


Abstract
Monoamine oxidase B (MAO-B) has been implicated in the pathogenesis of Alzheimer’s disease (AD) and other neurodegenerative disorders. Increased MAO-B expression in astroglia has been observed adjacent to amyloid plaques in AD patient brains. This phenomenon is hypothesized to lead to increased production of hydrogen peroxide and reactive oxygen species (ROS), thereby contributing to AD pathology. Therefore, reduction of ROS-induced oxidative stress via inhibition of MAO-B activity may delay the progression of the disease. In the present study we report the pharmacological properties of sembragiline, a novel selective MAO-B inhibitor specifically developed for the treatment of AD, and on its effect on ROS-mediated neuronal injury and astrogliosis in MAO-B transgenic animals. Sembragiline showed potent and long-lasting MAO-B-selective inhibition and did not inhibit MAO-A at doses where full inhibition of MAO-B was observed. Such selectivity should translate into a favorable clinical safety profile. Indeed, sembragiline neither induced the serotonin syndrome when administered together with the serotonin precursor l-5-hydroxytryptophan in combination with antidepressants such as fluoxetine, nor potentiated the pressor effect of tyramine. Additionally, in experiments using a transgenic animal model conditionally overexpressing MAO-B in astroglia, sembragiline protected against neuronal loss and reduced both ROS formation and reactive astrogliosis. Taken together, these findings warrant further investigation of the potential therapeutic benefit of MAO-B inhibitors in patients with AD and other neurologic disorders.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that currently affects 46 million people worldwide and is projected to affect 131.5 million by 2050 (Alzheimer's Disease International, 2015). Dementia due to AD is characterized by a significant loss of cognitive function, involving memory impairment, language disturbance, and visual-spatial disturbance, as well as difficulties with organization, reasoning, or judgment (McKhann et al., 2011). As the disease progresses, daily functioning decreases and neuropsychiatric symptoms increase (Alzheimer's Disease International, 2013).
Pathologically, AD is characterized by the accumulation of extracellular senile plaques containing amyloid-β (Aβ) peptides and intracellular neurofibrillary tangles, which, together with cerebral atrophy and neuronal cell death, comprise the hallmark features of the disease (Alzheimer's Disease International, 2013). The AD brain is also characterized by the presence of an “inflammatory” response, even at prodromal stages. These responses result in activation of astroglia and microglia cells, which can activate several signaling pathways (Wyss-Coray, 2006; Avila-Munoz and Arias, 2014) to produce inflammatory signals such as cytokines and reactive oxygen species (ROS), leading to oxidative stress (Sofroniew, 2009; Avila-Munoz and Arias, 2014). Markers of oxidative stress are observed early in AD, indicating that ROS may participate in the cascade of events leading to neuronal dysfunction (Meraz-Rios et al., 2013).
Monoamine oxidase (MAO), an enzyme localized to the outer mitochondrial membrane (Cohen and Kesler, 1999), plays an important role in the metabolism of monoamine neurotransmitters as well as other amines (Cai, 2014). Oxidative deamination by MAO produces hydrogen peroxide (H2O2), a mediator of oxidative stress. MAO exists in two isoforms, MAO-A and MAO-B, which are coded by two distinct genes, and have different tissue distribution patterns and different substrate selectivity (Thomas, 2000). MAO-B, the predominant form found in the human brain (Thomas, 2000), inactivates neurotransmitters, including dopamine, trace amines such as 2-phenylethylamine (PEA), and potentially other neuromodulatory amines such as polyamines (Youdim and Riederer, 1993; Cai, 2014).
MAO-B expression is increased in the hippocampus and cerebral cortex of AD patients compared with healthy controls (Parnetti et al., 1994; Reinikainen et al., 1988; Emilsson et al., 2002) and increased levels (>3-fold) of active MAO-B are found in reactive astrocytes around amyloid plaques (Saura et al., 1994; Gulyas et al., 2011). This upregulation of MAO-B in astrocytes is hypothesized to result in excessive metabolism of monoamines and increased production of H2O2 and oxygen radicals, and thus could conceivably contribute to the neurodegenerative processes taking place in AD (Riederer et al., 2004; Rahman, 2007). Such a mechanism appears to be an early event in AD and persists during disease progression (Kennedy et al., 2003; Carter et al., 2012; Leuzy et al., 2015).
Given the putative role of MAO-B in AD, inhibition of MAO-B activity may be expected to reduce oxidative stress and neurodegeneration, thus potentially delaying the progression of the disease. In addition, MAO-B inhibition may regulate levels of neuromodulatory amines that may be beneficial for behavioral symptoms. Indeed, the moderately selective irreversible MAO-B inhibitor selegiline was found to have a beneficial effect on cognitive symptoms in patients with moderate to severe AD (Sano et al., 1997). Nonetheless, the treatment effect of selegiline in AD remains controversial due to small numbers of patients included in most trials. In a Phase II clinical trial, treatment with lazabemide, a potent and selective MAO-B inhibitor, resulted in a 20–40% reduction in decline in cognitive function compared with placebo (Magni and Meibach, 1999). Furthermore, in two Phase III trials in patients with a diagnosis of probable AD, lazabemide was superior to placebo at slowing the deterioration in cognitive function and other measures over 1 year (Magni and Meibach, 1999). Although these unpublished results were suggestive of a treatment effect on symptom progression, the development of lazabemide was halted due to potential toxicity concerns. This prompted the development of sembragiline as a follow-on MAO-B inhibitor.
This paper describes the pharmacological and pharmacokinetic (PK) properties of sembragiline (referred to as R04602522, RG1577, and EVT 302 in previous communications) as well as its effect on cerebral neuromodulatory amines levels and its antioxidative and neuroprotective effects in a transgenic mouse model overexpressing MAO-B in astroglia.
