


Abstract
The central renin angiotensin system (RAS) is one of the most widely investigated cardiovascular systems in the brain. It is implicated in a myriad of cardiovascular diseases. However, studies from the last decade have identified its involvement in several neurologic abnormalities. Understanding the molecular functionality of the various RAS components can thus provide considerable insight into the phenotypic differences and mechanistic drivers of not just cardiovascular but also neurologic disorders. Since activation of one of its primary receptors, the angiotensin type 1 receptor (AT1R), results in an augmentation of oxidative stress and inflammatory cytokines, it becomes essential to investigate not just neuronal RAS but glial RAS as well. Glial cells are key homeostatic regulators in the brain and are critical players in the resolution of overt oxidative stress and neuroinflammation. Designing better and effective therapeutic strategies that target the brain RAS could well hinge on understanding the molecular basis of both neuronal and glial RAS. This review provides a comprehensive overview of the major studies that have investigated the mechanisms and regulation of the brain RAS, and it also provides insight into the potential role of glial AT1Rs in the pathophysiology of cardiovascular and neurologic disorders.
Introduction
The renin angiotensin system (RAS) is a cardioregulatory, peptidergic, hormonal system that is involved primarily in the regulation of blood pressure. Its pivotal function is to elevate blood pressure in hypotensive states. In response to a drop in blood pressure, low salt concentration, or low blood volume, the juxtaglomerular apparatus in the kidneys triggers the RAS cascade, which ultimately culminates in a significant antinatriuretic, antidiuretic, and vasoconstrictive effect (Hall, 1986; Montani and Van Vliet, 2004). Components of the RAS, as will be discussed in the succeeding sections, are found in virtually every organ/tissue of the body. Hence, their functions go beyond regulation of blood pressure. Of significant interest is the action of the RAS in the brain. An overactive brain RAS has been demonstrated to be a characteristic feature of multiple cardiovascular diseases. However, evidence shows that the underlying pathologic mechanisms of the RAS can be extrapolated to several neurologic disorders as well. An increase in oxidative stress, proinflammatory mediators, and a decrease in anti-inflammatory cytokines are tightly intertwined with the progression of both cardiovascular and neurologic disorders. The RAS plays a pivotal role in controlling these drivers of cardiovascular and neurologic disorders.
Classic and Nonclassic RAS and Therapeutic Interventions
The RAS is composed of several different components that encompass precursor and active peptides, enzymes, and receptors. Renin, secreted from the kidneys, converts angiotensinogen (AGT), secreted from the liver, to angiotensin (Ang) I. Ang I is converted into Ang II by the action of the angiotensin-converting enzyme (ACE), an enzyme produced in the lungs and blood vessels (Montani and Van Vliet, 2004). It is now widely accepted that Ang II is secreted by a diverse group of cell types, and its source is not restricted to only liver AGT (Ganong, 1994; Paul et al., 2006). The former is part of the local RAS, whereas the latter constitutes the systemic RAS. Optimal functioning of both the systemic and local RAS is critical for overall cardiovascular homeostasis (Lavoie and Sigmund, 2003). Ang II, the major effector peptide of the RAS, is a circulating hormone that has a major physiologic and pathophysiological bearing on cardiovascular functions. Although most of their functions converge to have one singular outcome (i.e., an elevation in blood pressure), the RAS also has a role in digestion, reproduction, and prenatal development (Paul et al., 2006).
The widely studied and documented actions of Ang II, such as aldosterone secretion and its vasoconstrictive and ionotropic effects, are due to its ability to interact with the angiotensin type 1 receptor (AT1R) (Fyhrquist et al., 1995). Ang II couples with AT1R, a pertussis toxin–insensitive G protein–coupled receptor (GPCR), to produce a spike in calcium levels. Elevations in calcium activate kinases, signaling pathways, and transcription factors and consequently cause several physiologic actions such as smooth muscle contraction and aldosterone synthesis (de Gasparo et al., 2000). By interacting with AT1R on renal, cardiac, and vascular cells, Ang II is able to increase aldosterone levels, elevate salt intake, cause sympathetic nervous system hyperactivation, have a positive ionotropic effect, and elicit potent vasoconstriction (Fyhrquist et al., 1995). Ang II also interacts with the angiotensin type 2 receptor (AT2R), which is known to elicit functions that are antagonistic to AT1R, such as a reduction of oxidative stress and neutralizing proinflammatory states (Stoll et al., 1995; Namsolleck et al., 2014). At a physiologic level, AT2R activation results in vasodilatory and cardioprotective effects (Li et al., 2012).
Several other RAS peptides that are both functionally similar and dissimilar to Ang II have also been identified, characterized, and studied (Paul et al., 2006). Ang III, Ang IV, and Ang (1-7) are physiologically active degradation products of Ang II. Ang III interacts with AT1R, whereas Ang IV and Ang (1-7) interact with their own cognate receptors, the Ang type 4 receptor (AT4R) and the Mas receptor, respectively (Varagic et al., 2008). Ang II is degraded by aminopeptidases to Ang III and Ang IV and by ACE2 to Ang (1-7) (Paul et al., 2006). Alternatively, Ang (1-7) can also be synthesized from Ang I by the action of neprilysin (Paul et al., 2006). Ang III exhibits functional similarity to Ang II, whereas Ang (1-7) counteracts the deleterious effects of Ang II (Ferrario et al., 1991). Owing to the anti-inflammatory and antioxidant properties of the Mas receptor, Ang (1-7) has been demonstrated to have potent cardioprotective and neuroprotective properties (Bennion et al., 2015). Ang IV also has cardioprotective and neuroprotective effects. Ang IV is not only known to elicit potent vasodilatory effects (Kramár et al., 1997; Hamilton et al., 2001), but it has also been demonstrated to diminish the production of several proinflammatory cytokines (Kong et al., 2015). Ang IV receptor activation is linked to an improvement in cognitive abilities such as learning and memory, and it has been suggested as a potential therapeutic target for Alzheimer disease (Wright and Harding, 2008; Royea et al., 2017).
