blue exclusion method. Test compounds were added to suspension human hepatocytes in Williams’ E medium buffer and incubated at 37°C in a humidified CO2 incubator (75% relative humidity, 5% CO2/air) for 4 hours. The final incubation contained 0.5 million cells/ml and 1 µM test compound in 15-µl total volume with 0.1% DMSO. At various time points (0, 3, 10, 30, 60, 120, and 240 minutes), an aliquot of the sample was taken and quenched with cold acetonitrile containing internal standard (CP-628374). The samples were centrifuged (Eppendorf, Hauppauge, NY) at 3000 rpm for 10 minutes at 4°C, and the supernatants were transferred to new plates, which were sealed before performing LC-MS/MS analysis. The apparent metabolic intrinsic clearance (CLint, met, app) was determined based on the substrate-depletion half-life estimated from the ratio of the peak area response of each compound to that of the internal standard, as described earlier (Di et al., 2012).
LC-MS/MS Analysis.
Sample analysis was conducted using LC-MS/MS system comprising an AB Sciex 5500 or 6500 triple-quadrupole mass spectrometer equipped with an electrospray source (AB Sciex, Framingham, MA), Agilent Technologies Infinity 1290 liquid chromatography-tandem mass spectrometry (Santa Clara, CA), and CTC Leap autosampler (Pflugerville, TX) programmed to inject 10 µl of sample onto an ACE 1.7-µm Excel C18-PFP 2.1 × 30 mm (Advanced Chromatography Technologies Ltd, Aberdeen, Scotland), Halo C18 2.7 µm, 100 Å 2.1 × 30 mm (Mac Mod, Chadds Ford, PA), or a Kinetex C18 2.6 µm, 100 Å, 3 × 30 mm (Phenomenex, Torrence, CA) analytical column. A binary gradient was used consisting of 0.1% (v/v) formic acid in water (mobile phase A) and 0.1% (v/v) formic acid in acetonitrile (mobile phase B) and monitored using the multiple reaction monitoring mode for the m/z transitions, as described in Supplemental Table 1.
Data Analysis.
In vivo hepatic intrinsic clearance (CLint, h) was calculated from human intravenous clearance data assuming the well stirred liver model in eq. 7 (Pang and Rowland, 1977): (7)where CLblood, h [= (CLplasma − CLrenal)/Rb] is the hepatic blood clearance obtained from intravenous total plasma clearance corrected for renal clearance and blood-to-plasma ratio (Rb), fu,b is the fraction unbound in blood [= the fraction unbound in plasma (fu,p)/Rb], and Qh is hepatic blood flow (20.7 ml/min per kilogram) (Kato et al., 2003).
(7)where CLblood, h [= (CLplasma − CLrenal)/Rb] is the hepatic blood clearance obtained from intravenous total plasma clearance corrected for renal clearance and blood-to-plasma ratio (Rb), fu,b is the fraction unbound in blood [= the fraction unbound in plasma (fu,p)/Rb], and Qh is hepatic blood flow (20.7 ml/min per kilogram) (Kato et al., 2003).
Predicted CLint, h values were calculated under three different scenarios: 1) considering total hepatic uptake alone (eq. 8) measured using plated human hepatocytes, 2) considering metabolic clearance alone (eq. 9) measured by substrate depletion in HLMs, and 3) extended clearance model (eq. 10) accounting for transporter-enzyme interplay using both uptake transporlt (from plated human hepatocytes) and metabolic (i.e., HLM) data (Sirianni and Pang, 1997; Liu and Pang, 2005; Shitara and Sugiyama, 2006; Camenisch and Umehara, 2012; Varma et al., 2014; Patilea-Vrana and Unadkat, 2016; Kimoto et al., 2017). A schematic of the stepwise approach used in IVIV extrapolation is given in Supplemental Fig. 1: (8)
(8) (9)
(9) (10)PSactive and PSpassive are sinusoidal intrinsic active uptake clearance and intrinsic passive uptake clearance, respectively. CLint, met represents the intrinsic metabolic clearance measured using HLMs coincubated with both NADPH and UDPGA. CLint, met is equal to the measured apparent intrinsic clearance (CLint, met, app) divided by microsomal binding (fu, mic) or hepatocyte binding (fu, hep). Active basolateral and canalicular efflux was assumed to be negligible (Sirianni and Pang, 1997; Shitara et al., 2013; Varma et al., 2014). The in vitro intrinsic values were scaled assuming the following: 1.5 × 106 hepatocytes/mg of measured protein, 118 × 106 hepatocytes/g of liver, 39.8 mg microsomal protein/g of liver, 24.5 g liver/kg of body weight (Varma et al., 2013). Relative expression factor (REF)—correction for the expression difference in the hepatocytes and human liver—of 1.8 and 2.0—were applied for OAT2 and OATP1B1 substrates, respectively, based on our previous published quantitative proteomics data (Kimoto et al., 2012; Vildhede et al., 2018). The hepatocyte lot (Hu8246) used for uptake studies showed expression levels of OAT2 similar to the median expression levels of 30 single-donor cryopreserved hepatocyte lots (Vildhede et al., 2018).