Materials and Methods
Sembragiline
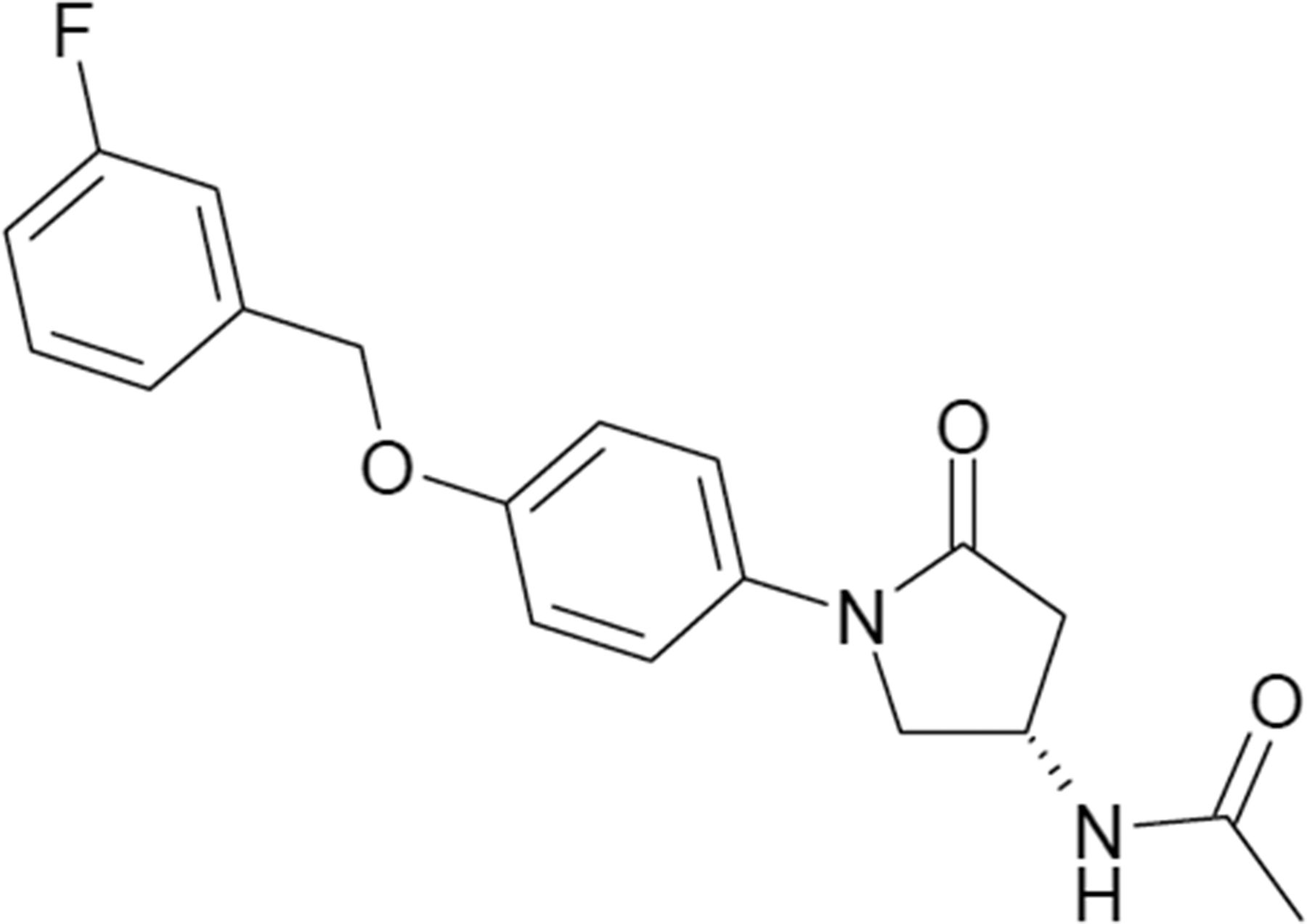
Figure 1 shows the chemical structure of sembragiline ((3S)-N-{1- [4-(3-fluorobenzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl}-acetamide), which was synthesized in the laboratory of F. Hoffmann-La Roche as a novel chemical structure.

The chemical structure of sembragiline.
Animal Experiments
Animal experiments were performed at several different locations (The Netherlands, United States, Germany, and Switzerland) in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health or local equivalent and were approved by the Institution’s Animal Care and use Committee or local equivalent. Experimental details are described in each relevant subsection.
In Vitro Pharmacological Profile of Sembragiline
Sembragiline, selegiline, and rasagiline were dissolved in dimethylsulfoxide at a concentration of 10 mM. Subsequent dilutions were made in assay buffer with a maximal final concentration of dimethylsulfoxide of 0.2% for the enzymatic assay and 0.5% for the binding assay.
Enzymatic Assay: In Vitro Evaluation of the Effect of Sembragiline, Selegiline, and Rasagiline on Human and Rat MAO-B and MAO-A.
The MAO-B and MAO-A enzymatic activity assay was carried out in a 96-well plate as described previously (Zhou and Panchuk-Voloshina, 1997). Membranes prepared from HEK 293 EBNA cells recombinantly expressing either human MAO-A or MAO-B (20 µg protein per well) were used for the human enzyme assay. For the rat enzyme assay, membranes prepared from rat brain (50 µg protein per well) were used. Because rat brain membranes contain both isoforms of MAO, the membranes used for the MAO-A assay were preincubated with the selective MAO-B inhibitor lazabemide (5 µM, see Supplemental Table 1 for relative potency and selectivity of the various MAO inhibitors used in this paper), whereas those used for the MAO-B assay were preincubated with the selective MAO-A inhibitor harmaline (5 µM). The assay was carried out in potassium phosphate buffer (0.1 M, pH 7.4). The plate was read with a spectrofluorimeter with excitation at 560 and emission at 590 nm. Blanks were prepared by addition of clorgyline or selegiline (10 µM final dilution for both) to the sample for the MAO-A and MAO-B assay, respectively. All assays were performed in duplicate in at least two separate experiments. IC50 values were determined using a four-parameter logistic equation from the percent inhibition values plotted against the logarithm of the concentration of the compound.
Binding Assay: Affinity of Sembragiline for Different Binding Sites.
The affinity of 10 μM sembragiline for other binding sites was assessed in a panel of >100 different receptors, enzymes, and ion channels (listed in Supplemental Table 2).
Biochemical and Behavioral Effects of Oral Sembragiline in Rats
Ex Vivo Measurement of Rat Brain and Liver MAO-B Enzymatic Activity.
Adult male Wistar rats received sembragiline via oral gavage in 0.3% Tween 80 in saline solution (0.9% NaCl) and were euthanized by decapitation under light anesthesia 2 hours after administration (for dose-response experiments) or at various time points after administration of 0.3 mg/kg sembragiline (for time course experiments). Brain homogenates were diluted 1/50 and 1/100 in phosphate buffer (KH2PO4/K2HPO4, 0.1 M, pH 7.3) for the assay of MAO-A and MAO-B, respectively. Liver homogenates were diluted in phosphate buffer 1/500 and 1/1000 for the assay of MAO-A and MAO-B, respectively. The enzymatic activity of MAO-A and MAO-B was measured in a 96-well plate using the method reported above. As rat brain and liver contain both isoforms of MAO, homogenates for MAO-A assays were preincubated with selegiline to inhibit MAO-B activity, whereas those to be measured in the MAO-B assay were preincubated with clorgyline to block MAO-A activity. Briefly, 200 µl of homogenate was incubated for 10 minutes at room temperature with 2 µl of either clorgyline 100 µM (for MAO-B assay, final concentration 1 µM) or selegiline 1 mM (for MAO-A assay, final concentration 10 µM) and with 5 μl “reaction mix” (0.6 mM AmplexRed, 40 U/ml peroxidase in phosphate buffer). At the end of the incubation, the reaction was started by addition of 5 µl of 4 mM 5-hydroxytryptamine (5-HT) (100 µM final concentration for MAO-A assay) or PEA 800 µM (20 µM final concentration, for MAO-B assay). Enzyme inhibition in treated animals was calculated as a percentage of the activity obtained in untreated control animals, and ID50 values were determined using a four-parameter logistic equation as described above.