Although a dysregulated RAS is one of the hallmarks of cardiovascular diseases (Veerasingham and Raizada, 2003), its functions are fundamental for survival under physiologic conditions. Through multiple mechanisms, Ang II controls and maintains blood pressure and blood volume within set boundaries. Although vasoconstriction is often viewed as its major mechanism, its ability to increase water retention and elevate sympathetic activity makes it a powerful and unique system in understanding the pathophysiology of cardiovascular diseases. Ang II–mediated hypertrophy and hyperplasia are characteristic features of multiple risk factors of cardiovascular diseases, such as hypertension and diabetes (Fyhrquist et al., 1995). Owing to the ubiquitous nature of AT1R expression in the body, overactivity of AT1R is linked to multiple organ and tissue dysfunctions. Ang II is a well established mitogen that is known to trigger hypertrophy and cell migration via AT1R activation (Ushio-Fukai et al., 1996; Takeuchi et al., 2006; Lee et al., 2007; Campos et al., 2012). Cardiac hypertrophy, renal disease, and endothelial dysfunction have all been demonstrated to have an augmented RAS component. Since both Ang II synthesis and AT1R activity are fundamental to RAS-mediated elevation in blood pressure, drugs that impede synthesis of Ang II, or those that antagonize the deleterious effects of AT1Rs, are the mainstays in the pharmacological management of numerous cardiovascular diseases, their risk factors, and their complications (Burnier and Zanchi, 2006; Atlas, 2007). In addition to its augmented ability to elevate blood pressure by multiple mechanisms in pathologic conditions, Ang II can also cause extensive damage to the heart, kidneys, and vasculature (Long et al., 2004; Montezano et al., 2014; Wang et al., 2014). For example, AT1R angiotensin receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACEIs) are routinely employed in the management of heart failure (Aranda and Conti, 2003; Atlas, 2007).
An alternative strategy is to counteract the effects of AT1R by activating receptors and systems that exhibit functional antagonism to the receptor. In this regard, several groups have explored the therapeutic potential of AT2R agonists, which exhibit vasodilatory and cardioprotective effects such as a reduction in inflammation and fibrosis as well as cause pro-oxidant states (Danyel et al., 2013; Matavelli and Siragy, 2015). Although AT2R activation has been shown to result in significant vasodilation, the AT2R agonist compound 21 (C21) did not alter blood pressure when administered alone in hypertensive and nonhypertensive rats (Carey et al., 2001; Bosnyak et al., 2010). C21 caused a significant reduction only in the presence of AT1R blockers. Owing to the relatively low expression of AT2Rs compared with AT1Rs, it could be that the AT1R tone is far greater than AT2R-mediated vasorelaxation under baseline conditions. In addition, the Ang (1-7)–ACE2–Mas axis has also been viewed as a potential therapeutic target to negate the deleterious effects of AT1R activation. Several in vitro and in vivo studies have demonstrated a significant protective role for this pathway in cardiac tissue under pathologic conditions (Zhong et al., 2010; Patel et al., 2012). The ability of Ang (1-7) to improve not only endothelial and vascular function (Sampaio et al., 2007; McKinney et al., 2014) but also the metabolic panel and to serve as a renoprotectant in diabetic states (Mori et al., 2014a,b) makes the Mas receptor an attractive druggable target. The schematic representation of the RAS pathway and the role of its receptors in regulation of cardiovascular functions is shown in Fig. 1.
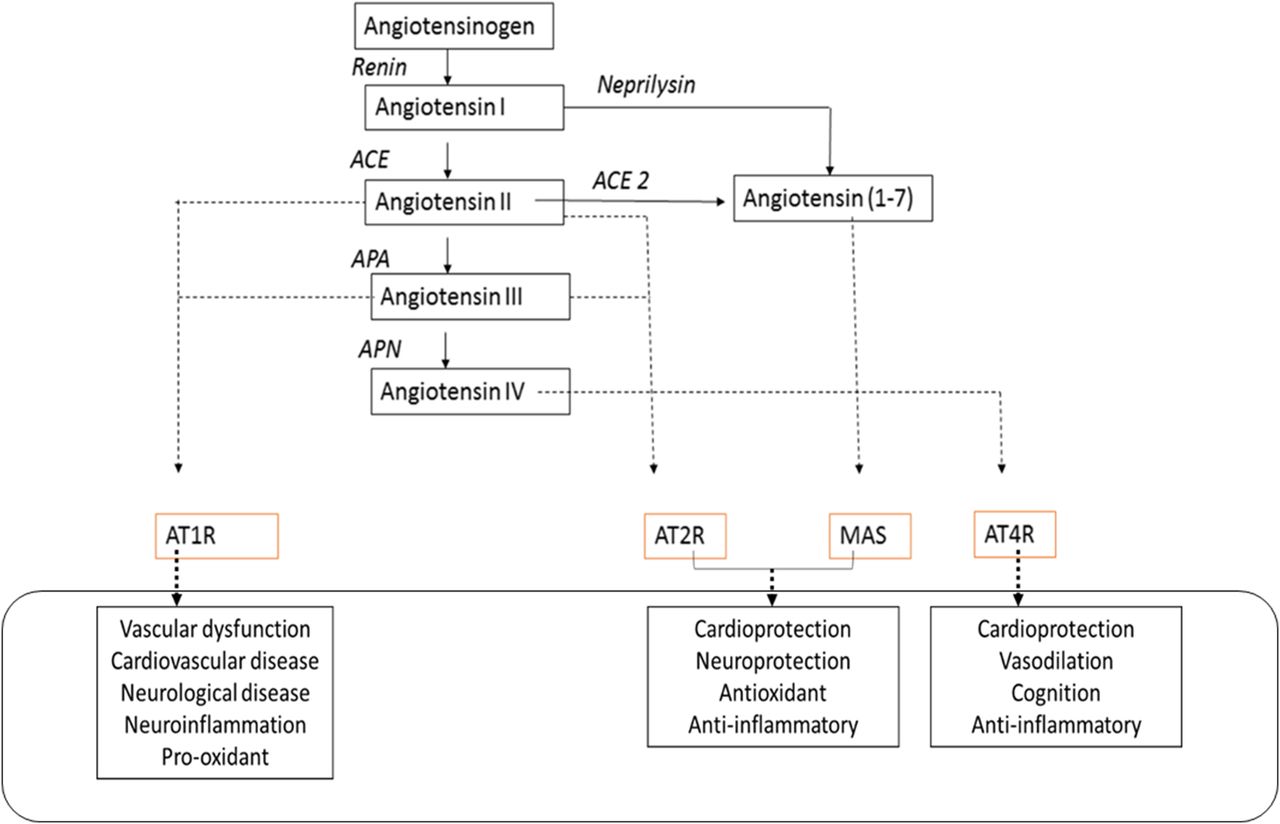
The RAS pathway and the role of its receptors in the regulation of cardiovascular functions. AGT is the precursor protein for the synthesis of the major Ang peptides. The actions of these peptides are mediated by the four receptors listed. APA, aminopeptidase A; APN, aminopeptidase N.