(10)PSactive and PSpassive are sinusoidal intrinsic active uptake clearance and intrinsic passive uptake clearance, respectively. CLint, met represents the intrinsic metabolic clearance measured using HLMs coincubated with both NADPH and UDPGA. CLint, met is equal to the measured apparent intrinsic clearance (CLint, met, app) divided by microsomal binding (fu, mic) or hepatocyte binding (fu, hep). Active basolateral and canalicular efflux was assumed to be negligible (Sirianni and Pang, 1997; Shitara et al., 2013; Varma et al., 2014). The in vitro intrinsic values were scaled assuming the following: 1.5 × 106 hepatocytes/mg of measured protein, 118 × 106 hepatocytes/g of liver, 39.8 mg microsomal protein/g of liver, 24.5 g liver/kg of body weight (Varma et al., 2013). Relative expression factor (REF)—correction for the expression difference in the hepatocytes and human liver—of 1.8 and 2.0—were applied for OAT2 and OATP1B1 substrates, respectively, based on our previous published quantitative proteomics data (Kimoto et al., 2012; Vildhede et al., 2018). The hepatocyte lot (Hu8246) used for uptake studies showed expression levels of OAT2 similar to the median expression levels of 30 single-donor cryopreserved hepatocyte lots (Vildhede et al., 2018).
Hepatic blood and plasma clearances are calculated assuming the well-stirred liver model (eq. 11), where CLblood, h = CLplasma,h/Rb: (11)Predictive precision and accuracy were assessed with average fold error (AFE) eq. 12 and bias eq. 13:
(11)Predictive precision and accuracy were assessed with average fold error (AFE) eq. 12 and bias eq. 13: (12)
(12) (13)N is the number of observations.
(13)N is the number of observations.
Results
Compound Selection.
In a previously published data set of 368 compounds spanning across six classes of ECCS—classified based on their ionization, molecular weight, and transcellular permeability—about 10% (n = 36) of compounds are binned in class 1A (Varma et al., 2015; El-Kattan et al., 2016). Human intravenous clearance and renal clearance data are available for all these drugs. Of these 36 high-permeability (>5 × 10−6 cm/s) and low-mol. wt. (<400 Da) acids/zwitterions, three compounds (milrinone, pralidoxime, and clinafloxacin) show predominantly renal clearance, whereas others involve hepatic clearance mechanisms. All the compounds were considered for the current study, with a few exceptions: control substances (hexobarbital and thiopental) and lack of compound availability (acenocoumarol, perindopril, and sinitrodil). A data set of 25 compounds with in vitro hepatic uptake measurements is available after a few compounds were dropped (acetylsalicylic acid, amniosalicylic acid, dichloracetic acid, diflunisal, milrinone, and valproic acid) owing to analytical challenges and/or low cellular accumulation in uptake studies. Of the 25, six compounds returned no measurable metabolic turnover in the HLM incubations under the experimental conditions used. Therefore, the final set of 19 compounds was considered for IVIV extrapolation of transporter-enzyme interplay via extended clearance model (Supplemental Fig. 1).
In Vitro Transport in OAT2- and OATP1B1-Transfected Cells.
ECCS 1A compounds were evaluated for their substrate affinity to OAT2 and OATP1B1 using transfected HEK293 cells. All the compounds studied showed uptake ratio (i.e., ratio of accumulation in transfected cells to wild-type cells) of more than 1.5 in OAT2-HEK cells, suggesting they are actively transported by OAT2, except clinafloxacin and nateglinide (Fig. 1). Whereas 85% of compounds showed an uptake ratio of >2, and compounds such as ibuprofen, meloxicam, R-warfarin, and sulfamethoxazole yielded uptake ratios of more than 5.

OAT2- and OATP1B1-mediated transport of ECCS 1A drugs in overexpressing cells. Uptake was measured in HEK293 cells transfected with OAT2 (A) and OATP1B1 (B) and presented as the ratio to uptake in wild-type HEK293 cells. Bars and error bars represent mean and S.D. (n = 3). Dashed and dotted vertical lines represent the uptake ratio of 1 and 1.5, respectively. Uptake ratio >1.5 was used as the criteria for transport activity.
