


Abstract
The selective estrogen receptor modulator tamoxifen increases extracellular dopamine in vivo and acts as a neuroprotectant in models of dopamine neurotoxicity. We investigated the effect of tamoxifen on dopamine transporter (DAT)–mediated dopamine uptake, dopamine efflux, and [3H]WIN 35,428 [(–)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane] binding in rat striatal tissue. Tamoxifen dose-dependently blocked dopamine uptake (54% reduction at 10 μM) and amphetamine-stimulated efflux (59% reduction at 10 μM) in synaptosomes. It also produced a small but significant reduction in [3H]WIN 35,428 binding in striatal membranes, indicating a weak interaction with the substrate binding site in the DAT. Biotinylation and cysteine accessibility studies indicated that tamoxifen stabilizes the outward-facing conformation of the DAT in a cocaine-like manner and does not affect surface expression of the DAT. Additional studies with mutant DAT constructs D476A and I159A suggested a direct interaction between tamoxifen and a secondary substrate binding site of the transporter. Locomotor studies revealed that tamoxifen attenuates amphetamine-stimulated hyperactivity in rats but has no depressant or stimulant activity in the absence of amphetamine. These results suggest a complex mechanism of action for tamoxifen as a regulator of the DAT. Due to its effectiveness against amphetamine actions and its central nervous system permeant activity, the tamoxifen structure represents an excellent starting point for a structure-based drug-design program to develop a pharmacological therapeutic for psychostimulant abuse.
Introduction
Dopamine plays a significant role in multiple neural processes, including motor control and reward processing. Dysfunction of the dopaminergic system leads to diverse disorders such as addiction, Parkinson disease, and schizophrenia (Abi-Dargham, 2014; German et al., 2015). Tamoxifen is a widely prescribed selective estrogen receptor modulator (SERM) used in the treatment and prevention of estrogen receptor–positive breast cancer (Jordan, 2014). It has long been known that the drug tamoxifen affects the dopaminergic system, yet the mechanisms by which it does so have remained unclear (Mikelman et al., 2017). Tamoxifen causes a small but significant increase in extracellular dopamine levels in the nucleus accumbens after peripheral administration (Chaurasia et al., 1998) and also inhibits amphetamine-stimulated hyperactivity (Einat et al., 2007; Cechinel-Recco et al., 2012; Pereira et al., 2014), which relies on elevation of extracellular dopamine levels in the striatum (French, 1986). Tamoxifen demonstrated neuroprotection against 1-methyl-4-phenylpyridinium (MPP+)- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in a mouse model of Parkinson disease (Obata and Kubota, 2001; Bourque et al., 2007; Obata and Aomine, 2009) and against methamphetamine-induced neurotoxicity (D’Astous et al., 2005). Both models of neurotoxicity selectively damage dopaminergic neurons and depend on uptake of the neurotoxin by the dopamine transporter (DAT) to exert their deleterious activities. Although the effects of tamoxifen on amphetamine-stimulated hyperactivity are believed to be the result of protein kinase C (PKC) inhibition (O’Brian et al., 1985; Einat et al., 2007), the neuroprotective effects of tamoxifen have largely been assumed to result from tamoxifen’s activity as an SERM. However, tamoxifen has many alternative mechanisms of action, including, but not limited to, binding to calmodulin (O’Brian et al., 1990) and the D2-like dopamine receptor (Hiemke and Ghraf, 1984; Toney and Katzenellenbogen, 1987). It is unclear whether any of these mechanisms are responsible for the effects of tamoxifen on the dopaminergic system. Therefore, we set out to better characterize the dopaminergic effects of tamoxifen and examine the mechanism by which it exerts these effects.
A logical query is the effect of tamoxifen on DAT function. Amphetamine, MPP+, and MPTP are all substrates for the DAT and require the transporter to enter the cell and exert their effects. The DAT is responsible for clearing dopamine from the extracellular space after its release and in this capacity is a crucial mechanism for regulating dopaminergic signaling (Jaber et al., 1997). Although tamoxifen was shown to block amphetamine-stimulated locomotor behavior (Einat et al., 2007) and we previously demonstrated that it inhibits DAT function independently of its action as an SERM (Mikelman et al., 2017), no studies reported to date have systematically examined the interaction between tamoxifen and the DAT.
The DAT facilitates the movement of dopamine across the plasma membrane through a series of conformational changes that can be simplified into the “alternating access” model of transport (Jardetzky, 1966). In this model, the DAT alternates among an outward-facing conformation, where dopamine binds to the primary substrate binding site (S1); a series of occluded conformations through which dopamine is transported across the membrane; and an inward-facing conformation that forms the transition between the two (Shi et al., 2008; Shan et al., 2011). More recently, a secondary site (S2), based on homology models with the bacterial leucine transporter and the Drosophila DAT (Nyola et al., 2010; Piscitelli et al., 2010; Penmatsa et al., 2015; Coleman et al., 2016), was proposed to play a role in transporter function. The existence of the site has been demonstrated with bivalent DAT ligands that demonstrate increased affinity compared with their monovalent counterparts, indicating the presence of two binding sites on the DAT (Schmitt et al., 2010). This secondary site is suggested to be an allosteric binding site for modulation of DAT conformation and dopamine transport (Shan et al., 2011).
Here we demonstrate that tamoxifen directly interacts with the DAT. We find that tamoxifen noncompetitively inhibits dopamine uptake and blocks amphetamine-stimulated dopamine efflux. We use cysteine accessibility assays and newly characterized “S2-defective” DAT mutants (Zhen and Reith, 2016) to demonstrate that tamoxifen stabilizes the outward-facing conformation of the DAT, potentially through an interaction with the S2 domain. This hypothesis is supported by computational docking experiments. Finally, we demonstrate that tamoxifen inhibits amphetamine-stimulated hyperactivity in vivo, yet exhibits no stimulant effects of its own. Our results demonstrate a heretofore unrecognized mechanism of action for tamoxifen.
Materials and Methods
Materials.