Crosstalk between different components of the RAS has also been reported. For instance, Ang II has been shown to upregulate ACE and downregulate ACE2 in kidney cells and neuronal cells (Koka et al., 2008; Xiao et al., 2013). Receptors belonging to RAS and also other molecular systems are known to negatively or positively regulate AT1R activity by several different modes of crosstalk. For instance, AT2R has been demonstrated to cause heterologous desensitization of AT1R through a protein kinase C–dependent mechanism and thus diminish its activity in transfected cells (Inuzuka et al., 2016). Interestingly, AT1R activity was reduced in the amygdala of mice brains that lacked the MAS receptor, further highlighting the crosstalk among the various components of the RAS (Von Bohlen und Halbach et al., 2000). Interestingly, a subpressor dose of Ang II has a similar angiogenic response to a low dose of Ang (1-7), further highlighting the complex interplay between various components of the RAS (Hoffmann et al., 2017). Although the cannabinoid type 1 receptor has been demonstrated to potentiate AT1R profibrinogenic activity by way of heterodimerization (Rozenfeld et al., 2011), it has also has been demonstrated to diminish AT1R’s ability to activate key signaling pathways in astrocytes (Haspula and Clark, 2017b). In addition, receptor interactions with adrenergic receptors also enable drugs such as β-blockers to influence AT1R activity (Barki-Harrington et al., 2003). Heterodimers between AT2R and MAS have been shown to be functionally relevant in astrocytes, whereby knockout of either of the receptors led to a diminished functionality of the other (Leonhardt et al., 2017).
Although AT1R is considered to be a prototypical plasma membrane receptor, evidence of Ang II binding sites that are localized intracellularly and bear similarity with AT1R has been reported in various tissues such as the liver (Booz et al., 1992; Tang et al., 1992). Evidence of an intracellular RAS in the brain comes from studies on a nonsecretory variant of renin that retains its enzymatic activity (Lee-Kirsch et al., 1999) and was also shown to modulate thirst and blood pressure in transgenic animals (Lavoie et al., 2006). In neurons, Ang receptors were recently identified on nuclei as well as on other organelles such as mitochondria, where they have a role in neuroprotection and respiration (Valenzuela et al., 2016; Villar-Cheda et al., 2017). It becomes important to understand the functional significance of intracellular binding sites for Ang II in brain cells such as astrocytes, since it was shown in cardiomyocytes that Ang II performed functions intracellularly that are different from those of extracellular Ang II (De Mello, 2008).
Brain RAS and Blood Pressure Control
Studies on borderline hypertensive humans and hypertensive animal models have confirmed the importance of an augmented central sympathetic activity in the pathogenesis of hypertension (Anderson et al., 1989; Korner et al., 1993; Fisher et al., 2009; Fisher and Paton, 2012). This has also led to the inception of the neuroadrenergic hypothesis for hypertension put forth by Grassi et al. (2010), which underscored the role of sympathetic hyperactivity in triggering and in perpetuating hypertensive conditions. Cardiovascular centers in the brainstem, rostral ventrolateral medulla (RVLM), and nucleus of the solitary tract (NTS) and in the hypothalamus and paraventricular nucleus (PVN) serve as both important convergence and divergence points for the regulation of blood pressure. Studies in hypertensive rat models characterized by an augmented sympathetic tone have revealed a dysfunction in cardiovascular centers (Allen, 2002; Ito et al., 2002). In addition, several other nuclei such as the subfornical organ (SFO), parabrachial nucleus, organum vasculosum of the lamina terminalis, and supraoptic nucleus (SON) also play a vital role in regulation of cardiovascular function.
Understanding the significance of the brain RAS is vital, since its augmentation has been reported and implicated in several cardiovascular diseases and conditions (Veerasingham and Raizada, 2003; Huang and Leenen, 2009; Campos et al., 2012). Although functional AT1Rs in the brain were identified in the 1960s (Bickerton and Buckley, 1961), the notion that brain cells could produce Ang II was suggested much later. Evidence of Ang II–synthesizing enzymes and Ang II precursors in brain cells provided the early foundation for the conception of an independently functioning brain RAS (Brooks and Malvin, 1979; Phillips, 1983; Campbell et al., 1984). In the central nervous system, AT1R levels are particularly greater in the cardiovascular centers of the hypothalamus and brainstem, further highlighting the importance of the RAS in the regulation of cardiovascular parameters (Phillips et al., 1993; Lenkei et al., 1995; Hu et al., 2002). Ang II, a potent dipsogen, can increase water intake by activating AT1Rs in the SFO, SON, and organum vasculosum of the lamina terminalis (Simpson et al., 1978; Qadri et al., 1993; Morris et al., 2002; Coble et al., 2014).
In addition, brain Ang II also has critical neuromodulatory functions, whereby it activates neuronal AT1Rs to alter synaptic strength and activity by modulating impulses generated by several neurotransmitters such as glutamate, GABA, and norepinephrine (Tsuda, 2012). The PVN and RVLM cardiovascular centers are especially critical for Ang II–mediated sympathoexcitation. By disinhibiting GABAergic neurons in the PVN, Ang II is able to stimulate neurons projecting from the PVN to the RVLM, thereby facilitating an increase in sympathetic activity (Cato and Toney, 2005; Li and Pan, 2005). AT1Rs present in the cardioregulatory centers of the brainstem are known to not only attenuate baroreflex sensitivity but also increase firing rates of sympathetic neurons (Matsumura et al., 1998; Matsuura et al., 2002). Viral transfection of a constitutively active form of the AT1R into the RVLM caused a spike in blood pressure (Allen et al., 2006), whereas inhibiting it resulted in a decrease in blood pressure in several animal models of hypertension (Ito et al., 2002; de Oliveira-Sales et al., 2010). These findings provide further evidence of its central role in hypertension. Ang II effects in the NTS, however, suggest a more complex mechanism of action. Low and high doses of Ang II elicited different responses, or even a lack of significant response, on blood pressure and heart rate regulation (Rettig et al., 1986; Mosqueda-Garcia et al., 1990; Paton and Kasparov, 1999). Ang II in the NTS was also shown to play a vital role in diminishing baroreflex sensitivity (Campagnole-Santos et al., 1988; Polson et al., 2007).