In contrast, only 4 of 25 compounds (bromfenac, entacapone, fluorescein, and nateglinide) showed an uptake ratio >1.5 in OATP1B1-HEK cells, implying that most of the compounds are not transported by OATP1B1; these four compounds are OAT2/OATP1B1 dual substrates, except nateglinide (OATP1B1 alone). Positive controls of OAT2 (cGMP) and OATP1B1 (rosuvastatin) showed robust signal, supporting the performance of the cell lines used. Diclofenac, tolbutamide, and R/S-warfarin were previously reported to be OAT2 substrates using transfected cells (Zhang et al., 2016; Bi et al., 2018a,b). On the other hand, fluorescein and nateglinide were previously shown to be OATP1B1 substrates (De Bruyn et al., 2011; Takanohashi et al., 2012; Izumi et al., 2016). For all the other compounds, the noted transporter substrate affinity was previously unknown to our best knowledge.
In Vitro Uptake in Human Hepatocytes.
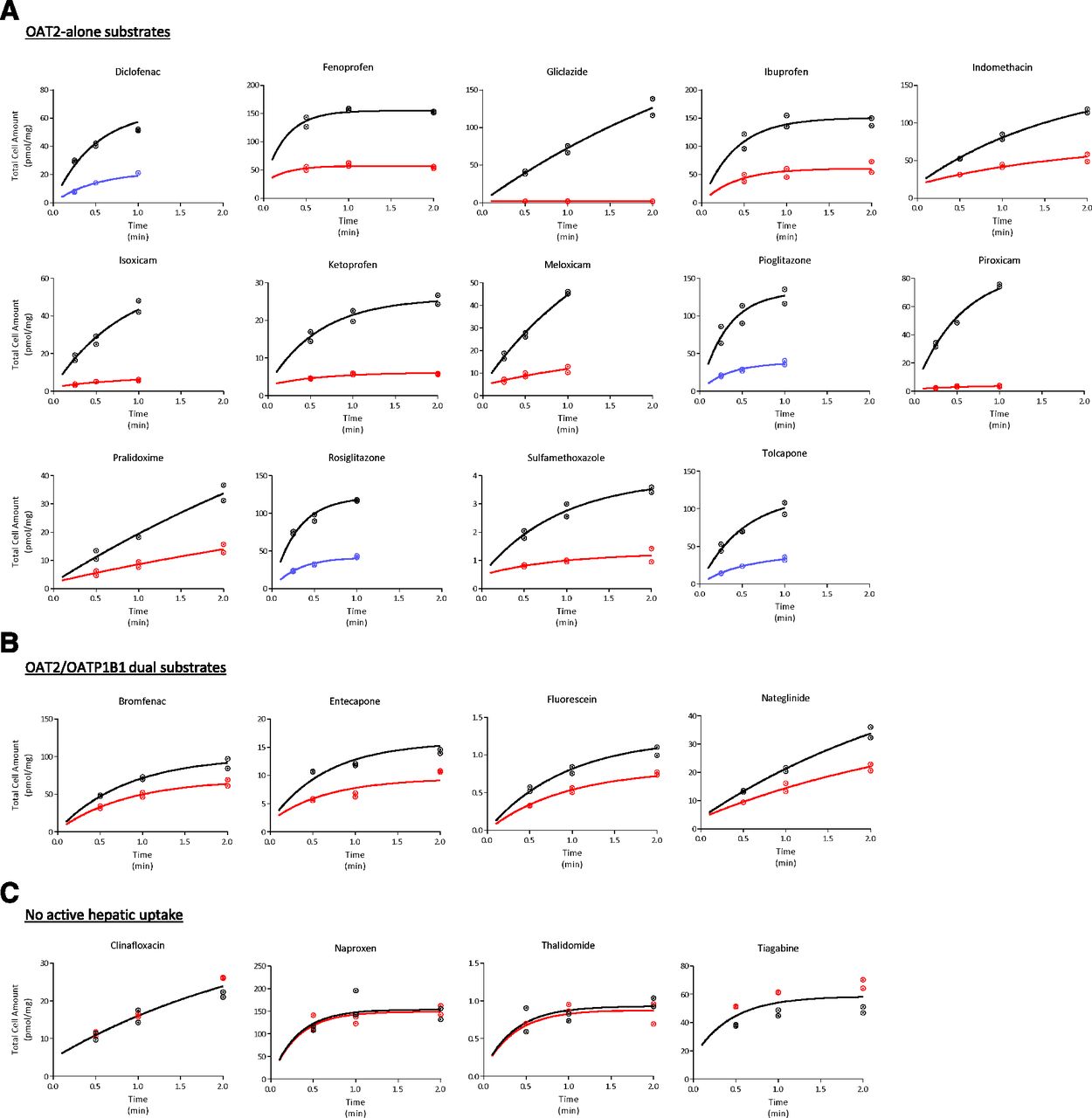
Figure 2 shows the time-dependent uptake of ECCS class 1A drugs by human hepatocytes plated in short-term culture. Rifamycin SV (1 mM), a pan-SLC inhibitor (including OAT2 and OATP1B1) (Bi et al., 2017, 2018a; Mathialagan et al., 2018), significantly reduced the uptake of all compounds, with a few exceptions. Clinafloxacin, naproxen, thalidomide, and tiagabine uptake was not altered by rifamycin SV under the experimental conditions used. In the case of diclofenac, pioglitazone, rosiglitazone, and tolcapone, the presence of rifamycin SV led to increased hepatocyte accumulation (data not shown). Although little is known about the stimulation of OAT2 activity, several previous studies have suggested plausible allosteric interaction for the substrate-dependent stimulation of the in vitro OATP activity in the presence of an inhibitor (Roth et al., 2011). Nonetheless, for these four drugs, hepatic uptake was significantly reduced when they were incubated on ice (4°C). Uptake time-course data were fitted to a two-compartment mathematical model to estimate the intrinsic active uptake clearance (PSactive) and intrinsic passive clearance (PSpassive), assuming 1 mM rifamycin SV (or 4°C) completely inhibited active uptake (Table 1). Estimated PSactive ranged from 0 (clinafloxacin) to 300 µl/min per milligram (pioglitazone), whereas PSpassive ranged from 0.2 (thalidomide) to 138 µl/min per milligram (rosiglitazone). Finally, compounds in the data set were categorized in three bins based on their transport characteristics observed with transfected cells and plated human hepatocytes: 1) OAT2 substrates with significant active uptake in human hepatocytes (referred to as OAT2-alone substrates, n = 17); 2) OAT2/OATP1B1 substrates with significant active hepatic uptake (OAT2/OATP1B1 dual substrates, n = 4); and 3) compounds with no measureable active transport in transfected and/or plated hepatocytes (i.e., no active hepatic uptake, n = 4). Nateglinide was recognized by only OATP1B1; however, it was included in the dual-substrates category for further data analysis. Generally, PSactive is higher than PSpassive for the OAT2-alone substrates in this set (Fig. 3A). Uptake clearance was lower for OAT2/OATP1B1 dual substrates compared with substrates for OAT2 alone.