Tamoxifen citrate and amphetamine hemisulfate were obtained from Sigma-Aldrich (St. Louis, MO). Tamoxifen citrate was dissolved in 100% dimethylsulfoxide (DMSO) at a concentration of 50 mM and diluted further for in vitro studies, or at 2 mg/ml in 3.5% DMSO and 10% Tween 80 in saline for in vivo studies. Cocaine hydrochloride was provided by the National Institutes of Health National Institute on Drug Abuse (Bethesda, MD). [3H]WIN 35,428 [(–)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane] and [3H]dopamine were purchased from PerkinElmer (Waltham, MA). All other chemicals were obtained from Sigma-Aldrich unless otherwise noted.
Animals.
All animal use procedures were approved by the University of Michigan University Animal Care and Use Committee and were in accordance with the National Institutes of Health guidelines. Male Sprague-Dawley rats aged 7–12 weeks were obtained from Harlan Laboratories (Indianapolis, IN). Rats were maintained under standard conditions on a 12-hour/12-hour light/dark cycle and were housed in groups of two or three.
Synaptosome Preparation.
Rat striata were homogenized in 10 volumes of homogenization buffer (0.32 M sucrose and 1 mM EDTA, pH 7.4). Homogenates were centrifuged at 1000g for 10 minutes and the supernatant was transferred to a fresh vial. The supernatant fraction was centrifuged at 15,000g for 15 minutes. The resulting pellet was resuspended in the appropriate buffer.
Suprafusion.
The synaptosome pellet was resuspended in Krebs-Ringer buffer (KRB) (145 mM NaCl, 2.7 mM KCl, 1.2 mM KH2PO4, 1.0 mM MgCl2, 10 mM glucose, 24.9 mM NaHCO3, 0.05 mM ascorbic acid, and 0.05 mM pargyline, pH 7.4). Synaptosomes were loaded into the reaction chambers of a Brandel Suprafusion apparatus (Brandel Inc., Gaithersburg, MD) and washed with KRB (with or without drug) for 1 hour at 37°C at 800 μl/min to allow for adequate treatment time and to reach a steady baseline. After the wash, 1-minute fractions were collected for a total of 14 fractions; 10 μM amphetamine sulfate was added to the buffer during fractions 7 and 8. Vehicle or tamoxifen was included in the buffer throughout fraction collection. An internal standard solution (final concentration: 50 mM perchloric acid, 25 μM EDTA, and 10 nM 2-aminophenol) was added to each sample in an approximately 1:20 dilution and samples were analyzed for dopamine content by high-performance liquid chromatography coupled with electrochemical detection (Thermo Scientific/ESA, Sunnyvale, CA). A small aliquot of synaptosomes was reserved and diluted 1:50 in internal standard solution. After 30-minute incubation at 4°C, the solution was centrifuged for 20 minutes at 15,000g. The supernatant was diluted 1:20 in KRB and measured for dopamine content. This value was used to calculate the total dopamine content of the synaptosomes.
[3H]Dopamine Uptake in Rat Striatal Synaptosomes.
Synaptosomes were resuspended in KRB as described above, aliquoted into 13- × 100-mm borosilicate glass test tubes, and incubated with tamoxifen or vehicle for 60 minutes at 37°C. Cocaine (100 μM) was used to measure nonspecific [3H]dopamine uptake. Unlabeled dopamine (20–300 nM) supplemented with 10 nM [3H]dopamine was added to the synaptosomes. The reaction was stopped at 3 minutes for saturation experiments and 30 seconds for kinetic experiments by the addition of 3 ml cold KRB followed by filtration through glass fiber filters (GF/C; Fisher Scientific) and washed twice more with cold KRB. Filters were dried and transferred to scintillation vials, and radioactivity was counted in 5 ml ScintiVerse cocktail (Thermo Fisher Scientific, Waltham, MA) using a Beckman LS5801 scintillation counter (Beckman Coulter, Brea, CA).
[3H]WIN 35,428 Binding in Membranes.
Synaptosomes were resuspended in buffer containing 30 mM sodium phosphate and 0.32 M sucrose, pH 7.4, and homogenized on ice for 3 seconds with a Polytron tissue homogenizer (BioSpec Products, Bartlesville, OK). The suspension was aliquoted into 13- × 100-mm glass test tubes containing tamoxifen or vehicle and [3H]WIN 35,428. Membranes were incubated for 2 hours at 4°C to allow the binding reaction to reach equilibrium and were then filtered, washed, and counted as described above. Nonspecific binding was determined by incubation with 30 μM nomifensine.
[3H]Dopamine Uptake in LLC-PK1 Cells.
LLC-PK1 (pig kidney epithelial) cells stably transfected to express wild-type (WT) and mutant human DATs (hDATs) with S2 site disruption were grown to confluence in a 24-well plate for 2 to 3 days (approximately 2 × 105 cells/well). To enhance DAT mutant expression, D476A- and I159A-hDAT–transfected cells were treated for 16 hours with 100 mM sodium butyrate prior to the experiment. Cells were washed three times with phosphate-buffered saline (PBS) and preincubated with vehicle or 10 µM tamoxifen prepared in 240 µl uptake buffer (122 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 15 mM Na2HPO4, 10 mM glucose, and 1 mM CaCl2) supplemented with 1 mM ascorbic acid for 1 hour at room temperature. [3H]Dopamine uptake assays were initiated by the addition of 30 µl of varying concentrations of unlabeled dopamine (final concentration ranging from 0 to 10 µM), followed quickly by the addition of 30 µl of 6–11 nM [3H]dopamine for a final per-well reaction volume of 300 µl. Nonspecific uptake was determined using 100 µM cocaine. Assays were conducted in 24-well plates for 5 minutes (WT cells) and 7 minutes (mutant cells) at 25°C, followed by extensive washing (3×) in ice-cold PBS. Cells were lysed with 5% ice-cold trichloroacetic acid for 30 minutes at 4°C and measured by liquid scintillation counting.
Biotinylation Assays to Determine Surface Levels of the DAT and Cysteine Accessibility.