Further evidence for a role of brain RAS in cardiovascular disorders came from studies in spontaneously hypertensive rats (SHRs), widely regarded as the best model to study essential hypertension. The brain RAS was demonstrated to be overactive in SHRs compared with their normotensive controls (Veerasingham and Raizada, 2003; Haspula and Clark, 2018). AT1R levels were observed to be higher in the brainstems of SHRs compared with their normotensive controls, Wistar Kyoto (WKY) rats (Gutkind et al., 1988; Hu et al., 2002). RAS activity was demonstrated to be markedly potentiated in the cardioregulatory regions of the brainstem and hypothalamus of SHRs compared with their normotensive controls (Casto and Phillips, 1985; Matsuda et al., 1987; Gutkind et al., 1988; Muratani et al., 1991; Stadler et al., 1992; Zhu et al., 1998; Hu et al., 2002; Ito et al., 2002; Sun et al., 2009; Haspula and Clark, 2017b; Negussie et al., 2017). Both synthesis and turnover of Ang II were also enhanced in SHRs compared with their normotensive controls (Ganten et al., 1983; Hermann et al., 1984). An overview of studies that have reported an elevation in AT1R activity in the brainstems of SHRs is shown in Fig. 2.

An overview of studies reporting AT1R elevation in the brainstems of SHRs (Gutkind et al., 1988; Muratani et al., 1991; Yang and Raizada, 1998; Hu et al., 2002; Matsuura et al., 2002; Sun et al., 2009; Haspula and Clark, 2017b; Negussie et al., 2017).
Apart from AT1R, other components of the RAS were also observed to be altered in rodent models of hypertension. ACE2 levels were reported to be markedly lower in the hypothalamus of SHRs compared with WKY rats, resulting in an augmented ACE–Ang II–AT1R axis (Wang et al., 2017). AT2R activation was observed to have a greater antihypertensive effect in hypertensive models compared with normotensive models (de Kloet et al., 2017; Steckelings et al., 2017). Ang (1-7) was demonstrated to restore normal baroreflex functioning and autonomic activity in transgenic (mRen2)27 hypertensive rats (Kangussu et al., 2015). In response to hypoxia, often accompanied by an elevation in sympathetic activity, ACE levels were shown to be elevated in the median preoptic nucleus (Faulk et al., 2017). Brain AT2R activation can negate the sympathoexcitatory response that is a consequence of AT1R-mediated sympathoexcitation and also renal volume expansion (Stegbauer et al., 2005; Gao and Zucker, 2011; Abdulla and Johns, 2017). Increased blood pressure and attenuated baroreflex sensitivity in the offspring of betamethasone-exposed sheep was shown to be a consequence of central RAS impairment, which was improved by Ang (1-7) (Hendricks et al., 2017). Ang (1-7) also negated the Ang II–mediated chronotropic response in the hypothalamic neurons of prehypertensive SHRs (Modgil et al., 2012).
At a molecular level, several mechanisms such as endoplasmic reticulum stress, mitochondrial dysfunction, and redox-sensitive transcriptional factors have all been attributed to brain-derived Ang II promoting neurogenic hypertension and baroreflex impairment (Nautiyal et al., 2013; Coble et al., 2015; Young and Davisson, 2015; Case et al., 2017). An increase in reactive oxygen species (ROS) levels in the brainstem cardioregulatory centers is a crucial step by which Ang II is able to augment multiple cardiovascular parameters such as sympathetic tone, heart rate, and water intake (Zimmerman et al., 2002, 2004; Nozoe et al., 2008; Chan and Chan, 2012). In a model of renovascular hypertension, increased levels of both oxidative stress markers and AT1R levels in the PVN and RVLM were demonstrated to be significant contributors to sympathoexcitation and hypertension (Oliveira-Sales et al., 2009; de Oliveira-Sales et al., 2010). Although NADPH oxidase and the mitochondrial electron transport chain serve as two potent sources of ROS, nitric oxide synthase (NOS) is also capable of generating ROS under a less stable and uncoupled state (Chan and Chan, 2012). The various NOS isoforms (endothelial, neuronal, and inducible NOS) were described as key mediators as well for Ang II effects on blood pressure and baroreflex gain (Paton et al., 2001; Cheng et al., 2010; de Oliveira-Sales et al., 2010). An increase in the levels of endothelial NOS, neuronal NOS, and inducible NOS was observed in the brainstem nuclei of hypertensive rats compared with their normotensive controls (Kishi et al., 2001; Edwards et al., 2004; Kimura et al., 2009; de Oliveira-Sales et al., 2010). Interestingly, nitric oxide (NO) levels were demonstrated to both neutralize (Zanzinger, 2002) as well as contribute to (Paton et al., 2001) Ang II–mediated elevation in ROS levels. Although a decrease in the levels of NO and NOS isoforms has been linked to sympathoexcitation, an increase in their levels has also been demonstrated to result in an increase in sympathetic activity (Chan et al., 2003; Hirooka et al., 2003). Losartan, an AT1R antagonist, reduced oxidative stress by acting on the mitochondrial electron transport chain (Sumbalová et al., 2010) as well as increasing levels of NO in the brainstem (Cheng et al., 2010). To add to the complexity of the sympathomodulatory role of the brainstem NOS, NOS activity was observed to be diminished under prehypertensive conditions but elevated under established hypertensive states (Qadri et al., 2003). A balance between the production of NO, ROS, and antioxidant levels is eminent for optimum regulation of sympathetic activity in the brainstem nuclei.