Time course of cellular uptake of ECCS 1A drugs in plated human hepatocytes. Drugs were binned in three categories: OAT2-alone substrates (A), OAT2/OATP1B1 dual substrates (B), and no active hepatic uptake (C). Data points and curves represent uptake in control conditions (black) in the presence of 1 mM rifamycin SV (red) and at 4°C (blue). Curves are data-fit to the two-compartment mathematical model. Uptake rates of tolbutamide and R/S warfarin were obtained from our previous studies (Bi et al., 2018a,b). Incubation concentration is 10 µM for fenoprofen, ibuprofen, pralidoxime, naproxen, and thalidomide and 1 µM for all others.
Summary of drug properties and in vitro transport and metabolism measurements of the 25 high-permeability–low-molecular-weight acid and zwitterion drugs evaluated

Comparison of passive hepatic uptake with the active uptake clearance measured using plated human hepatocytes (A). Comparison of passive hepatic uptake measured using plated human hepatocytes with the intrinsic metabolic clearance measured in microsomal incubations (B). Circles represent OAT2-alone substrates, triangles represent OAT2/OATP1B1 dual substrates, and squares represent drugs with no measurable active uptake in human hepatocytes. Solid and dashed diagonal blue lines indicate unity and 3-fold range, respectively.
In Vitro Metabolic Clearance in HLMs and Primary Human Hepatocytes.
Intrinsic clearance of all compounds was measured by substrate-depletion method using pooled HLMs with both NADPH and UDPGA cofactors in the incubations (Table 1). The measured intrinsic clearance (CLint, met, app) (not corrected for the microsomal binding (fu,mic)) spanned a broad range from 1.9 (meloxicam, tolbutamide) to 140 µl/min per milligram of protein (diclofenac). A few compounds (gliclazide, isoxicam, piroxicam, sulfamethoxazole, and R/S-warfarin) did not show measurable substrate depletion during the 180-minute incubations. In our previous study, apparent metabolic clearance in hepatocytes corrected for cell-to-media concentration (Kpuu) was available for R/S-warfarin (Bi et al., 2018a). Microsomal binding was determined by equilibrium dialysis, and the values ranged from no binding to approximately 50% bound. It is noteworthy that the PSpassive measured in plated hepatocytes is equal or greater than microsomal metabolic clearance for 10 of 13 (∼76%) OAT2-alone substrates (Fig. 3B).