Determination of Sembragiline in Rat Plasma.
Blood samples were taken from each rat at the time of euthanasia. Plasma was obtained from blood samples by treatment with heparin sodium and centrifugation at 4°C. Plasma concentrations of sembragiline were determined using a selective high-performance liquid chromatographic method combined with ion-spray tandem mass spectrometry (PE SCIEX API 2000) with a lower limit of quantification of 5 ng/ml.
PEA-Induced Hyperlocomotor Activity Potentiation Test.
Locomotor activity was monitored with a Digiscan Animal Activity Monitoring system (model RXYZCM Omnitech Electronics, Columbus, OH) inside a Plexiglas test box. Both sembragiline and PEA (Sigma-Aldirch, Buchs, Switzerland) were suspended in 0.3% Tween 80 dissolved in saline solution (0.9% NaCl in distilled water). Sembragiline or vehicle were administered orally to male adult Wistar rats (100–110 g; CrlGlxBrlHan: Charles River, WI) at doses of 0.1, 0.3, 1, or 3 mg/kg by mouth. Thirty minutes later, the rats received an intraperitoneal injection of PEA 20 mg/kg and were placed in the test box. After 30 minutes of habituation, total horizontal activity was evaluated for 60 minutes. Groups of six rats were used for each treatment condition. Animals were used only for a single experiment. Data were analyzed with analysis of variance (ANOVA) followed by Dunnett’s post hoc tests.
Effect of Sembragiline on Neuromodulatory Amines.
Adult male Sprague-Dawley rats (294–408 g; Harlan, Horst, The Netherlands) were dosed with vehicle [0.9% saline with Tween 80 (2 drops Tween per 10 ml saline)] or sembragiline at 3 mg/kg orally and euthanized by overdose of pentobarbital 2 hours postadministration. The cortex, striatum, hippocampus, and hypothalamus were rapidly dissected and stored at –80°C until analysis. Solvent for extraction of the analytes from tissue matrix consisted of 0.01% ascorbic acid and 50% acetonitrile in ultrapurified H2O and was added to each sample at a volume of 4 ml/g. The sample was homogenized and the resulting homogenate centrifuged. The supernatant was used as tissue extract for quantification of PEA, spermidine, and spermine as described in Supplemental Material. Treatment effects on each analyte in each brain area were compared with vehicle, using t tests.
Effect of Sembragiline on the Hypertensive Effect of Tyramine.
Arterial blood pressure and heart rate were measured in wakeful and freely moving adult male specific pathogen-free Wistar rats (290–320 g RCC, Fullinsdorf, Switzerland) through a catheter inserted in the left femoral artery. After surgery to insert the catheter, the rat was transferred to the test box and the catheter was connected to a PowerLab data recording unit for the continuous measurement of blood pressure. Baseline arterial blood pressure values were measured during the first hour of the experiment, after which animals received the test drug or vehicle. The two compounds tested were sembragiline and RO0411049 (Da Prada et al., 1990), a selective MAO-A inhibitor used as an active control; control animals were treated with vehicle (0.3% Tween 80 in saline solution). One hour later, each rat was treated with tyramine hydrochloride (10 mg/kg) and blood pressure was further monitored for 60 minutes. All compounds were administered orally (gavage) at a volume of 2 ml/kg. Data were analyzed using ANOVA followed by a post hoc Bonferroni comparison.
Effect on l-5-Hydroxytryptophan-Induced Behavioral Effects.
The first experiment investigated the interaction of sembragiline with l-5-hydroxytryptophan (l-5-HTP). Clorgyline (1, 3, 10 mg/kg), selegiline (1, 3, or 10 mg/kg), or sembragiline (1, 3, or 10 mg/kg) were orally administered to male Wistar rats (Charles River), followed 1 hour later by l-5-HTP (50 mg/kg s.c.). Clorgyline and selegiline were dissolved in saline. Sembragiline was dissolved in 0.2% Tween 80. l-5-HTP was dissolved in 300 μl 0.5 N HCl and diluted with saline to obtain the correct concentration. Animals were observed for the occurrence of specific behaviors for 1 hour as shown in Supplemental Table 3, by experienced raters blinded with respect to the treatment conditions. The total frequency of individual behaviors was added into two compound scores: the 5-HT syndrome and the other behavior scores. As there were no significant differences in other behavior scores (frequencies ranging from 2 to 10), only the frequencies of total 5-HT syndrome scores will be presented here.
The second experiment investigated the interaction of sembragiline or selegiline with the antidepressants fluoxetine, citalopram and venlafaxine. Rats were treated with sembragiline or selegiline for 6 days at 3 mg/kg by mouth. Immediately after the last injection, rats received an intraperitoneal injection with either vehicle or fluoxetine (30 mg/kg i.p.; Sigma-Aldrich, St. Louis, MO), citalopram (30 mg/kg i.p.; TRC, C505000), or venlafaxine (10 mg/kg i.p.; synthesized in-house) dissolved in 0.2% Tween 80 v/v 0.9% saline. One hour later, the animals received an injection with l-5-HTP (50 mg/kg s.c.), after which they were observed for 1 hour for the occurrence of specific behaviors as described above.
The cumulative 5-HT syndrome scores were first checked for normality using the Kolmogorov-Smirnov test. As all groups were found to significantly deviate from normality, the nonparametric Kruskal-Wallis test for K-independent samples (with the Mann-Whitney U test for two independent samples as a post hoc test) was performed to determine significant differences between treatments.
PK of Sembragiline
Absorption, Distribution, Metabolism, and Excretion Methods.
PK characterization of sembragiline was conducted in male Wistar rats and male beagle dogs. In both species, single doses of sembragiline were administered either intravenously or orally by gavage in a vehicle of 1% methylcellulose aqueous suspension. For ascending-dose studies, sembragiline was administered as suspension (thixotropic vehicle containing polysorbate 80, hydroxyethylcellulose, methylparaben, and propylparaben) at doses of 5.0, 50, and 150 mg/kg in rats; N = 3 per dose group. Blood samples (∼0.2 ml per time point) were collected without anesthesia from the jugular vein catheter at predose, 0.5, 1, 3, 5, 8, 24, 32, 48, 56, 72, and 80 hours postdose. The collection tubes contained EDTA/NaF as anticoagulant and stabilizer. After centrifugation, plasma samples were stored at –20°C until analysis.
Plasma concentrations were determined by liquid chromatography-mass spectrometry after collecting blood samples at multiple time points up to 24 hour after single-dose administration.
In vitro plasma protein binding of sembragiline was determined by centrifugal ultrafiltration after addition of drug to human, dog, or rat plasma. Results were compared with values obtained by equilibrium dialysis at pH 7.4 and 37°C. The free fraction (fup) and the percentage of free and bound drug were calculated as follows: fup, Cb/Cp (Fup, unbound fraction of drug in plasma; Cb, drug concentration on the buffer side at dialysis end; Cp, drug concentration in plasma).