In addition to an elevation in ROS levels, various GPCR scaffold proteins, kinase signaling pathways, redox-dependent and independent transcription factors, ion channels, and tyrosine kinase receptors are also key players in brain AT1R-mediated pathologic effects (Shapiro et al., 1994; Yang et al., 1996; Huang et al., 1998; Zhu et al., 1999; Clark and Gonzalez, 2007; Agarwal et al., 2013; Xiao et al., 2013; Coble et al., 2014; Bali and Jaggi, 2016; Farag et al., 2017). AT1R downregulation mediated by β-arrestin 1 overexpression in the SFO has been demonstrated to reduce blood pressure and sympathetic activity in SHRs compared with WKY rats (Sun et al., 2018). Dysfunctions of cardiovascular parameters in response to centrally administered Ang II have been demonstrated to occur through mitogen-activated protein kinase (MAPK)–dependent, as well as MAPK-independent, pathways such as phosphoinositide 3-kinase (PI3K) (Yang et al., 1996). PI3K has also been demonstrated to be crucial for Ang II–mediated depression of baroreflex function in the NTS of SHR brainstems (Sun et al., 2009). Although PI3K was demonstrated to be a critical mechanism for AT1R-mediated elevation of RVLM neuronal activity in SHRs compared with their normotensive controls (Seyedabadi et al., 2001; Veerasingham et al., 2005), extracellular signal–regulated kinase (ERK) 1/2 was involved in Ang II–mediated inhibition of neuronal NOS activity (Cheng et al., 2010). Activation of ERK1/2 also plays a key role in the regulation of AT1R turnover in neuronal cultures (Yang et al., 1997). P38 MAPKs were shown to be involved in Ang II–mediated regulation of ACE and ACE2 activities in neuronal cells (Xiao et al., 2013). Both p38 and ERK MAPK pathways are also involved in Ang II–mediated activation of caspase 3 in RVLM neurons, the latter effect resulted in sympathoexcitation in the spontaneously hypertensive stroke-prone rat (Kishi et al., 2010). Transcription factors such as nuclear factor κB (NF-κB) that are crucial to Ang II–mediated deleterious effects, such as an increase in the proinflammatory states in cells (Agarwal et al., 2013), are also responsible for AT1R-mediated homologous upregulation (Haack et al., 2013). The role of these intracellular mediators in glial cells is described in a later section.
RAS and catecholaminergic interactions are well established mechanisms by which central Ang II is capable of eliciting sympathoexcitation and dipsogenic effects (Gelband et al., 1998; Tsuda, 2012). Ang II has been shown to elevate both adrenaline (norepinephrine) release and uptake in brain regions involved in the regulation of cardiovascular functions, and the latter functionality has also been shown to be elevated in SHR brain neurons (Qadri et al., 1991; Yang et al., 1996). In addition, Ang II via the AT1R and α-adrenoreceptors in the SON has been shown to increase vasopressin release (Qadri et al., 1993). The dependence of Ang II on the mineralocoticoid receptor in mediating oxidative stress in the SFO has also been demonstrated (Wang et al., 2015). The effects of Ang II on other neurotransmitters and neuromodulators are mentioned in a subsequent section. An overview of the brainstem AT1R and its role in the regulation of various cardiovascular parameters is shown in Fig. 3.

An overview of the roles of brainstem AT1R in the regulation of cardiovascular parameters.
Significance of the Glial RAS in Cardiovascular Disease
Overt excitatory impulses from the RVLM presympathetic neurons are ascribed as a major cause for sympathoexcitation that is observed in essential hypertension and cardiovascular diseases (Kumagai et al., 2012). Several theories have been attributed to this phenomenon. Imbalances in the levels of activating and restraining inputs to the RVLM (Smith and Barron, 1990; Ito et al., 2000), pro-oxidant and proinflammatory states in the RVLM (Wu et al., 2012), and imbalances between the excitatory and inhibitory neurotransmitters in the RVLM (Kishi et al., 2002) are some of the mechanisms that have been investigated. ATP is demonstrated to be an extremely important neurotransmitter in mediating the excitatory components of the RVLM (Marina et al., 2013) and the chemoreflex input of the NTS neurons (Braga et al., 2007). Purinergic signaling is a key signaling system that regulates cardiovascular function by exerting autonomic modulatory influences (Gourine et al., 2009). Purinergic signaling is also employed by astrocytes to communicate not only with other astrocytes but also with neurons (Fields and Burnstock, 2006). Brainstem hypoxia, which is commonly observed in hypertension and cardiovascular diseases, is also known to be a favorable environment for ATP release from astrocytes (Marina et al., 2015, 2016). Using rat models of heart failure and hypertension, the roles of astroglial ATP were demonstrated to be exceedingly important in the pathogenesis of the aforementioned disorders (Marina et al., 2016).
Components of the RAS are expressed not only in neuronal cells but also in glial cells, and both of these brain cells have a role in regulating cardiovascular functions (Morimoto et al., 2002). Higher levels of AT1Rs were reported in neonatal rat astrocytes compared with neuronal cells (Sumners et al., 1991), highlighting the significance of astrocytes in mediating the effects of Ang II. Astrocytes are theorized to be the major source of AGT in the brain (Deschepper et al., 1986; Stornetta et al., 1988). In SHRs, an increase in the levels of AGT, prior to the development of hypertension, was identified in the brain (Tamura et al., 1996). Interestingly, our laboratory has demonstrated an increase in the levels of AGT from brainstem astrocytes in response to Ang II (Gowrisankar and Clark, 2016c). In addition, we observed an increase in ACE and a decrease in ACE2 in response to Ang II treatment (Gowrisankar and Clark, 2016b). Hence, the balance between the synthesizing and degradative enzymes could be altered, potentially favoring Ang II mobilization from astrocytes. This could explain the augmentation of RAS activity observed in the brainstems of SHRs.
Interestingly, neuroinflammatory cytokines were also reported to be elevated in the cardiovascular centers of the brainstem and hypothalamus (Agarwal et al., 2011). Not only do astrocytes and microglial cells have fundamental roles in the homeostatic regulation of cytokines in the brain, but cytokines themselves can influence glial cell functions (John et al., 2003). Impaired resolution of neuroinflammatory conditions could well lead to an impairment of cardioregulatory regions of the brain. In support of this view, SHRs treated with an inhibitor of S100 calcium-binding protein B, a proinflammatory and an apoptotic marker of astrocytes (Higashino et al., 2009), resulted in significant blood pressure reduction. Prominent astrogliosis was reported in SHRs by multiple groups, further highlighting the role of astrocytes in the pathogenesis of hypertension (Tomassoni et al., 2004).