Biotinylation assays were adapted from Hong and Amara (2010). Synaptosomes were incubated with vehicle, 10 µM tamoxifen, or 100 µM cocaine in KRB for 1 hour at 37°C. The reactions were transferred to ice and washed with cold KRB. The vehicle- and tamoxifen-treated samples were divided in two. One set (vehicle-, cocaine-, and tamoxifen-treated) was further incubated with 5 mg/ml PEG (Thermo Scientific) in PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBSCM), pH 7.1, for 45 minutes at 4°C in the continued presence of vehicle, cocaine, or tamoxifen. These samples were used to assess cysteine accessibility on the DAT. The other set (vehicle- and tamoxifen-treated) was incubated with 2 mg/ml sulfo-NHS-biotin (Thermo Scientific) in PBSCM, pH 7.4, under the same conditions. These samples were used to assess surface levels of the DAT after treatment with tamoxifen. The remaining PEG or sulfo-NHS-biotin was quenched by adding 500 mM cysteine or 1 M glycine, respectively, in PBSCM at 4°C for 15 minutes. Synaptosomes were centrifuged for 10 minutes at 16,000g and washed once more with the quenching solution. After centrifugation for 10 minutes at 16,000g, maleimide-PEG2-biotin–treated synaptosomes were resuspended in lysis buffer [10 mM Tris, 150 mM NaCl, and 1 mM EDTA, pH 7.5, containing protease inhibitors (Roche Applied Science, Penzberg, Germany)]. Sulfo-NHS-biotin–treated samples were resuspended in solubilization buffer (50 mM Tris, 150 mM NaCl, and 1% Triton × 100, pH 7.4, containing protease inhibitors) and lysed for 1 hour at 4°C, followed by 10-minute centrifugation at 12,000g. The supernatant was incubated with a 50% slurry of streptavidin agarose beads (Thermo Scientific) overnight at 4°C. The beads were washed once with 400 µl respective buffers and twice with 600 µl PBS. Biotinylated proteins were eluted with SDS-PAGE sample buffer; a 20-µl sample of lysate was prepared for electrophoresis as a control. All samples were heated at 70°C for 10 minutes and separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with anti-DAT, monoclonal antibody mab16 (Dr. Roxanne Vaughan, University of North Dakota, Grand Forks, ND), anti–Na+/K+-ATPase (1:1000 dilution; Cell Signaling Technology, Danvers, MA), or anti–glyceraldehyde 3-phosphate dehydrogenase (1:6000; Cell Signaling Technology) with horseradish peroxidase–conjugated mouse secondary antibody and developed using chemiluminescence. Blots were incubated with primary antibody overnight and washed with Tris-buffered saline/Tween 20, were then incubated with secondary antibody for 1 hour at room temperature and washed with Tris-buffered saline/Tween 20, and finally, were developed with chemiluminescent reagent. The Na+/K+-ATPase primary antibody was used at 1:1000 dilution and secondary anti–rabbit horseradish peroxidase was used at 1:3000.
Locomotor Assays.
Locomotor activity was evaluated in testing chambers (41 × 25.4 × 20.3 cm) equipped with a photocell beam array. Activity was quantified as the number of beam breaks in a designated period of time. Rats were allowed to acclimate in the chamber for 2 hours prior to administration of 5 mg/kg tamoxifen citrate (3 mg/kg tamoxifen) or vehicle (2 mg/ml in 3.5% DMSO and 10% Tween 80 in saline i.p.) and monitored for an additional 3 hours. This procedure was repeated the next day. On the third day, rats were allowed to acclimate in the chambers for 2 hours prior to administration of 1 mg/kg amphetamine (1 mg/ml in saline i.p.) or the equivalent volume of saline and activity was monitored for an additional 3 hours. No tamoxifen was given on the third day.
Computational Modeling and Ligand Docking into LeuT-Based hDAT.
Tamoxifen was docked into an hDAT homology model constructed based on the crystal structure of the bacterial neurotransmitter sodium symporter protein LeuTAa in the tricyclic-bound occluded conformation (Zhou et al., 2007; Protein Data Bank identifier 2QJU). The in silico hDAT homology model was constructed as previously described (Schmitt and Reith, 2011; Reith et al., 2012; Schmitt et al., 2013). Cotransported Na+ ions were initially placed based on their location in the LeuT crystal and the Cl− ion was initially placed at a position corresponding to the side-chain carboxyl moiety of Glu290 in the occluded LeuT structure [see Forrest et al. (2007) and Zomot et al. (2007) for further discussion]. Flexible ligand docking and energy minimization were performed using the Molecular Operating Environment (MOE) 2014.09 program (Chemical Computing Group, Montreal, QC, Canada). For docking, energy-minimized ligand structures were imported into MOE and docked at the vestibular S2 binding pocket. During the docking procedure, a minimum of 100,000 ligand poses were generated and sorted based on London dG and GBVI/WSA dG free-energy scoring functions. We selected a top-scoring, low-energy ligand/DAT conformation and then performed successive rounds of energy minimization with the AMBER12:EHT forcefield (a hybrid all-atom forcefield that uses AMBER ff12 for parameterization of protein atom charges and extended Hückel theory to assign partial charges to small molecules). The AMBER forcefield was parameterized with a nonbonded attenuation/cutoff interval of 10–12 Å, which smoothly tapers nonbonded (e.g., van der Waals) interactions over the interval and neglects the energetic contribution of potential nonbonded interactions for atoms greater than 12 Å apart. The Generalized Born implicit solvation model implemented in MOE was used to approximate solvent effects (Labute, 2008). Ligand atoms, ions, and atoms of all ligand-adjacent residues (peptide backbone and side-chain atoms of residues located within a 5-Å radius) were allowed to move freely and more distant residues were weakly tethered to their initial position (1 kcal mol−1 Å−1 force constant) during successive rounds of energy minimization. For the final minimization round, the tethering constant for distal atoms was increased to 10 kcal mol−1 Å−1 and the convergence gradient was set at 0.001 kcal mol−1 Å−1.
Statistical Analysis.
All statistical analyses were carried out using GraphPad Prism 6 software (GraphPad Software Inc., San Diego, CA). Data are plotted as the mean ± S.E.M. Significance was set at P < 0.05. Comparisons between multiple groups were made with one- or two-way analysis of variance (ANOVA), with post hoc tests as stated. Where only two groups were analyzed, unpaired or paired two-tailed t tests were used. In kinetic uptake and saturation binding assays, nonlinear regression was used to determine the appropriate parameters. When kinetic data were compared, comparison of fits in nonlinear regression was used to determine whether curves differed from each other. The null hypothesis was that the best-fit parameters for the values did not differ. A conclusion of statistical significance represents a rejection of the null hypothesis and indicates a difference between designated values. In these experiments, values are given with or without the 95% confidence intervals (CIs).