Cytochrome P450 Inhibition by Sembragiline.
Human liver microsomes prepared from a pool of 10 individuals (to account for interindividual differences) were obtained from Gentest (Woburn, MA). Cytochrome P450 (CYP)3A4, CYP1A2, CYP2D6, CYP2C9, and CYP2C19 cDNA expressed in insect cell microsomes was obtained from Gentest.
A 96-well plate assay, initially described by Crespi et al. (1997), was used for IC50 determination of CYP450 inhibition using recombinant human isoenzymes and fluorescent substrates. Specific substrates used for assessing CYP450 inhibition in human microsomes were as follows: midazolam (CYP3A4), testosterone (CYP3A4), nifedipine (CYP3A4), (S)-mephenytoin (CYP2C19), diclofenac (CYP2C9), bufuralol (CYP2D6), and tacrine (CYP1A2 and CYP1A1).
Ex Vivo Effect of Sembragiline on the Formation of Peroxide in Brain Sections from Individuals with AD
Generation of hydrogen peroxide by human MAO-B was measured on cryosectioned postmortem AD brain sections from the superior temporal gyrus of AD patients. Brain blocks from three different subjects were used: a female subject, 78 years old, Braak stage VI; a male subject, 84 years old, Braak stage V; and a female subject, 89 years old, Braak stage V. All brain blocks were obtained from the Departments of Neuropathology of the Sun Health Research Institute (University of Arizona, Tucson, AZ) with informed consent obtained for the use of human brain tissue and for access to medical records for research purposes. Tissue was obtained and used in a manner compliant with the ethical standards of the Committee on Human Experimentation of the institution in which the experiments were performed and in accordance with the Declaration of Helsinki and was approved by the local Institutional Review Board.
Cryosections (10 µm) were mounted on silane-subbed slides [glass slides were immersed in freshly prepared 2% 3-aminopropyltriethoxysilane (A3648, Sigma) in acetone for 30 seconds, then dipped twice each in acetone and distilled H2O] and air dried at 42°C. Shandon cover plates (72-110-017, Thermo Scientific, Waltham, MA) were used for the histochemical detection of H2O2. Sections were incubated overnight at 4°C in the following buffer: 50 mM Tris-HCl buffer with 0.1% horseradish peroxidase (HRP, P6782, Sigma) and 0.01% 3,3′-diaminobenzidine∙4HCl (DAB, D5637, Sigma) with 3 mM NaN3 (6688, Merck) and 0.3% nickel-ammonium-sulfate (09885, Fluka) with 0.2% Triton-X (93420, Fluka), pH 7.6, at 300 μl/slide. β-phenylethylamine-HCl (1 mM; P6513, Sigma) was added in the presence or absence of 100 µM selegiline.
Before immunofluorescence staining, sections were rinsed in phosphate-buffered saline (PBS) with 0.2% Triton-X (assay buffer) 4× 2 minutes, treated with acetone (−20°C for 2 minutes), rinsed with PBS, and blocked for 15 minutes with a hydrogen peroxide block (TA-125-HP, ThermoScientific, Waltham, MA). Sections were then rinsed with PBS and blocked for 15 minutes with 1% BSA (735086, Böhringer, Germany) +1% ovalbumin (05440, Fluka) +1% goat serum (G9023, Sigma). Sections were incubated with 1:400 mouse monoclonal anti-glial fibrillary acidic protein (GFAP) conjugated to Cy3 (clone G-A-5, Accurate Chem., BYA63771) for 1 hour followed by four PBS rinses. Mouse monoclonal anti-Aβ antibody (5 µg/ml) conjugated to AlexaFluor488 (clone BAP-2, Roche, Switzerland) was incubated with the sections for 1 hour followed by four PBS rinses and nuclear staining with 1 µg/mL DAPI (D9542, Sigma) for 5 minutes. Sections were then rinsed in PBS for 30 minutes.
Autofluorescence of lipofuscin was suppressed with 4 mM CuSO4 (61230, Sigma) in 50 mM ammonium acetate (09690, Fluka) buffer, pH 5.0, followed by two PBS rinses, and slides were mounted with fluorescence mounting medium (S3023, DAKO, Agilent Technologies, Basel, Switzerland). Sections were imaged on a Zeiss AxioPlan2 epifluorescence microscope with a Hamamatsu C5810 camera, and images were processed by Adobe Photoshop software. The violet-brown DAB precipitate (H2O2 signal) was imaged in brightfield and signals converted to white color before combination with fluorescence images.
Effect of Sembragiline in Transgenic Mice Overexpressing Human MAO-B
These experiments were performed in a doxycycline-inducible astrocytic MAO-B mouse model (14–16 months old). For this study, a doxycycline-inducible transgenic mouse model overexpressing MAO-B in astrocytes generated at the Buck Institute was used. As reported elsewhere, this mouse model shows many hallmark neuropathological features of age-related human neurodegenerative disease (Mallajosyula et al., 2008; Siddiqui et al., 2011).
Doxycycline was administered in the feed for a period of 2 weeks at 0.5 g/kg/day (Research Diets, New Brunswick, NJ) to induce maximal MAO-B levels. Controls received doxycycline-free feed. The mice were concomitantly treated with sembragiline (3 mg/kg once daily) or vehicle via oral gavage during this 2-week period.
Differences among the means for all experiments described were analyzed using two-way ANOVA with time or treatment as the independent factor. The Newman-Keuls post hoc test was employed when differences were observed by analysis of variance testing (P < 0.05).
Effects of Sembragiline on ROS Production.
Production of ROS was assessed using dichlorofluorescein diacetate (DCF) fluorescence in dopaminergic striatal synaptosomes isolated using a modified immune-magnetic protocol (Mallajosyula et al., 2008). ROS levels were measured using the fluorescent probe DCF (Molecular Probes, Eugene, OR). DCF was loaded directly into media containing synaptosomes at 5 mM for 30 minutes. After loading, synaptosomes were washed with PBS, counted, and loaded into 96-well plates. Fluorescence was measured on a Molecular Devices fluorescent plate reader.
Effects of Sembragiline on Neuronal Cell Loss and Astrogliosis.
The effects of sembragiline on dopaminergic neuron loss were assessed via stereological assessment of the numbers of neurons positive for tyrosine hydroxylase (TH+) in the substantia nigra (SN). Cryostat-cut sections (40 μm) were made through the SN, hippocampus, and cortex. SN sections were immunolabeled with either dopaminergic (anti-TH antibody, 1:500 dilution, Chemicon)- or microglial [anti-Iba1 (ionizing calcium-binding adaptor molecule 1) antibody, 1:500 dilution, DAKO]-specific antibodies. Cortical and hippocampal sections were stained with astrocytic-specific antibodies (anti-GFAP, 1:500 dilution, DAKO). This was followed by incubation with either biotinylated horse anti-rabbit IgG (1:200, Vector Laboratories) in combination with DAB reagent for TH or fluorescent donkey anti-rabbit IgG conjugated to Alexafluor555 (red) or Alexafluor488 (green) (1:500, Life Technologies, Carlsbad, CA) for Iba1 and GFAP, according to the manufacturer’s instructions. Slides were decoded, and SN TH+ cell counts were performed using an unbiased dissector method (McCormack et al., 2002).