Additionally, we measured apparent intrinsic clearance after substrate depletion in suspension human hepatocyte incubations. Values ranged from 1.6 (gliclazide) to 66.5 µl/min per million cells (diclofenac) (Table 1). For OAT2-alone substrates, where the substrate depletion was measurable in both systems (n = 15), hepatocellular CLint, met values were on average 2.6-fold greater than the microsomal CLint, met (Supplemental Fig. 2).
IVIV Extrapolation to Human Hepatic Clearance.
A summary of human hepatic clearance predictions from the in vitro data are presented in Table 2; and the predicted hepatic clearance values (CLint, h and CLplasma, h) are plotted versus the observed clearance values in Fig. 4. IVIV extrapolation was evaluated assuming well stirred considerations under three variations: 1) uptake-determined clearance, where total hepatic uptake (PSactive.REF + PSpassive) was assumed to be the rate-determining step (eq. 8); 2) metabolism-determined clearance, where liver microsomal clearance was scaled alone (eq. 9); and 3) transporter-enzyme interplay, where hepatic transport and microsomal metabolic clearance were integrated using extended clearance model (eq. 10) (Supplemental Fig. 1).
In vitro-in vivo extrapolation of transport and metabolism data assuming hepatic clearance is determined by total uptake, metabolism-alone, and extended clearance model
Clearance predictions from pooled hepatocytes substrate-depletion studies were also summarized.

IVIV extrapolation to predict human hepatic clearance of ECCS 1A drugs. Observed versus predicted hepatic clearance assuming (A) total uptake clearance alone measured using plated human hepatocytes, (B) metabolic clearance alone measured using HLMs, and (C) extended clearance model considering active and passive uptake transport and microsomal metabolic clearance. Left and right panels represent hepatic intrinsic clearance and hepatic plasma clearance, respectively. Circles represent OAT2-alone substrates, triangles represent OAT2/OATP1B1 dual substrates, and squares represent drugs with no measurable active uptake in human hepatocytes. Solid, dotted, and dashed diagonal blue lines indicate unity, 2-fold, and 3-fold range, respectively. AFE and bias (red diagonal lines) of OAT2-alone substrates are shown.
For OAT2-alone substrates, AFE for CLint,h predictions based on uptake-determined clearance is ∼8.2, where most of the compounds are overpredicted (bias ∼6.4) (Table 3). Metabolism-determined clearance alone underpredicted (AFE ∼5.2 and bias ∼0.20) in vivo CLint, h for all compounds, with a few exceptions (pioglitazone, and pralidoxime). Only 5 of 13 (38%) predictions are within 3-fold of observed values; however, prediction accuracy and bias markedly improved when using extended clearance model (AFE∼1.9 and bias ∼0.90). About 8 (62%) and 11 (85%) of the 13 predictions of OAT2-alone substrates are within 2-fold and 3-fold error of the observed values, respectively. Similar predictive performance could be seen for the plasma hepatic clearance predictions (Table 3).
Summary of predictive performance of various in vitro-in vivo extrapolation approaches employed
Bias was calculated for only OAT2-alone drugs (eq. 13).
OAT2/OATP1B1 dual substrates were considerably underpredicted in all scenarios. A scaling factor of 10.6 for active hepatic uptake (instead of OATP1B1 REF of 2.0) in the extended clearance model (eq. 10), which was previously reported by our group (Varma et al., 2014; Kimoto et al., 2017), improved clearance predictions for dual substrates to some extent (Supplemental Fig. 3). Because of limited microsomal stability data, no conclusions could be drawn for compounds with no active uptake in plated human hepatocytes.
Finally, direct scaling of intrinsic clearance measured by substrate depletion in suspension human hepatocytes reasonably predicted hepatic clearance (Fig. 5; Table 3). AFE and bias are ∼2.3 and 0.63, respectively, for the OAT2-alone substrates, with ∼47% and 73% predictions within 2-fold and 3-fold error, respectively. For OAT2/OATP1B1 dual substrates, this method underpredicted in vivo CLint,h on an average of about ∼10-fold.