Although the importance of the astroglial RAS has been underscored by several studies, the molecular mechanisms underlying these effects have not yet been well investigated. Several groups have demonstrated the ability of proinflammatory cytokines to regulate sympathetic activity (Winklewski et al., 2015). The idea that a dysregulated neuroinflammatory state, in the cardiovascular centers of the brainstem, could contribute to sympathoexcitation was suggested by Paton and colleagues less than a decade ago (Waki et al., 2008). This was based on gene expression studies in the NTS of SHRs, where they observed altered inflammatory states, specifically in the levels of junctional adhesion molecule 1, monocyte chemotactic protein 1, and interleukin (IL)-6 (Waki et al., 2008; Paton and Waki, 2009). Prominent neuroinflammatory states were observed at very early stages of hypertension in SHRs, indicating a causative factor in the pathogenesis of essential hypertension (Waki et al., 2008). Whether an increase in proinflammatory cytokines, in the cardiovascular centers of the brain, was an important mediator of Ang II–mediated sympathoexcitation was investigated by Kang et al. (2009). They demonstrated that chronic infusion of Ang II in Sprague-Dawley rats caused sympathoexcitation, which was characterized by a proinflammatory and a pro-oxidant state in the PVN. Blockade of AT1Rs and NF-κB was also demonstrated to normalize proinflammatory cytokines as well as sympathetic activity, further authenticating the role of neuroinflammation and the RAS in sympathoexcitation (Kang et al., 2009). Chronic infusion of Ang II resulting in a prominent inflammatory status in the brain vasculature was shown to occur via an increase in ROS levels (Zhang et al., 2010). Since glial cells play a critical role in the regulation of neuroinflammatory and oxidative states in the brain, glial AT1Rs could well be important factors in the augmentation of sympathetic activity. In support of this, ablation of astroglial AT1Rs in the brainstem has been demonstrated to result in an improvement in the symptoms of heart failure by normalization of sympathetic activity (Isegawa et al., 2014).
The link between the glial RAS, neuroinflammatory states, and sympathoexcitation was discussed in a review by Shi et al. (2010b). They conceptualized that glial AT1R activation results in an increase in the levels of proinflammatory cytokines, which can then act as neuromodulators of synaptic activity (Shi et al., 2010b). It is plausible that mobilization of cytokines from glial cells can alter neuronal activity, since low levels of cytokines can alter neuronal activity (Waki and Gouraud, 2014). Definitive evidence of the role of inflammatory cytokines and Ang II–mediated elevation in sympathetic nervous system activity came from studies in the hypothalamus by the same group (Shi et al., 2010a). Chronic Ang II infusion in the PVN resulted in an increase in proinflammatory cytokines and a decrease in anti-inflammatory cytokines, which then caused an elevation in blood pressure (Shi et al., 2010a). This effect could be blocked by minocycline, indicating that this effect was mediated by microglial AT1Rs (Shi et al., 2010a). Circulating Ang II has also been shown to gain access to the cardiovascular centers in the brain by promoting blood-brain barrier breakdown (Vital et al., 2010; Biancardi and Stern, 2016). Both microglial AT1R-mediated elevation in oxidative stress and proinflammatory cytokines as well as molecular alterations in the brain microvasculature are viewed as the major mechanisms (Vital et al., 2010; Biancardi and Stern, 2016). Microglial cells were transformed predominantly into the M1 phenotype when exposed to Ang II, which represents a proinflammatory and a cytotoxic version of microglia, highlighting the role of microglial AT1Rs in promoting neuroinflammation and neurotoxicity (Rodriguez-Perez et al., 2016; Labandeira-Garcia et al., 2017). Further support came from another study investigating the crosstalk between the AT1R and Toll-like receptor 4 in the PVN. Ang II–mediated increases in ROS production were demonstrated to be due to an interaction between AT1R and Toll-like receptor 4 in microglial cells in the PVN (Biancardi et al., 2016). A synergistic effect on sympathetic activity has also been reported between proinflammatory cytokines and Ang II in the PVN (Shi et al., 2011). In addition to Ang II, other components of the RAS (e.g., prorenin) have also been highlighted as a potential inducer of neuroinflammation by microglial activation (Shi et al., 2014; Zhu et al., 2015). Interestingly, Ang (1-7) has not only been demonstrated to counteract the effects of prorenin, but it also induces microglial cells to assume an anti-inflammatory phenotype under basal conditions (Liu et al., 2016).
Microglial cell activation is often followed by astroglial activation in several neurologic disorders (Liu et al., 2011b). Hence, microglia may initiate the inflammatory response, and astrocytes may aid in perpetuating the proinflammatory states that have been reported in cardiovascular nuclei of the SHR brainstem and hypothalamus (Agarwal et al., 2011). Not just Ang II but other RAS peptides can also elicit proinflammatory states by interacting with the astroglial AT1R. PVN astrocytes isolated from SHRs, when treated with prorenin, resulted in an augmented increase in proinflammatory cytokines (Rodríguez et al., 2015). Ang III was also observed to have potent proinflammatory effects in normotensive brainstem astrocytes (Kandalam et al., 2015).
Our group observed an elevation of pro-oxidant and proinflammatory cytokines IL-1 and IL-6 in brainstem astrocytes, isolated from SHRs and Wistar rats, in response to Ang II treatment (Gowrisankar and Clark, 2016a; Haspula and Clark, 2017a). Baseline levels of the neuroinflammatory cytokines were also elevated in SHR brainstem astrocytes (Waki et al., 2008; Gowrisankar and Clark, 2016a; Haspula and Clark, 2017a). Interestingly, we observed an elevation in the levels of both pro- and anti-inflammatory cytokines in these cells (Haspula and Clark, 2017a). Since astrocytes are known to play a role in neutralizing oxidative stress and proinflammatory states, an increase in anti-inflammatory status could well be a compensatory/protective mechanism that needs to be further investigated. Studies from our laboratory have focused on uncovering the molecular pathways triggered by astroglial AT1R under hypertensive and normotensive conditions. Ang II via AT1R elevates ROS as well as activates key signal transduction pathways such as the ERK, p38, and Janus kinase/signal transducer and activator of transcription pathways. These are important systems that are critical to astroglial functions such as cell proliferation as well as to cardiovascular functions such as mobilization of inflammatory cytokines from astrocytes (Kandalam and Clark, 2010; Clark et al., 2013; Alanazi et al., 2014; Negussie et al., 2016; Haspula and Clark, 2017b).