Results
Pharmacological Profile of Sembragiline
Sembragiline Is a Potent and Selective MAO-B Inhibitor.
Sembragiline inhibits enzymatic activity of human MAO-B with a greater than 600-fold selectivity for MAO-B versus MAO-A; similar results were obtained using rat enzymes (Table 1). For comparison purposes, Table 1 also reports the potencies of rasagiline and selegiline, two MAO-B inhibitors used for the treatment of Parkinson’s disease. Sembragiline exhibited higher selectivity compared with either of these compounds. Additionally, in binding assays at the concentration of 10 µM, sembragiline displayed no affinity for more than 100 different receptors, enzymes, and ion channels (listed in Supplemental Table 2), demonstrating a lack of off-target activities.
Inhibition of human and rat MAO-B and MAO-A by different compounds
Data are means ± S.D. of 2–8 independent experiments.
Sembragiline Inhibits Brain MAO-B Activity in the Rat after Oral Administration.
Sembragiline produced a dose-dependent inhibition of MAO-B activity in the brain and liver of rats with an ID50 of 0.11 (0.10–0.13, 95% CI) and 0.036 (0.027–0.049, 95% CI) mg/kg, respectively, 2 hours after oral dosing. Complete brain MAO-B inhibition was achieved at doses of 0.3–1 mg/kg sembragiline (Fig. 2). No inhibition of MAO-A was observed at any of the doses tested. Determination of sembragiline concentrations in terminal plasma indicated an 80% inhibition of brain MAO-B activity at 170 ng/ml of sembragiline (data not shown).

MAO-B inhibition in the rat brain and liver by sembragiline. After oral administration of sembragiline to rats, dose-dependent MAO-B inhibition was observed in the brain (A) and in the liver (B) without inhibition of MAO-A activity. Data represent mean ± S.D., n = 3.
Sembragiline Is a Long-Lasting MAO-B Inhibitor.
After a single oral administration to rats at a dose of 0.3 mg/kg, sembragiline rapidly entered the brain and produced maximal MAO-B inhibition (∼ 90%) 2 hours after administration. This degree of inhibition remained constant for more than 6 hours and then slowly began to decline, returning to pretreatment level after approximately 72 hours, hence demonstrating the reversibility of MAO-B inhibition by sembragiline. No effect was observed on brain MAO-A enzymatic activity (Fig. 3).

MAO-B inhibition by sembragiline over time in vivo. Long-lasting MAO-B inhibition was observed after single oral administration of sembragiline (0.3 mg/kg) to rats. No effect was observed on MAO-A activity. Data represent mean ± S.D., n = 3.
After repeated administration of sembragiline at 0.3 mg/kg once daily for 5 days, the time course of brain MAO-B inhibition and recovery was virtually identical to that observed after a single administration, suggesting that the compound does not accumulate in the brain (Supplemental Fig. 1). Again, no effect was observed on MAO-A activity at any time point or upon repeated dosing, which provides further in vivo confirmation that sembragiline is a selective MAO-B inhibitor.
Effect of Sembragiline on PEA-Induced Hyperlocomotor Activity.
Administration of an MAO-B inhibitor together with PEA leads to an increase in locomotor activity due to elevation of PEA brain levels and consequent stimulation of dopaminergic neurotransmission (Braestrup et al., 1975). This test represents a simple in vivo pharmacodynamics model to assess the efficacy of MAO-B inhibitors and establish PK/pharmacodynamic relationships. Sembragiline treatment potentiated the locomotor effects of PEA [F(4, 25) = 20.33; P < 0.0001] in rats, indicating an in vivo inhibition of MAO-B. As shown in Fig. 4, the lowest sembragiline dose that significantly potentiated the PEA-induced locomotor activity was 1 mg/kg and was further increased at 3 mg/kg.

The effect of sembragiline on PEA-induced locomotor activity. In rats, sembragiline given orally at the doses indicated (mg/kg) potentiates the hyperlocomotor activity induced by the MAO-B substrate 2-phenylethylamine (PEA). Data represent mean ± standard error of the mean (S.E.M.); n = 6. ***P < 0.001 versus vehicle.
Effect of Sembragiline on the Levels of Trace Amines, Polyamines, and Other Neuromodulatory Amines.
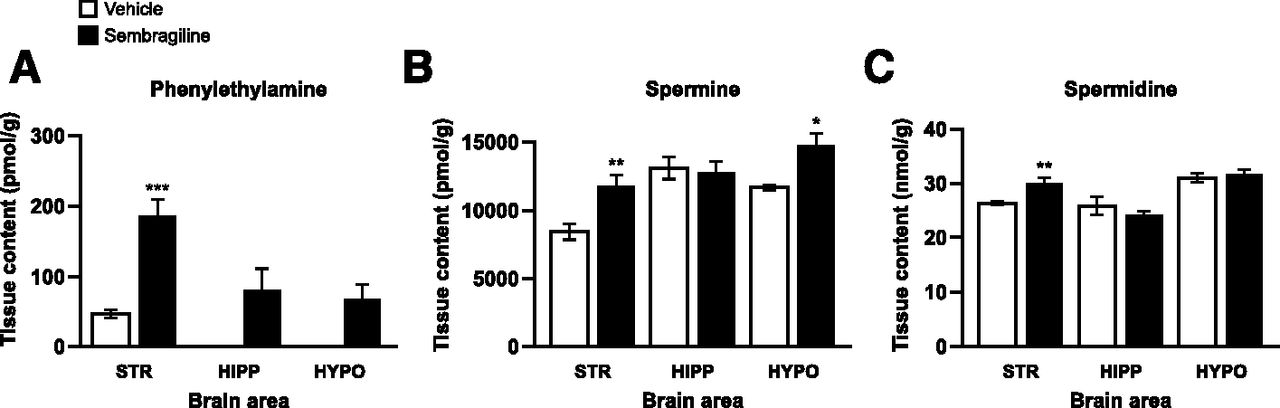
After oral administration of 3 mg/kg sembragiline or vehicle to rats, we investigated the effect of sembragiline on neuromodulatory amines that have been reported to be substrates for MAO-B. The brain tissue concentration of these amines was evaluated in the striatum, hippocampus, and hypothalamus (Fig. 5, A–C).