Comparison of hepatic clearance predicted from intrinsic clearance measured using substrate depletion in human hepatocytes with the observed in vivo hepatic clearance. Left and right plots represent hepatic intrinsic clearance and hepatic plasma clearance, respectively. Circles represent OAT2-alone substrates, triangles represent OAT2/OATP1B1 dual substrates, and squares represent drugs with no measurable active uptake in human hepatocytes. Solid, dotted, and dashed diagonal blue lines indicate unity, 2-fold, and 3-fold range, respectively. AFE and bias (red diagonal lines) of OAT2-alone substrates are shown.
Discussion
This study, using a diverse set of 25 drugs, provided evidence for the functional role of OAT2-mediated hepatic uptake in the pharmacokinetics of high-permeability–low-molecular-weight acid and zwitterion drugs (i.e., ECCS 1A drugs). These drugs generally undergo extensive hepatic metabolism by CYP2C, UGTs, and other enzymes and are eliminated from the body primarily as metabolites (Varma et al., 2015; El-Kattan et al., 2016). Here, most ECCS 1A drugs demonstrated substrate affinity to OAT2 and active hepatic uptake. Clearly, hepatic clearance of OAT2-alone substrates was well predicted when using an extended clearance model, assuming transporter-enzyme interplay; however, scaling HLM clearance alone, assuming metabolism-determined clearance, showed the tendency of systemic underprediction (∼5-fold). To our knowledge, this is the first study to suggest an OAT2 role in the hepatic uptake clearance and thus the pharmacokinetics for most high-permeability–low-moleculat-weight acid and zwitterion drugs. These findings are of clinical importance as many such drugs are widely prescribed in a variety of therapeutic areas, including inflammation, diabetes, Parkinson disease, and thrombosis.
Based on the in vitro mechanistic studies and PBPK analyses, we have demonstrated that OAT2 plays a key role in the overall hepatic clearance of tolbutamide and R/S-warfarin, which are ECCS class 1A drugs (Bi et al., 2018a,b). We therefore set out to investigate the role of OAT2 in the hepatic clearance for an unbiased set of 25 drugs in this class. Except for clinafloxacin and nateglinide, all drugs showed OAT2-mediated transport in transfected cells. These drugs also showed significant active uptake in primary human hepatocytes plated in short-term cultures, with a few exceptions under the experimental conditions used (clinafloxacin, naproxen, thalidomide, and tiagabine). Collectively, these in vitro studies provided functional evidence for active hepatic uptake driven by OAT2 for most high-permeability–low-molecular-weight acids/zwitterions. Further work is needed to understand the role of OAT2 in the clearance of other classes of drugs.
We used pooled liver microsomes to estimate in vitro metabolic intrinsic clearance by following substrate-depletion time course. Previous studies have shown that CLint, met obtained from substrate-depletion and metabolite-formation approaches are often comparable (Jones and Houston, 2004; Di et al., 2013). The presence of both cofactors, NADPH and UDPGA, in the incubations of alamethicin-activated microsomes allowed simultaneous assessment of oxidation and glucuronidation pathways (Fisher et al., 2000; Kilford et al., 2009). Moreover, values obtained here were generally consistent with the reported CLint values measured using liver microsomes and hepatocytes (Obach, 1999; Brown et al., 2007); however, the microsomal CLint,met underpredicted in vivo CLint,h by an average of ∼5-fold (bias ∼0.20). This observation is also consistent with several previous reports highlighting that the in vitro CLint,met systematically underpredict the clearance of acid drugs that undergo extensive metabolism (Obach et al., 1997; Obach, 1999; Brown et al., 2007; Wood et al., 2017). No trend was noted between the predictive accuracy and the known primary enzyme involved (CYP2C vs. UGT). The current study suggests lack of consideration to OAT2-mediated hepatic uptake as a major source for IVIV disconnect noted previously in this physicochemical space.