Brain RAS and Neurologic Disorders
Higher prevalence rates of hypertension have been reported in neurologic impairments characterized by prominent neuroinflammation, such as cognitive decline (Skoog et al., 1996; Tzourio, 2007; Nelson et al., 2014; Muela et al., 2017). Some conflicting data exist relating to the correlation between hypertension and cognitive decline. Although some studies have shown a strong correlation between the two, others have shown no correlation or even a negative correlation (Morris et al., 2000; Qiu et al., 2003; Reitz et al., 2007; Warchol-Celinska et al., 2015). Since activation of brain AT1Rs leads to an increase in proinflammatory and pro-oxidant states, it comes as little surprise that blockade of brain AT1Rs was investigated as a therapeutic strategy for several neurologic disorders (Wright et al., 2013; Mascolo et al., 2017). Evidence points to neutralization of the brain RAS as a treatment option in disorders pertaining to cognitive decline and memory loss (Mogi et al., 2012; Bodiga and Bodiga, 2013). Centrally acting ARBs were observed to have a greater efficacy than other antihypertensives in memory preservation in older hypertensive individuals (Ho and Nation, 2017). An observational case-control study assessing the efficacy of ACEIs on cognitive functions in elderly individuals also had favorable results (Gao et al., 2013). Furthermore, in a recently published study, an improvement in cognitive functions was observed with centrally acting ARBs in individuals diagnosed with Alzheimer disease (Fazal et al., 2017). ARBs also showed favorable neuropathological outcomes in hypertensive individuals with cognitive impairments (Hajjar et al., 2012). Considering that a strong correlation exists between cognitive decline and hypertension (Skoog et al., 1996; Tzourio, 2007; Nelson et al., 2014; Muela et al., 2017), hyperactivity of brain RAS, often associated with hypertension, could well be a hallmark of not only cardiovascular but also neurologic disorders as well.
An overactive brain RAS has also been linked to other neurodegenerative diseases such as Parkinson disease (Wright et al., 2013; Laudisio et al., 2017). In a prospective case-controlled study conducted on hypertensive individuals on ACEIs and ARBs, a reduced incidence of Parkinson disease was reported (Lee et al., 2014). In a recently published study, the ARB azilsartan was demonstrated to counteract apoptosis of dopaminergic neurons in a rat model of Parkinson disease (Gao et al., 2017). Telmisartan, an ARB that has peroxisome proliferator–activated receptorγ activating ability, has been shown to reduce neuronal loss and inflammation in a mouse model of Alzheimer disease (Saavedra, 2012). Several preclinical studies have highlighted the role exogenous and endogenous Ang II in worsening Alzheimer disease pathophysiology, highlighting the potential deleterious role of AT1R in neurologic indices (Ongali et al., 2014; Takane et al., 2017). In addition to AT1R, other components of the RAS have also been shown to be altered in neurologic disorders. ACE2 activity was observed to be markedly reduced in the postmortem brain tissue of individuals diagnosed with Alzheimer disease (Kehoe et al., 2016). Prospective cohort analysis in males diagnosed with Alzheimer disease revealed a significant improvement in the progression of the disease in individuals taking ARBs compared with other cardiovascular drugs (Li et al., 2010). By employing animal models, the RAS has also been demonstrated to worsen autoimmune disorders such as multiple sclerosis (MS) by activating transforming growth factor β (Lanz et al., 2010). Imbalances in cerebrospinal fluid Ang II levels, along with an impairment in perivascular astrocytes, were observed in patients with MS (Matsushita et al., 2010). Additionally, immunohistochemistry analysis revealed that plaques from the brains of patients with MS showed a strong upregulation in AT1R levels (Platten et al., 2009). Blocking of Ang II production was demonstrated to suppress the neuroinflammatory phenotype induced by activation of NF-κB via AT1R (Platten et al., 2009). Owing to the potential neuroprotective effect of ARBs, blocking of AT1R was shown to have promise in traumatic brain injury (Villapol et al., 2015). Since strong evidence exists linking centrally and noncentrally acting ACEIs or AT1R blockers with improvement of cognitive function, dementia, and neurodegeneration (Saxby et al., 2008; Mogi and Horiuchi, 2009; Davies et al., 2011), it is imperative that we understand and fully elucidate the mechanisms by which Ang II contributes to neuronal damage. Since astrocytes have a central role in brain homeostasis by regulating levels of cytokines and ROS, hyperfuctional astroglial AT1Rs may be a prominent feature of not only cardiovascular but also neurologic disorders.
Astrocytes from brain regions other than the brainstem, such as the cerebellum, are also responsive to Ang II treatment (Clark et al., 2013). Ang II caused a significant increase in the proinflammatory cytokine, IL-6, and ROS levels in astrocytes isolated from the cerebellum from both Wistar rats and SHRs (Gowrisankar and Clark, 2016a). Ang II–mediated ROS elevation and proinflammatory conditions are associated with neurodegeneration and also astrocyte senescence (Lanz et al., 2010; Liu et al., 2011a; Min et al., 2011). In addition, the SHR, which is characterized by a hyperactive brain RAS, is routinely employed as a model for attention deficit hyperactivity disorder (ADHD) (Adriani et al., 2003). Traits that are often observed in individuals with ADHD, such as shorter attention spans, inability to focus, and hyperexcitability, are also observed in the SHR, making it an ideal model to investigate the etiology of ADHD (Adriani et al., 2003). Similar to individuals with ADHD, SHRs are also characterized by cerebellar atrophy and cerebellar impairment (Yun et al., 2014). Evidence of a greater incidence of learning disabilities has also been reported in children with ADHD diagnosed with primary hypertension than in those without (Adams et al., 2010). These studies further highlight the importance of neuroinflammation in the pathogenesis of not only cardiovascular disorders but neurologic impairments as well. Considering that the RAS is a premium hormonal system that is augmented in the brains of SHRs, it is surprising to observe a paucity of studies investigating the effects of the RAS in ADHD. Nevertheless, the ability of Ang II to promote a proinflammatory state in different regions of the brain may lead to significant alteration in brain functions, eventually leading to neurologic disorders or an exacerbation of several neurologic conditions. A schematic of the potential role of glial AT1Rs in cardiovascular and neurologic diseases is shown in Fig. 4.

A schematic of potential roles of astroglial AT1Rs in cardiovascular and neurologic diseases. Ang II activates AT1Rs leading to the stimulation of a host of intracellular mediators. These mediators lead to an increase in proinflammatory cytokines that can trigger a diverse array of events in the central nervous system. AP-1, activator protein-1; BBB, blood-brain barrier; DAG, diacylglycerol; IP3, inositol trisphosphate; JNK, Jun N-terminal kinase; PIP2, phosphatidylinositol biphosphate; PKC, protein kinase C; PLC, phospholipase C.