The effect of oral administration of sembragiline (3 mg/kg) to rats on brain content of phenylethylamine (A), spermine (B), and spermidine (C) in the striatum (STR), hippocampus (HIPP), or hypothalamus (HYPO). For (A), detection of PEA in the hippocampus and hypothalamus was below sensitivity for vehicle-treated animals. Data are expressed as mean ± SEM, n = 5. *P < 0.05, **P < 0.01 and ***P < 0.001.
A significant increase in striatal PEA content was observed in sembragiline-treated animals compared with vehicle-treated animals (P < 0.001) (Fig. 5A). It is well known that PEA content in certain brain regions is very low. In agreement with this, the content in the hippocampus and hypothalamus for vehicle-treated animals were all below the lower limit of quantification, although a trend of increase was observed in response to sembragiline.
Similarly, a significant increase in striatal content of both spermine and spermidine was observed in sembragiline-treated animals compared with the vehicle-treated animals (P < 0.01 for both amines; Fig. 5, B and C). In addition, spermine content in the hypothalamus was also significantly increased with sembragiline compared with vehicle (P < 0.05). A significant increase in the levels of telemethylhistamine (a metabolite of the neurotransmitter histamine), but no corresponding effect on those of histamine, was observed in the striatum and hypothalamus (data not shown).
Finally, no effect of sembragiline was observed on other trace amines (tryptamine, m- and p-tyramine and p-octopamine) or amino acids (arginine, lysine, ornithine) (data not shown).
Safety Profile of Sembragiline
Lack of Potentiation of the Hypertensive Effect of Tyramine in the Rat.
Potentiation of the pressor effect of dietary tyramine, potentially resulting in hypertensive crises (so called “cheese effect”; Da Prada et al., 1990), is a safety concern for MAO-A and moderately selective MAO-B inhibitors. Administration by mouth of 30 mg/kg sembragiline to rats had no effect on blood pressure (time 60–120 minutes in Fig. 6). The average plasma levels of sembragiline before administration of tyramine (time 55 minutes) and at the end of the experiment (time 120 minutes) were 3621 and 4730 ng/ml, respectively—well above the concentrations necessary to fully inhibit brain MAO-B (see Fig. 2). Conversely, when rats were pretreated with the selective, but poorly reversible, MAO-A inhibitor RO0411049 at a dose of 10 mg/kg by mouth, subsequent administration of tyramine produced a significant elevation of blood pressure consistent with a previous study (Da Prada et al., 1990). These results show that sembragiline, even at a dose in excess of those necessary to completely inhibit MAO-B in the brain (see above), has little or no liability to potentiate the pressor effect of tyramine and is therefore unlikely to cause hypertensive crisis due to dietary tyramine in human patients. Therefore, dietary restriction of tyramine-rich foods is likely not required with sembragiline.
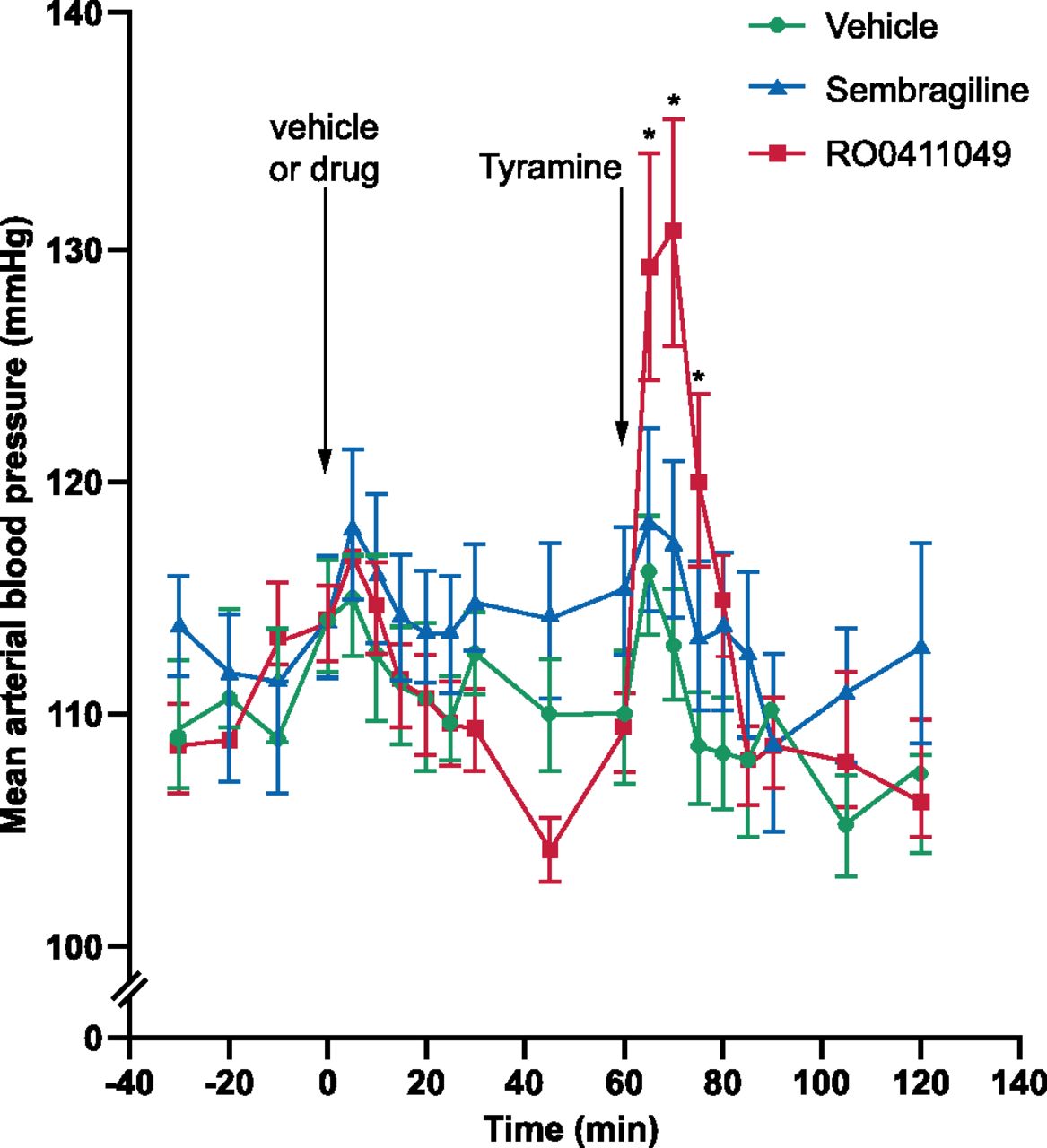
The effect of sembragiline on the hypertensive effect of tyramine in rats. Graph illustrates the effect of oral administration of the poorly reversible MAO-A inhibitor RO0411049 (10 mg/kg) and sembragiline (30 mg/kg) on rat arterial blood pressure. Drugs were administered orally at time 0 minutes and tyramine was administered at time 60 minutes as indicated. Time points represent mean ± S.E.M.; n = 8. *P < 0.05. Green circles represent vehicle; blue triangles, sembragiline; and red squares, RO0411049 treatment.