Results
Tamoxifen Noncompetitively Inhibits [3H]Dopamine Uptake in Rat Striatal Synaptosomes.
To determine whether tamoxifen interacted with the DAT, we first analyzed the effect of tamoxifen on [3H]dopamine uptake into rat striatal synaptosomes. As shown in Fig. 1A, tamoxifen significantly inhibited [3H]dopamine uptake in rat striatal synaptosomes. A repeated-measures one-way ANOVA indicated a significant treatment effect for tamoxifen [P < 0.0007, F(1.693, 6.773) = 27.49, n = 5]. Kinetic analysis demonstrated that this blockade was due to a decrease in the Vmax of [3H]dopamine uptake from vehicle values, indicating noncompetitive inhibition (Fig. 1B), with no significant change in KM. Vmax values for [3H]dopamine uptake in picomoles/0.5 minute per milligram protein for vehicle and tamoxifen-treated tissue were 47.6 (95% CI, 35.83–59.3) and 24.4 (95% CI, 18.09–30.65), respectively [F(1, 44) = 9.185, P < 0.005]. KM values for vehicle and tamoxifen-treated tissue were 1421 nM (95% CI, 633.6–2208) and 834 nM (95% CI, 263.3–1405), respectively, and did not significantly differ [F(1, 44) = 1.263, P = 0.267]. Similar results were found for the effect of tamoxifen on the kinetics of DAT in hDAT-N2A cells (Mikelman et al., 2017).

Tamoxifen impairs dopamine uptake. (A and B) Synaptosomes from the striatum of male Sprague-Dawley rats were incubated for 1 hour at 37°C with vehicle or indicated concentrations of tamoxifen, then treated with 310 nM (A) or indicated concentrations of [3H]DA and incubated for an additional 3 minutes (A) or 30 seconds (B). (A) Data are represented as the mean ± S.E.M. (n = 5). *P < 0.05; **P < 0.01 compared with vehicle (post hoc Dunnett multiple-comparison tests). (B) Data are represented as the mean ± S.E.M. (n = 5). **P < 0.01; ***P < 0.001 (post hoc Sidak multiple-comparisons test). DA, dopamine; TMX, tamoxifen.
Tamoxifen Attenuates Amphetamine-Stimulated Dopamine Efflux.
The dose-dependent effect of tamoxifen on amphetamine-stimulated dopamine efflux was determined using suprafusion of rat striatal synaptosomes to determine whether tamoxifen would affect dopamine efflux to the same degree that it affects dopamine uptake. A one-way ANOVA indicated a significant treatment effect [P < 0.002, F(3,16) = 8.309, n = 4–8] for tamoxifen (Fig. 2).
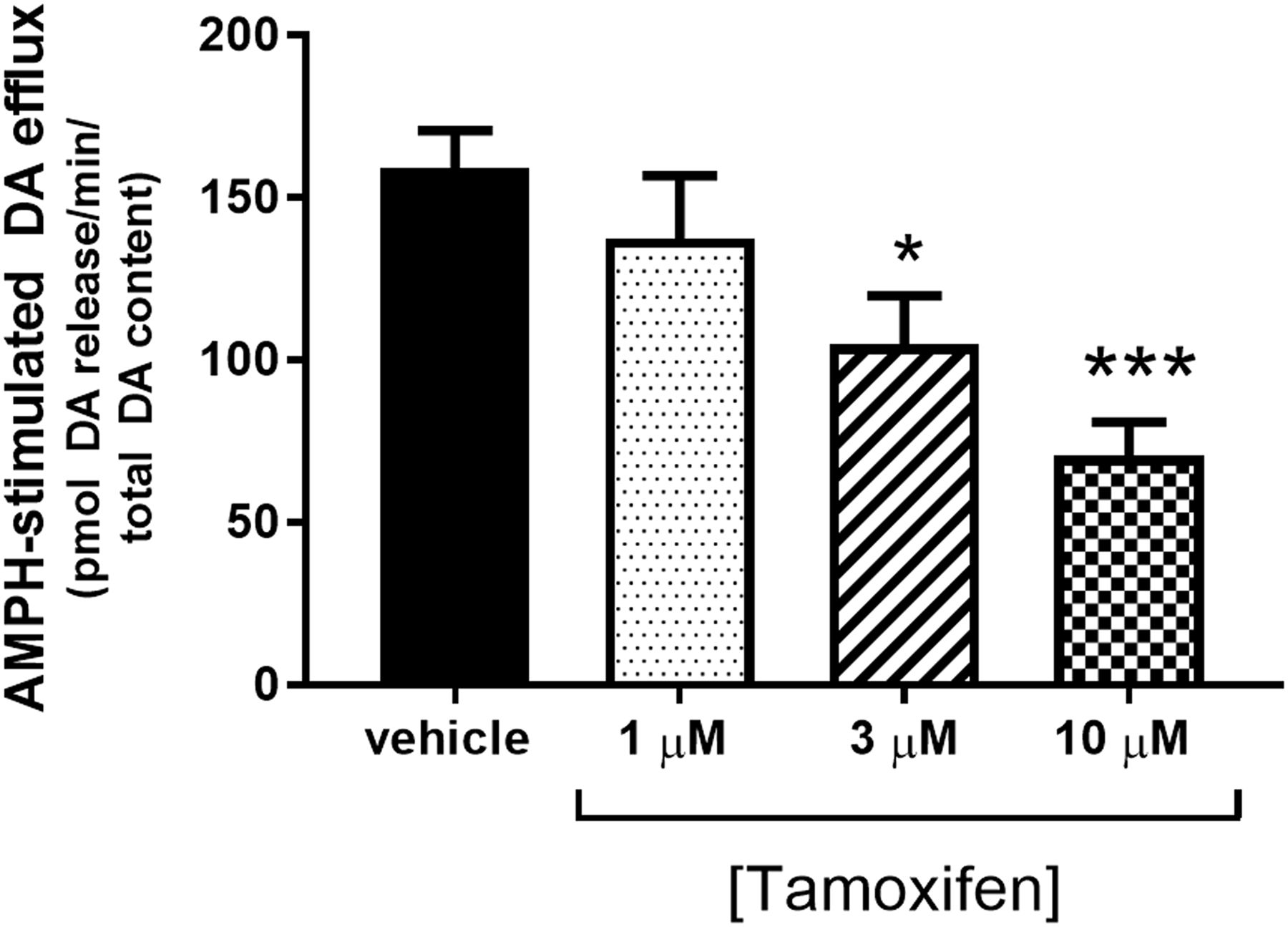
Tamoxifen attenuates amphetamine-stimulated dopamine efflux. Striatal synaptosomes were perfused for 1 hour at 37°C with vehicle or various concentrations of tamoxifen as described in the Materials and Methods. Amphetamine (10 μM) was included in the perfusate during fractions 7 and 8. Data were calculated as the area under the curve after treatment with amphetamine. Data are represented as the mean ± S.E.M. (n = 3–5). *P < 0.05; ***P < 0.001 compared with vehicle control (post hoc Dunnett multiple-comparisons test). Baseline release of dopamine in picomoles of dopamine/total dopamine was as follows: vehicle, 6.8 ± 2.0; 1 µM tamoxifen, 7.1 ± 1.8; 3 µM tamoxifen, 8.9 ± 1.2; and 10 µM tamoxifen, 4.9 ± 0.3. AMPH, amphetamine; DA, dopamine.