Exogenous administration of Ang II in the rat striatum was demonstrated to cause an increase in dopamine and serotonin metabolites, 3,4-dihydroxyphenylacetic acid and 5-hydroxyindoleacetic acid, respectively (Mendelsohn et al., 1993). A decrease in acetylcholine levels in the rat entorhinal cortex was also observed (Barnes et al., 1989), further highlighting the importance of the RAS in not only cardiovascular disorders but also neurologic disorders. Although AT1R is known to be associated with an increase in oxidative and inflammatory status, the role of AT2R in neuronal functions cannot be overlooked. The administration of both an AT2R agonist and an AT1R antagonist was shown to lower 3,4-dihydroxyphenylacetic acid levels (Mertens et al., 2010). Activation of AT2R has been reported to improve memory performance (Matavelli and Siragy, 2015) and to also confer neuroprotection in middle cerebral artery occlusion–induced stroke (Joseph et al., 2014; Min et al., 2014). The mechanisms could be due to its role in neuronal differentiation, excitability, regeneration, apoptosis, and reduction in oxidative stress and inflammation (Côté et al., 1999; Gendron et al., 2003; Min et al., 2014). AT2R is shown to be mostly expressed on neuronal cells and not glial cells (Bennion et al., 2017). It is mostly the neuronal AT2R that is responsible for the cerebroprotective effects conferred by AT2R agonism after middle cerebral artery occlusion–induced stroke (Bennion et al., 2017).
In addition to AT1Rs and AT2Rs, AT4R has also been identified to have a significant impact on cognition. Ang IV and other AT4R ligands have been shown to significantly improve several different facets of memory and learning performance in rats (Wright et al., 1993; Lee et al., 2004; Wright and Harding, 2008). An elevation in acetylcholine release and facilitation of long-term potentiation, independent of glutamergic signaling, were reported to be a few of the key mechanisms of AT4R’s ability to improve cognitive ability (Wright and Harding, 2008).
Conclusion and Future Perspectives
At a molecular level, elevations in ROS and inflammatory cytokines are identified as an overarching paradigm for AT1R-mediated effects (Mehta and Griendling, 2007). Convincing evidence in the last decade has underscored the role of centrally expressed proinflammatory and pro-oxidant mediators in the progression of cardiovascular diseases and as hallmarks/risk factors for these diseases (Kishi et al., 2004; Wu et al., 2012; Haspula and Clark, 2018). By employing animal models, it was shown that elevations in central sympathetic outflow could be corrected by counteracting proinflammatory and pro-oxidant states (Shi et al., 2010a; Winklewski et al., 2015). The past decade has seen a significant increase in the number of studies highlighting the importance of the brain RAS in neurologic disorders such as dementia (Mogi et al., 2012) and MS (Lanz et al., 2010) and in neurodegenerative disorders such as Alzheimer and Parkinson diseases (Wright et al., 2013). Neuroinflammation and oxidative stress appear to be the common threads that connect the brain RAS to both cardiovascular and neurologic disorders (Zhang et al., 2010; Saavedra, 2012). AT1R-mediated elevation in ROS and multiple proinflammatory mediators are key factors in Ang II driving the progression of several pathologic conditions. Similar to many GPCRs, AT1Rs are known to crosstalk with other receptors, including those involved in mediating neuroprotection such as the Mas receptor (Von Bohlen und Halbach et al., 2000), AT2R (Inuzuka et al., 2016), and cannabinoid type-1 receptor (Rozenfeld et al., 2011; Szekeres et al., 2012; Haspula and Clark, 2016, 2017b). Hence, the pathologic significance of AT1R crosstalk with other receptors and systems present in different regions of the brain needs to be further explored. However, evidence of brain AT1R-mediated changes in receptors and systems that play a crucial role in neuroprotection has not been extensively researched. Understanding AT1R-mediated interactions with GPCRs that are known to be both functionally similar, as well as those that are deemed to be functionally antagonistic to AT1Rs, could aid in identifying the important players in Ang II–mediated neuroinflammation and oxidative stress, leading to the identification of therapeutic strategies that could be given in combination with central RAS antagonists. In addition, ARBs that are known to exhibit neuroprotective properties, independent of AT1R blockade, could well have potential for the treatment of neurologic disorders that are characterized by excessive inflammation. ARBs such as telmisartan are known to have pleiotropic effects apart from negating AT1R-induced oxidative stress and inflammation in the brain. Telmisartan has already been identified to have neuroprotective roles in preclinical studies conducted in mouse models of Alzheimer disease and stroke (Thoene-Reineke et al., 2011; Saavedra, 2012; Torika et al., 2017). However, clinical studies have confirmed a lack of effectiveness of telmisartan in conferring neuroprotection in subacute stroke (Sare et al., 2013). Since neuroprotection has been demonstrated for ARBs such as candesartan and telmisartan, it could be worthwhile to investigate these drugs not only for cardiovascular disorders but also for neurologic outcomes (Benicky et al., 2009; Noda et al., 2012; Villapol and Saavedra, 2015). Whether ARBs such as telmisartan that target neuroinflammation through multiple modes of mechanisms have beneficial roles in neurologic disorders such as Alzheimer and Parkinson diseases remains to be seen.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Haspula, Clark.
Footnotes
- Received February 28, 2018.
- Accepted May 8, 2018.
This work was supported by Nova Southeastern University [President’s Faculty Research and Development Grant 335309 and Health Professions Division Grant 335585].
Abbreviations
- ACE
- angiotensin-converting enzyme
- ACEI
- angiotensin-converting enzyme inhibitor
- ADHD
- attention deficit hyperactive disorder
- AGT
- angiotensinogen
- Ang
- angiotensin
- ARB
- angiotensin receptor blocker
- AT1R
- angiotensin type 1 receptor
- AT2R
- angiotensin type 2 receptor
- AT4R
- angiotensin type 4 receptor
- C21
- compound 21
- ERK
- extracellular signal–regulated kinase
- GPCR
- G protein–coupled receptor
- IL
- interleukin
- MAPK
- mitogen-activated protein kinase
- MS
- multiple sclerosis
- NF-κB
- nuclear factor κB
- NO
- nitric oxide
- NOS
- nitric oxide synthase
- NTS
- nucleus of the solitary tract
- PI3K
- phosphoinositide 3-kinase
- PVN
- paraventricular nucleus
- RAS
- renin angiotensin system
- ROS
- reactive oxygen synthase
- RVLM
- rostral ventrolateral medulla
- SFO
- subfornical organ
- SHR
- spontaneously hypertensive rat
- SON
- supraoptic nucleus
- WKY
- Wistar Kyoto
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}