Lack of Potentiation of l-5-HTP and Pharmacological Interaction with Serotoninergic Antidepressants.
The serotonin syndrome is thought to arise from toxicity due to excessive synaptic serotonin. Combination of either nonselective or MAO-A-selective MAO inhibitors with serotoninergic agents such as selective serotonin reuptake inhibitor (SSRI) antidepressants may induce this syndrome with a potentially fatal outcome (Gillman, 2006). To investigate this potential toxicity, sembragiline was tested in a rodent model of serotonin syndrome, where serotonin production is increased by the administration of the serotonin precursor l-5-HTP.
When administered to rats 60 minutes before treatment with l-5-HTP (50 mg/kg s.c.), a single oral administration of up to 10 mg/kg sembragiline did not potentiate l-5-HTP-induced behavior observed during the 60-minute period. Because the Kolmogorov-Smirnov test showed a non-normal distribution (Z = 22.48, N = 93, P < 0.001), the nonparametric Kruskal Wallis test was performed, which confirmed the lack of a significant effect for sembragiline (χ2 = 0.5, df = 3, P > 0.5). In contrast, administration of the irreversible MAO-A inhibitor clorgyline (3 mg/kg by mouth) greatly potentiated l-5-HTP-induced behavior as expected, producing severe signs typical of the serotonin syndrome (χ2 = 19.8, df = 3, P < 0.001). No effect was observed with the MAO-B preferential inhibitor selegiline (3 mg/kg by mouth) (Fig. 7A) (χ2 = 2.9, df = 3, P > 0.4).

The effect of sembragiline on the potentiation of l-5-HTP-induced behavioral effects in rats. (A) 5-HT syndrome scores after acute oral administration of sembragiline (indicated doses), selegiline (Sele; 3 mg/kg), or the MAO-A inhibitor clorgyline (Clorg; 3 mg/kg) are displayed. (B) Graph displays the effect of repeated (3 mg/kg for 6 days) oral administration of sembragiline or selegiline with intraperitoneal coadministration of fluoxetine (30 mg/kg) or vehicle on 5-HT syndrome behavior. Data represent mean ± S.E.M.; n = 9 or 10 [except n = 3 for clorgyline group (A) because the effects were so severe that no further animals were treated in agreement with local regulations]. *P < 0.05 versus control; #P < 0.05 versus selegiline alone.
In subchronic treatment experiments, no potentiation of l-5-HTP-induced behavior was observed after oral administration of sembragiline for 6 days at 3 mg/kg. Again, as the data were not normally distributed, a Mann Whitney U test was performed, showing no significant effects (Z = −0.6, P > 0.5). In addition, no significant potentiation of l-5-HTP-induced behavior was observed with sembragiline after coadministration with the SSRI fluoxetine (30 mg/kg i.p.) (Fig. 7B; Z = −1.7, P > 0.2). No effect on l-5-HTP-induced behavior was observed also after coadministration with the SSRI citalopram (30 mg/kg i.p.) and the serotonin-norepinephrine reuptake inhibitor venlafaxine (10 mg/kg i.p.) (data not shown). Conversely, in selegiline-treated rats, l-5-HTP-induced behavioral signs were significantly enhanced by administration of fluoxetine (Fig. 7; Z = −3.7, P < 0.001) as previously observed (Riederer and Youdim, 1986); this confirmed that selegiline also inhibits MAO-A upon repeated administrations.
PK Profile of Sembragiline in Preclinical Species
After oral administration, sembragiline was absorbed with a time to peak concentration (Tmax) of 3.2 hours in rats and 2.0 hours in dogs and showed variability of 20–30% coefficient of variation in both species. Oral bioavailability was complete at low doses, suggesting low first-pass extraction and rapid absorption of sembragiline (Table 2). The mean (±S.D.) maximum plasma concentration (Cmax) of sembragiline increased linearly with dose in rats from 1350 (±420), 13,800 (±3320), and 24,900 (±8060) ng/ml for single oral doses of 5, 50, and 150 mg/kg, respectively. The mean (±S.D.) area under the curve (0–24 hours) values increased dose proportionally from 14,500 (±666) at 5 mg/kg to 175,000 (±35,600) at 50 mg/kg and to 409,000 (±79,700) h·ng/ml at 150 mg/kg.
Pharmacokinetic profile of sembragiline in rat and dog after intravenous and oral administration
Sembragiline has a low potential for drug-drug interactions, with no significant inhibition of CYP450 isoenzymes (IC50 > 50 µM for 1A1, 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) or induction of CYP3A4, CYP1A2, and CYP2B6 (data not shown). In vitro plasma protein binding exhibited a low interspecies difference and was largely concentration independent; the mean calculated free fraction was 17.4–18.1% in humans, 21.3–23.7% in dogs, and 11.4–14.6% in rats.
Reduction of ROS Production and Neuroprotective Effects
Inhibition of MAO-B Blocks Production of Hydrogen Peroxide in AD Brain Sections.
To investigate MAO-B-mediated H2O2 production around Aβ plaques, brain sections taken from patients with AD were incubated with the MAO-B substrate PEA with or without the MAO-B inhibitor selegiline (100 µM). Representative results obtained in sections taken from a Braak VI stage subject are shown in Fig. 8. Formation of H2O2, visualized using HRP and DAB, appeared as a white precipitate and were prominently localized around plaques, indicating discrete formation of ROS. The presence of H2O2 was completely abolished in the brain sections incubated with selegiline, which blocked MAO-B enzymatic activity (Fig. 8). Similar results were obtained in brain sections taken from the other two subjects with AD (results not shown). These results provide direct evidence for in situ formation of ROS mediated by MAO-B and therefore for the potential of MAO-B inhibition in reducing oxidative stress around Aβ deposits and senile plaques; these structures are considered the main triggering event eliciting a cascade of cellular consequences leading to neurotoxicity, synaptic dysfunction, and ultimately to AD (Selkoe and Hardy, 2016).

Ex vivo activity of MAO-B revealed by H2O2 histochemistry on cryosections from post-mortem human AD brain (Braak VI, superior temporal gyrus). Overlay images of H2O2 histochemistry (HRP/DAB, inverted signal) and immunofluorescence using monoclonal antibodies against Aβ and GFAP. (Left) High-density of MAO-B leads to the formation of H2O2, visualized using HRP and DAB and appearing as a white precipitate (white arrowheads) around amyloid plaques (green). GFAP+ astroglia are shown in bright red. (Right) Inhibition of MAO-B using selegiline at 100 µM blocks the formation of H2O2.
Reduction of ROS Formation, Neurodegeneration, and Astrogliosis in Transgenic Mice.