Tamoxifen Does Not Affect Surface Expression of the DAT.
Tamoxifen could be decreasing surface DAT levels throughout the hour incubation, accounting for the reduction in dopamine uptake and efflux. We examined this possibility using a surface biotinylation assay. There was no change in biotinylated DAT levels in synaptosomes treated with tamoxifen compared with vehicle (Fig. 3A), indicating no change in surface expression of DAT. Representative immunoblots for DAT in biotinylated and lysate samples are shown in Fig. 3B. Na+, K+-ATPase is included as a loading control for the biotinylated samples.

Tamoxifen does not affect surface expression of the DAT. Rat striatal synaptosomes were incubated for 1 hour with 10 μM tamoxifen or vehicle prior to biotinylation of surface proteins with sulfo-NHS-biotin. After avidin-biotin pulldown, DAT content in biotinylated fractions and lysates was quantified by Western blotting. (A) Biotinylated transporter/total transporter in lysate. (B) Representative Western blots showing the biotinylated fraction blotted for DAT protein and Na+/K+-ATPase, and the corresponding total lysate. Calculations of the ratio (± S.E.M.) of the optical densities of biotinylated transporter–Na+/K+-ATPase were 1.09 ± 0.2 for vehicle and 0.96 ± 0.1 for tamoxifen (data not shown) (n = 3). TMX, tamoxifen; Veh, vehicle.
Tamoxifen Inhibits Binding of [3H]WIN 35,428 to the DAT.
We next probed whether the effects of tamoxifen on dopamine uptake and efflux were the result of a direct interaction with the DAT. [3H]WIN 35,428 is a cocaine analog that binds to the substrate binding site of the DAT. The effect of tamoxifen on the binding of [3H]WIN 35,428 to the DAT was measured in rat striatal membranes. In a competition binding assay, a repeated-measures one-way ANOVA indicated a significant effect of treatment [P < 0.0001, F(4, 28) = 11.4] (Fig. 4A). In a saturation binding assay, kinetic analysis indicated that tamoxifen significantly decreased the Bmax of [3H]WIN35,418 binding to the transporter [vehicle vs. tamoxifen in picomoles per milligram protein, 4.1 (95% CI, 3.69–4.47) vs. 3.5 (95% CI, 3.16–3.80), P < 0.05], but had no effect on Km compared with vehicle [vehicle vs. tamoxifen, 11.4 nM [95% CI, 8.493–14.28] vs. 14.3 nM (95% CI, 10.98–17.53)] (Fig. 4B).

Tamoxifen inhibits [3H]WIN 35,428 binding to the DAT in rat striatal membranes. Rat striatal membranes were incubated with [3H]WIN 35,428 with or without tamoxifen or vehicle for 3 hours at 4°C. Nonspecific binding was determined with 30 μM nomifensine. (A) Membranes were incubated with 4 nM [3H]WIN 35,428 without and with various concentrations of tamoxifen (n = 8). **P < 0.01; ***P < 0.001 (post hoc Dunnett multiple-comparisons test). (B) Membranes were incubated with 10 μM tamoxifen and various concentrations of [3H]WIN 35,428 to equilibrium. Data are represented as the mean ± S.E.M. (n = 6). **P < 0.01; ****P < 0.001 (post hoc Sidak multiple-comparisons test). TMX, tamoxifen.
Tamoxifen Increases Biotinylation of Extracellular Cysteines in a Cocaine-Like Manner.
The partial reduction in [3H]dopamine uptake and amphetamine-induced dopamine efflux have been reported in a number of allosteric modulators of the DAT (Pariser et al., 2008). To explore this notion for tamoxifen, we determined whether an interaction with tamoxifen might be affecting DAT conformation. Previous work demonstrated that cocaine enhances the availability of cysteine-306 in the DAT for modification by maleimide-PEG2 biotin, and that this correlates with an increase in the outward-facing conformation of the DAT (Hong and Amara, 2010). We assessed whether tamoxifen would similarly affect biotinylation by maleimide-PEG2. As shown in Fig. 5A, incubation with 10 μM tamoxifen increases biotinylation of extracellular cysteines over that of vehicle in a manner similar to cocaine [one-way ANOVA, P = 0.012, F(2, 6) = 10.11, n = 3]. Representative immunoblots of DAT in surface and lysate samples reveal an increase in surface but not total DAT as compared to vehicle control (Fig. 5B). Glyceraldehyde 3-phosphate dehydrogenase is shown as a loading control for the lysate.

Tamoxifen stabilizes the outward-facing conformation of the DAT in a cocaine-like manner. Rat striatal synaptosomes were incubated for 1 hour with 100 μM cocaine, 10 μM tamoxifen, or vehicle prior to biotinylation of surface cysteines with maleimide-PEG2-biotin. DAT content in biotinylated fractions was quantified by Western blotting. (A) Biotinylated transporter/total transporter in lysate. *P < 0.05 compared with vehicle control (post hoc Dunnett multiple-comparisons test). (B) Representative Western blot showing the biotinylated DAT protein, its corresponding total lysate, and the content of GAPDH in the lysate (n = 3). Coc, cocaine; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; OD, optical density; TMX, tamoxifen; Veh, vehicle.