Our recent mass spectrometry–based targeted proteomics studies indicated about 1.8-fold higher abundance of OAT2 in human liver tissue samples (n = 52) compared with the expression in single-donor cryopreserved hepatocytes (n = 30) (Vildhede et al., 2018). Similarly, in our previous studies, ∼2-fold higher OATP1B1 expression was observed in liver tissues relative to the levels in cryopreserved hepatocytes (Kimoto et al., 2012). Therefore, REFs of 1.8 and 2.0 were applied when scaling in vitro active uptake to predict hepatic clearance for OAT2-alone substrates and OAT2/OATP1B1 substrates, respectively. We examined the sensitivity of OAT2 REF (1.8 vs. 1.0 – later assuming no correction for transporter abundance) and noted only a marginal effect on the overall predictive performance of extended clearance model (AFE 1.9 vs. 2.0; bias 0.9 vs. 0.6). However, consistent with many earlier reports (Jones et al., 2012; Ménochet et al., 2012; Varma et al., 2012b, 2014; Li et al., 2014), a scaling factor much larger than the OATP1B1 expression differences was needed to recover hepatic clearance of OATP1B1 substrates. To this end, a previously derived empirical scaling factor for the active uptake clearance (SFactive ∼10.6) (Varma et al., 2014) provided closer predictions for the four OAT2/OATP1B1 substrates in the current data set (Supplemental Fig. 3). Collectively, direct IVIV extrapolation of OAT2-alone substrates suggests that OAT2 functional activity, unlike OATP1B1, is well preserved in cryopreserved human hepatocytes.
Depletion of parent was also assessed in pooled human hepatocytes, and the predictive performance of this system is better than that from liver microsomes (Fig. 5; Table 3). Interestingly, hepatocytes on average yielded ∼2.6-fold greater substrate depletion than that measured in microsomal incubations (Supplemental Fig. 3). This is likely due to greater free cell-to-media concentrations (Kpuu) driven by OAT2-mediated uptake in the hepatocytes. In our previous studies, we observed a Kpuu of ∼4 to 5 for R/S-warfarin in the suspension human hepatocytes (Bi et al., 2018a). Although further studies are needed to understand the reasons leading to differences in the measurements between the two systems, this study suggests that substrate-depletion in suspension human hepatocytes can be used as a pragmatic screening tool to predict hepatic clearance in this physicochemical space.
“Rapid equilibrium” between blood and liver compartments is often presumed for high-permeability drugs, resulting in the hypothesis that metabolism is the rate-determining process in their hepatic clearance. Transcellular permeability of the OAT2-alone substrates in our data set, measured across monolayers of Madin-Darby canine kidney (MDCK) low- efflux cells, ranged from about 8 (sulfamethoxazole) to 35 × 10−6 cm/s (fenoprofen). In the same experimental conditions, permeability of ECCS class 1B drugs (high-permeability–high-molecular-weight acids/zwitterions) span a similar range, ∼5.5 (atorvastatin, pitavastatin) to 16 × 10−6 cm/s (repaglinide) (Varma et al., 2015; El-Kattan et al., 2016). ECCS class 1B drugs are well proven to involve OATP-mediated hepatic uptake in their systemic clearance (Shitara et al., 2013; Varma et al., 2017a). Indeed, much evidence has been presented for “uptake-determined” clearance for highly permeable and metabolically labile drugs such as atorvastatin, bosentan, and glyburide (Zheng et al., 2009; Watanabe et al., 2010; Maeda et al., 2011; Yoshikado et al., 2017). Under similar principles, the current study demonstrated that OAT2-mediated active uptake contributes to the systemic clearance of high-permeability–low-molecular-weight acids/zwitterions. No correlation was apparent between the transcellular permeability across MDCK cell monolayers and passive uptake clearance measured in human hepatocytes (in the presence of rifamycin SV or 4°C) for the compounds in the current dataset (Supplemental Fig. 4). Drugs in our data set have a median acid pKa of ∼4.4 (range, 2.2–7.9), implying that they exist in >99.9% ionized form (at physiologic pH 7.4), a form that is expected to have negligible passive diffusion across the cell membrane (Avdeef, 2001). This is consistent with the low passive uptake clearance and implies dependence on the active transport mechanism to achieve high total hepatic uptake noted in the hepatocytes. Whereas cell membrane composition difference across the systems may contribute to the observed poor correlation (Simons and Ikonen, 1997), the role of uptake transporters cannot be ruled out in the transcellular permeability model (Dobson and Kell, 2008; Ahlin et al., 2009; Kell et al., 2011). Overall, potential role of uptake transporters in hepatic clearance should be duly evaluated for the acids and zwitterions irrespective of the membrane permeability across cell culture models (e.g., MDCK or Caco-2).
We observed a minimal overlap in the substrate specificity for OAT2 versus OATP1B1, which is in general agreement that OATPs preferentially accept high-MW (>400 Da) acids/zwitterions (class 1B) as substrates (Varma et al., 2012a). Of the four OATP1B1 substrates identified in the ECCS 1A space, nateglinide and fluorescein were previously reported to be OATPs substrates (De Bruyn et al., 2011; Takanohashi et al., 2012; Izumi et al., 2016), whereas this is the first report suggesting that bromfenac, an nonsteroidal anti-inflammatory drugs for ocular inflammation, and entacapone, an catechol-O-methyltransferase inhibitor used in Parkinson disease, involve OATP1B1-mediated hepatic uptake clearance. Clinical drug-drug interaction studies with OATP inhibitors rifampicin and cyclosporine may be helpful to further define uptake-determined clearance in these cases. The MW of these four OATP1B1 substrates ranges from 305 to 334 Da. While the occurrences are low, this study suggests that low-MW acids/zwitterions may involve OATP1B1-mediated clearance.