Tamoxifen Decreases Amphetamine-Stimulated Locomotor Activity But Not Basal Locomotion.
The decrease in dopamine uptake capacity caused by tamoxifen might be expected to lead to an increase in extracellular dopamine levels. Tamoxifen elicited small but significant amounts of extracellular dopamine in vivo as detected via microdialysis (Chaurasia et al., 1998). To test whether this increase in extracellular dopamine could cause tamoxifen to have stimulant effects in vivo, we followed the tamoxifen dosing protocol established by Einat et al. (2007). Animals were given an intraperitoneal injection of 3 mg/kg tamoxifen or vehicle once each day for 2 days while acclimating to locomotor beam break boxes. On the third day, animals were given an injection of saline. Locomotor activity was measured as the number of beam breaks in the 2-hour period preceding and after each injection. Although locomotor activity after intraperitoneal injection with saline or drug was significantly higher than the activity during habituation in both groups and across all days, there was no significant difference in locomotor activity between tamoxifen- and vehicle-treated animals (Fig. 6). Following the same experimental protocol, rats were pretreated with tamoxifen over 2 days; however, they received an injection of amphetamine (1 mg/kg, i.p.) without tamoxifen on day 3. In contrast to the lack of effect of tamoxifen on basal locomotor activity, a two-way repeated-measures ANOVA indicated a significant effect for pretreatment on amphetamine-stimulated hyperactivity [P < 0.005, F(1, 14) = 11.83], time [P < 0.0001, F(22, 308) = 11.67], and interaction between time and pretreatment [P = 0.002, F(22, 308) = 2.57] (Fig. 7).

Tamoxifen does not affect normal locomotor activity in rats. Locomotor activity of male Sprague-Dawley rats was monitored for 2 hours before and 2 hours after intraperitoneal injection. On days 1 and 2, animals received either 5 mg/kg tamoxifen citrate or an equivalent volume of vehicle. On day 3, all animals received saline. Data are represented as the mean ± S.E.M. (n = 8). Hab, XXX; Sal, saline; TMX, tamoxifen; Veh, vehicle.

Tamoxifen pretreatment attenuates amphetamine-stimulated hyperactivity. Male Sprague-Dawley rats were pretreated with 5 mg/kg tamoxifen citrate or vehicle 48 and 24 hours prior to administration of amphetamine (1 mg/kg). Saline was administered at 40 minutes and amphetamine (1 mg/kg i.p.) was administered at time point 100. Data are represented as the mean ± S.E.M. (n = 8). *P < 0.05; **P < 0.01; ***P < 0.001 (post hoc Sidak multiple-comparisons test). AMPH, amphetamine.
Potential Interaction of Tamoxifen with S2 Residues of hDAT.
Binding of [3H]WIN 35,428 is generally believed to occur at the S1 site on the DAT (Loland et al., 2008; Reith et al., 2012). Tamoxifen’s relatively small effect on [3H]WIN 35,428 binding is not commensurate with its larger effect on dopamine uptake and amphetamine-induced efflux and indicates that an interaction between tamoxifen and the DAT may be occurring somewhat distally from the primary S1 substrate. This distal “S2” site is hypothesized to facilitate binding of substrate to the primary substrate site (Nyola et al., 2010) and possibly even drive the translocation of the dopamine molecule across the membrane (Shan et al., 2011). A potential interaction with the S2 site was assessed with recently described mutants containing disruptions in the S2 site by mutation of Asp476 and Ile159 to Ala (D476A and I159A, respectively) (Zhen and Reith, 2016). Previously, we reported that the Kd for binding of [3H]WIN 35,428 to the DAT in WT and the two S2 mutants was the same, whereas the ability of dopamine in competing for [3H]WIN 35,428 binding was reduced in the mutants (Zhen and Reith, 2016). This is consonant with an allosteric effect of S2 site disruption on S1 site functionality. In addition, compounds that bind to the S2 site would be expected to block permeation of substrate by obstructing passage through S2. As observed in our previous work (Zhen and Reith, 2016), disruption of the S2 site greatly reduced [3H]dopamine uptake activity, as shown in the absolute Vmax values (Table 1; results for vehicle treatment). These reductions are far greater than the modest decreases observed in surface expression in the S2 mutants, and previous observations indicated a considerably lower dopamine turnover in these mutants compared with WT (Zhen and Reith, 2016).
Effect of tamoxifen on dopamine uptake kinetics in WT and S2 mutant DAT
Data are expressed as the mean ± S.E.M. for the number of experiments, each assayed in triplicate, indicated in parentheses on the top of each column. For the TMX groups, percent values listed in parentheses are the mean ± S.E.M. of values expressed as the percentage of vehicle value obtained on the same day. For the vehicle group, the average value is set at 100%.
Consistent with our results in synaptosomes, tamoxifen (10 µM) appreciably reduced the Vmax of [3H]DA uptake in WT DAT–LLC-PK1 cells (Table 1; see values in parentheses as % WT for easier comparison). In contrast, tamoxifen did not alter the Vmax in cells containing either D476A- or I159A-hDAT. This is consonant with the idea that D476 plays an essential role in tamoxifen binding to DAT and, when it is mutated, the compound loses its inhibitory property. A similar role for I159 in tamoxifen binding is postulated. Km values for WT-, D476A-, and I159A-DAT were not altered by tamoxifen (Table 1), consistent with our previous observations of noncompetitive inhibition of dopamine uptake by tamoxifen.
Computational Modeling of the Interaction Between Tamoxifen and the DAT.
Computational docking experiments were carried out to provide insight into the above findings. For this purpose, tamoxifen was docked into the S2 site of the LeuT-based hDAT by computational methods described above and in previous publications (Schmitt and Reith, 2011; Reith et al., 2012). The docking model suggests that tamoxifen occupies the majority of the S2 site, resting just above the “aromatic lid” formed by F155, F320, and Y156 that partitions the S1 and S2 sites (Fig. 8A). Being a highly lipophilic, polyaromatic compound, tamoxifen does not possess many polar functional groups that would serve as hydrogen bond donors/acceptors. As such, the most significant interaction observed is a hydrogen bond between the protonated amine of tamoxifen and the carboxyl side chain of D476 in the DAT (Fig. 8B). In addition, a less prominent aromatic interaction (H/π-bond) occurs between one of the tamoxifen rings and W84. Overall, the docking model suggests an interaction between the protonated amine of tamoxifen and negatively charged D476 in the S2 site of DAT.

Tamoxifen readily docks in the S2 pocket of hDAT modeled after LeuT. The in silico hDAT model based on LeuT was constructed as described in the Materials and Methods. (A) Final energy-minimized pose of DAT/tamoxifen complex after flexible docking of tamoxifen at the DAT secondary (S2) substrate binding site. Selected binding pocket residues are labeled and rendered as sticks; bound tamoxifen (also shown in sticks) is highlighted in yellow. Cotransported sodium and chloride atoms are labeled and rendered as orange and green spheres, respectively. (B) Two-dimensional interaction diagram of tamoxifen bound at the S2 site of DAT. The interaction map depicts respective DAT residues located within 4.5 Å of the bound tamoxifen molecule (hydrophobic residues are colored green and polar residues are purple). The most significant non–van der Waals DAT/ligand interactions are indicated with dotted lines and a symbol depicting the chemistry of the interaction formed: side-chain hydrogen bond between D476 and amine nitrogen of tamoxifen (green), and an aromatic H/π-bond interaction between one of the tamoxifen rings and W84 (green, hexagon H).
Discussion
Here we demonstrate, for the first time to our knowledge, that tamoxifen inhibits normal function of the DAT in an atypical manner. These conclusions are based on our results demonstrating that tamoxifen attenuates both [3H]dopamine uptake and amphetamine-stimulated dopamine efflux in vitro and hyperactivity in vivo, yet fails to induce hyperlocomotion in vivo. Inhibition of dopamine uptake by tamoxifen is noncompetitive and independent of changes in surface DAT levels. Additional analysis demonstrates that tamoxifen may be interacting with the DAT at an allosteric S2 site.
The observation that tamoxifen inhibits dopamine uptake without increasing locomotor activity is reminiscent of the effects of atypical DAT blockers. An atypical DAT blocker is defined as a compound that inhibits dopamine uptake, yet has no stimulant effects of its own (Reith et al., 2015) and reduces the locomotor-stimulating effects of cocaine and amphetamine (Velázquez-Sánchez et al., 2010). Some atypical DAT blockers inhibit the self-administration and conditioned place preference of cocaine without exhibiting any reinforcing characteristics in the absence of cocaine (Ferragud et al., 2009, 2014; Hiranita et al., 2009). Peripheral administration of tamoxifen (1.5 mg/kg s.c.) increased baseline levels of dopamine in rat striatum by 25%–35% as measured by in vivo microdialysis (Chaurasia et al., 1998), but this elevation is small compared with the 600% increase in dopamine over baseline attained by 1.0 mg/kg amphetamine (Carpenter et al., 2017). This leaves the possibility that the atypical DAT blockers may serve as effective substitution therapies for psychostimulant abuse (Tanda et al., 2009) due to their ability to antagonize psychostimulant action without exhibiting their own abuse liability (Schmitt et al., 2013). Because tamoxifen exhibits blockade of DAT function without psychostimulant properties of its own, there is a potential that many of the beneficial effects of atypical blockers may be seen with tamoxifen as well.
Tamoxifen also chemically resembles compounds shown to be allosteric inhibitors of the DAT (Pariser et al., 2008). The compounds share some ring structures, such as di- and triphenyl moieties. The increase in cysteine availability on the DAT elicited by tamoxifen indicates that tamoxifen stabilizes the outward-facing conformation of the DAT, similar to cocaine. If tamoxifen were allosterically eliciting this effect, [3H]WIN 35,428 binding would increase, as was observed by Hong and Amara (2010) for cocaine. However, tamoxifen noncompetitively reduces [3H]WIN 35,428 binding to the DAT, albeit weakly. This is consonant with the possibility that tamoxifen, when bound to S2, reduces dopamine uptake, either by hindering its passage through the permeation pathway at the location of S2 or perhaps by impeding the movement of extracellular vestibule gating residues theorized to play an integral role in “actuating” the transport cycle of neurotransmitter sodium symporter proteins (Shi et al., 2008; Shan et al., 2011). In contrast, drugs acting at the primary S1 recognition site (e.g., cocaine; Loland et al., 2008) are known to be competitive dopamine uptake inhibitors, altering the Km of dopamine uptake with substrate affinity for the transporter being one of the determinants of the overall Km value (Zimányi et al., 1989; Bönisch, 1998). Thus, ligands that exclusively bind at the S2 site could be expected to reduce the turnover rate (Vmax) for the complete symporter substrate translocation cycle without necessarily altering the absolute affinity (Kd) of ligands that exclusively bind at the S1 site.
It is important to emphasize that our transport and in silico docking data do not prove that tamoxifen binds in the S2 pocket of hDAT. It is, however, an interesting possibility that fits with the biochemical results obtained. The evidence strongly suggests that tamoxifen readily docks within the S2 pocket, leaving the putative [3H]WIN 35,428 binding site largely unaltered. This is consistent with the finding that the effect of tamoxifen is lost in the D476A mutant, as this mutation would eliminate the hydrogen bond between the side chain of D476 and the amine nitrogen of tamoxifen. The effect of the I159A mutation is more difficult to reconcile, because the docking model did not suggest a direct interaction between the side chain of I159 and tamoxifen. It is possible that removal of the bulky aliphatic side chain of isoleucine expands the S2 binding pocket, weakening the binding of tamoxifen; however, further evidence would be required to fully support this conclusion. However, the Vmax of [3H]dopamine uptake through I159A-hDAT is very low, so the lack of effect of tamoxifen on the Vmax should be interpreted with caution.
The discovery that tamoxifen blocks the DAT could explain previous findings concerning tamoxifen and amphetamine. Tamoxifen acts as a neuroprotectant in methamphetamine- and MPP+-induced dopaminergic neurotoxicity. This was largely believed to be due to the estrogen receptor–modulating effects of tamoxifen, since estrogen is neuroprotective as well. However, estrogen is neuroprotective against methamphetamine-induced neurotoxicity in female mice only, whereas tamoxifen is neuroprotective in both male and female mice (Bourque et al., 2007), indicating the presence of an alternative nonestrogenic-mediated mechanism. Both methamphetamine and MPP+ require uptake through the DAT to induce dopamine neurotoxicity; thus, in light of our results, the “neuroprotective” effects of tamoxifen could simply be the result of decreased DAT-mediated uptake.