Although a plethora of drug-drug interaction and pharmacogenetic studies have shown the relevance of CYP2C and UGT mechanisms, our findings will open the field to consider OAT2-mediated hepatic uptake as a source of variability in the pharmacokinetics and pharmacodynamics of drugs in the ECCS 1A class. According to extended clearance concept (Shitara et al., 2013; Patilea-Vrana and Unadkat, 2016; Li et al., 2014; Varma et al., 2015), the extended clearance model (eq. 10) is reduced such that the hepatic clearance can be approximated by PSuptake × CLmet/PSpassive, when PSpassive > CLint, met, which is often the case for the compounds in the current data set (Fig. 3B). Therefore, functional variability in OAT2 and enzymatic metabolism can lead to variability in pharmacokinetics for these drugs. For instance, genetic polymorphism in CYP2C8 and CYP2C9 was associated with systemic clearance of many ECCS 1A drugs, including ibuprofen, piroxicam, tolbutamide, and S-warfarin (Rettie et al., 1994; Kirchheiner et al., 2002; García-Martín et al., 2004; Perini et al., 2005). For the last two drugs, we previously demonstrated that a PBPK model with permeability-limited hepatic disposition (transporter-enzyme interplay) quantitatively describes the pharmacogenetic effects when considering genotype, phenotype, and fraction metabolism by CYP2C9 (Bi et al., 2018a,b). Our preliminary literature search, however, revealed limited knowledge about the clinically relevant OAT2 inhibitors and functional polymorphic variants of SLC22A7 (gene-encoding OAT2) (Bi et al., 2018a). Further studies are needed in these areas to assess the role of OAT2 in clinical settings. This assessssment is important because changes in uptake or metabolic clearance will have a proportional impact on hepatic clearance, and a simultaneous change in both mechanisms in same direction can result in a marked change in clinical pharmacokinetics. Moreover, OAT2-mediated uptake can lead to high free liver-to-plasma concentrations (Kpuu), which may contribute to the liver-specific pharmacologic and/or toxicologic activities. It will be of interest to understand the association between liver Kpuu and drug-induced hepatotoxicity noted for several of the OAT2-alone substrates in our data set (e.g., diclofenac, ibuprofen. pioglitazone, piroxicam, rosiglitazone, tolcopone, etc.) (Boelsterli, 2003; Morgan et al., 2010; Chen et al., 2011).
In conclusion, our systemic evaluation provided robust evidence for the role of a previously unrecognized OAT2-mediated hepatic uptake in the clearance of several high-permeability–low-molecular-weight acid and zwitterion drugs. For this class of drugs or new chemical entities (ECCS class 1A), uptake transport characterization and considerations to transporter-enzyme interplay are important for predicting clinical pharmacokinetics and assessing variability owing to drug-drug interactions and other intrinsic and extrinsic factors.
Authorship Contributions
Participated in research design: Kimoto, Mathialagan, Tylaska, Niosi, Lin, Tess, Varma.
Conducted experiments: Kimoto, Mathialagan, Tylaska, Niosi, Lin, Carlo.
Performed data analysis: Kimoto, Mathialagan, Tylaska, Niosi, Lin, Tess, Varma.
Wrote or contributed to the writing of the manuscript: Kimoto, Mathialagan, Tylaska, Niosi, Lin, Carlo, Tess, Varma.
Footnotes
- Received July 12, 2018.
- Accepted August 15, 2018.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AFE
- average fold error
- CLint
- intrinsic clearance
- CLint
- h, intrinsic hepatic clearance
- CLint
- met, intrinsic metabolic clearance
- CLplasma
- h, plasma hepatic clearance
- ECCS
- extended clearance classification system
- HBSS
- Hanks’ balanced salt solution
- HLM
- human liver microsome
- IVIV
- in vitro-in vivo
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MDCK
- Madin-Darby canine kidney (cells)
- OAT
- organic anion transporter
- OATP
- organic anion-transporting polypeptide
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetic
- PSactive
- active uptake clearance
- PSpassive
- passive clearance
- REF
- relative expression factor
- SLC
- solute carrier
- UGT
- uridine 5′-diphospho-glucuronosyltransferase
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}