It is also important to consider tamoxifen’s SERM activity in the context of the above results. It is still unclear whether the effects of tamoxifen on the estrogen receptor in a dopamine neuron are predominantly estrogenic or antiestrogenic. Estrogen increases amphetamine-stimulated dopamine efflux in striatal tissue (Becker, 1990). However, at concentrations where it binds the estrogen receptor, tamoxifen is unable to block this effect of estrogen on amphetamine-stimulated efflux, nor affect efflux on its own, indicating that it does not have antiestrogenic effects in the dopamine neuron (Xiao et al., 2003). In addition, although tamoxifen inhibits breast cancer cell growth at 100 nM through estrogen receptor–dependent mechanisms (Coezy et al., 1982), we fail to see any effect of tamoxifen on dopaminergic processes until we approach concentrations 10–100 times higher. Finally, we previously demonstrated that the effects of tamoxifen on the DAT in hDAT-N2A cells are unaltered by blockade of the estrogen receptors, indicating that tamoxifen is exerting its effects on the DAT independently of its actions at the estrogen receptors (Mikelman et al., 2017). Taken together, these results strongly indicate a division between estrogen and tamoxifen and their effects on the DAT.
Although the concentrations of tamoxifen used in these studies are higher than those needed to block estrogen receptors, they are not outside the ranges of concentrations found in brain tissue in the pharmacokinetic literature (Lien et al., 1991a,b; Robinson et al., 1991; Kisanga et al., 2003). Furthermore, although we found that tamoxifen has a similarly high IC50 for inhibition of PKC (1 μM in a cell based assay, data not shown), the dosing regimen used here was sufficient to decrease amphetamine-stimulated, PKC-mediated phosphorylation of growth-associated protein GAP-43 in rat striatum in vivo (Einat et al., 2007), indicating that tamoxifen can reach sufficient levels in the brain in vivo to be comparable to our in vitro studies.
Contrary to previously published work (Pereira et al., 2014), we were unable to see an effect of tamoxifen on amphetamine-stimulated hyperactivity unless the animals were treated over 2 days (data not shown). These results are puzzling considering the observation that a 1-hour treatment with tamoxifen in vitro is sufficient to produce a robust inhibition of the DAT, but they are consistent with the data of Einat et al. (2007). Tamoxifen, given peripherally at 3.0 mg/kg, elicited a 35% rise in striatal extracellular dopamine in vivo within an hour (Chaurasia et al., 1998), but this small increase may be too small to elicit locomotor behavior. One explanation could reside in the pharmacokinetic properties of tamoxifen. The half-life (t1/2) of tamoxifen is long (primary t1/2 of 11 hours and secondary t1/2 of 62 hours; Fromson et al., 1973; Robinson et al., 1991), and it may take time and repeated dosing to build up to relevant concentrations of active metabolite in the brain. The time to steady state in the rat is 4 days and tissue levels increase with repeated daily dosing (Robinson et al., 1991). Despite being central nervous system permeant, lower accumulated concentrations of tamoxifen are found in the brain compared with other key tissues such as the liver, lung, and fat (Lien et al., 1991a). The predominant metabolite of tamoxifen in the rat and human brain is N-desmethyltamoxifen (Lien et al., 1991a), but this metabolite has no effect on dopamine uptake or efflux at DAT (Mikelman et al., 2017). In contrast, the brain levels of 4-hydroxytamoxifen, a metabolite that inhibits DAT, are very low (Lien et al., 1991a). Alternatively, and just as likely, there could be a compensatory change in another component, perhaps relating to changes in gene expression, that regulates the behavioral activity. In this regard, we found that peripheral administration of a non–estrogen receptor binding analog of tamoxifen needed to occur at least 18 hours prior to testing before its reduction of self-administration of amphetamine was detected (Carpenter et al., 2017).
In conclusion, we demonstrated that tamoxifen significantly impairs DAT function in vitro. In vivo, however, this effect on DAT functionality appears to only have behavioral relevance in the presence of a nonphysiologic stimulus such as amphetamine. This effect provides an explanation for many of the results seen previously in studies of tamoxifen and dopaminergic signaling. In addition, we believe that the DAT-modulating functions of tamoxifen exhibit potential as a pharmacological treatment of psychostimulant abuse and that a further investigation of the tamoxifen structure is warranted.
Acknowledgments
We thank Dr. Roxanne Vaughan (University of North Dakota) for the generous donation of monoclonal antibody mAb16.
Authorship Contributions
Participated in research design: Mikelman, Guptaroy, Gnegy.
Conducted experiments: Mikelman, Guptaroy, Schmitt, Jones, Zhen.
Performed data analysis: Mikelman, Guptaroy, Schmitt, Jones, Zhen, Reith, Gnegy.
Wrote or contributed to the writing of the manuscript: Mikelman, Schmitt, Jones, Zhen, Reith, Gnegy.
Footnotes
- Received February 1, 2018.
- Accepted July 26, 2018.
↵1 Current affiliation: Cell Therapy and Cell Engineering Facility, Memorial Sloan Kettering Cancer Center, New York, New York.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant T32GM007767 (to S.R.M.)] and the National Institutes of Health National Institute on Drug Abuse [Grants R01DA11697 (to M.E.G.), T32DA007267 (to S.R.M.), and R01DA019676 (to M.E.A.R.)].
Abbreviations
- ANOVA
- analysis of variance
- CI
- confidence interval
- DAT
- dopamine transporter
- DMSO
- dimethylsulfoxide
- hDAT
- human dopamine transporter
- KRB
- Krebs-Ringer buffer
- MOE
- Molecular Operating Environment
- MPP+
- 1-methyl-4-phenylpyridinium
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NHS
- N-hydroxysulfo-succinimide
- PBS
- phosphate-buffered saline
- PBSCM
- phosphate-buffered saline containing 0.1 mM CaCl2 and 1 mM MgCl2
- PEG
- polyethylene glycol
- PKC
- protein kinase C
- SERM
- selective estrogen receptor modulator
- t1/2
- half-life
- WIN 35,428
- (–)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane
- WT
- wild type
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}