The MAO-B transgenic animal model described by Mallajosyula et al. (2008) in which MAO-B is specifically and conditionally overexpressed in astroglial cells using a tetracycline-responsive bidirectional promoter, provides a useful translational animal model for investigating the effect of MAO-B overexpression in vivo. Notably, in this model, the increase in MAO-B upon treatment with the tetracycline derivative doxycycline is astroglia specific and in the same order of magnitude as that observed in AD brain (Saura et al., 1994; Mallajosyula et al., 2008).
As reported previously (Mallajosyula et al., 2008), increased expression of astrocytic MAO-B resulted in an increased generation of ROS and a loss of dopaminergic neurons in the SN (Fig. 9, A and B). Administration of sembragiline resulted in significantly reduced levels of ROS and prevented decreased dopaminergic (TH+) neuron numbers in the SN, when compared with vehicle-treated mice. Notably, doxycycline treatment does not produce any change in ROS production and the number of TH+ neurons and reactive astrocyte number in wild-type mice (Mallajosyula et al., 2008).

The effect of sembragiline on oxidative stress (A) and neurodegeneration (B). MAO-B mice with and without induction of MAO-B overexpression were treated with 3 mg/kg oral sembragiline or vehicle daily for 14 days. ++P < 0.01 and +++P < 0.001 versus vehicle-treated control mice; *P < 0.05 versus vehicle-treated MAO-B overexpression-induced mice; **P < 0.01 versus vehicle-treated control mice. SN, substantia nigra. Data represent mean ± S.D., n = 6.
Interestingly, overexpression of MAO-B in astrocytes induced reactive astrogliosis not only in brain regions where neurodegeneration takes place, such as the SN and the striatum, but also in regions not affected by neurodegenerative processes like the cortex and hippocampus. Notably, inhibition of MAO-B by sembragiline treatment significantly reduced astrogliosis as assessed by the number of activated astroglia (GFAP+) cells, both in the cortex (Fig. 10) and in the hippocampus (Supplemental Fig. 2). This result suggests that MAO-B enzymatic activity, and presumably formation of ROS, plays a role in the processes leading to activation of astrocytes.

The effect of sembragiline on astrogliosis. MAO-B mice with and without induction of MAO-B overexpression were treated with 3 mg/kg oral sembragiline or vehicle daily for 14 days. Images in (A) are representative of immunocytochemistry in the cortex. Scale bar is 100 μM at 10× magnification. GFAP+ cortical cell count was quantified in B. +++P < 0.001 versus vehicle-treated control mice; *P < 0.05 versus vehicle-treated MAO-B overexpression-induced mice. Data represent mean ± S.D., n = 3.
Discussion
In this paper, we report on the pharmacological characterization of sembragiline, a novel oral MAO-B inhibitor, which originated from an AD therapeutics program. Sembragiline is a follow-on from lazabemide, a MAO-B inhibitor that showed encouraging data but whose development was halted due to the emergence of adverse events. The pharmacological profile of sembragiline (potency, selectivity, and in vivo activity) and its PK properties were evaluated. To further support the rationale for MAO-B as source of ROS contributing to AD pathology, we also examined the effect of sembragiline treatment on ROS production, neuroprotection, and astrogliosis in an animal model that mimics the localization of MAO-B overexpression in the human AD brain. These studies characterize sembragiline as an orally available, potent, and selective inhibitor of MAO-B with excellent brain penetration.
Selectivity of sembragiline for MAO-B was demonstrated by the lack of effect on MAO-A and on a wide variety of receptors, ion channels, or other enzymes. Sembragiline is active in vivo at low doses as shown by the potentiation of locomotor activity of PEA in a behavioral animal model of MAO-B inhibition and was not active in an in vivo model of MAO-A inhibition, l-5-HTP potentiation. Notably, selegiline (Braestrup et al., 1975) and lazabemide (Skuza and Rogoz, 1991) also potentiate the locomotor activity in rats and mice, respectively. Taken together, these data confirm the MAO-B selectivity of sembragiline.
In addition, the time course experiments indicate that sembragiline is a long-acting but reversible MAO-B inhibitor, because rat brain MAO-B was inhibited for >6 hours with complete recovery 72 hours after oral administration. Previous clinical trials carried out with the MAO-B inhibitors selegiline (Sano et al., 1997) and lazabemide demonstrated a therapeutic effect in AD patients at doses producing almost complete MAO-B inhibition (Bench et al., 1991; Fowler et al., 1994). In the rat, a dose of approximately 0.3 mg/kg by mouth or plasma concentrations of 170 ng/ml of sembragiline are required to achieve 80% inhibition of brain MAO-B. A recent PET study in healthy volunteers and AD patients using 11C-labeled deuterated selegiline as ligand for MAO-B showed that, with repeated dosing, virtually complete enzyme occupancy was achieved at 1 mg sembragiline daily in both populations (Sturm et al., 2017). Thus oral doses in the rat ranging from 0.3 to 3 mg/kg produced brain MAO-B inhibition comparable to that produced in humans at the doses tested in the Phase II studies, 1 and 5 mg daily (Nave et al., 2017).
The clinical use of MAO inhibitors has been associated with potential liabilities that have limited their use. These include the potentiation of tyramine pressor effect, resulting in hypertensive crisis (termed “cheese effect” due to the high content of tyramine in fermented cheeses) and the serotoninergic syndrome when given in combination with serotoninergic agents such as SSRIs. Because these liabilities are linked to MAO-A inhibition (Youdim et al., 2006), it is important to assess the degree of selectivity of MAO-B inhibitors in vivo and whether they may be liable to produce these potentially fatal toxic events. Notably, marketed MAO-B inhibitors, i.e., selegiline and rasagiline, are only preferential MAO-B inhibitors and also inhibit MAO-A to a substantial extent after chronic treatment (Elsworth et al., 1978; Riederer and Youdim, 1986; Holford et al., 1994); clinical pharmacology studies have been performed for rasagiline to address the tyramine potentiation potential (Goren et al., 2010).
Sembragiline neither potentiated tyramine pressor effects nor induced the serotonin syndrome (Jacobs, 1976) in animals, even if administered at doses in excess of those necessary to completely inhibit MAO-B in the brain. This suggests that, due to its high selectivity for MAO-B, sembragiline may be administered without dietary restriction and in combination with serotoninergic agents, such as SSRI antidepressants, which may be prescribed to patients with AD (Tsuno and Homma, 2009). Hence, the high selectivity of sembragiline for MAO-B both in vitro and in vivo and sembragiline’s lack of effect on MAO-A enhance the safety profile of sembragiline. A clinical pharmacology study conducted in healthy volunteers has confirmed that sembragiline, at doses up to four times its anticipated clinical dose, does not produce a clinically relevant increase in blood pressure after ingestion of tyramine (Roche, data on file). In addition, no changes in 3-methoxy-4-hydroxyphenylglycol, the deaminated metabolite of the MAO-A substrate adrenaline, were observed in plasma of human subjects treated with sembragiline (Roche, data on file), whereas rasagiline reduced the formation of this metabolite, indicating some degree of MAO-A inhibition at clinical doses (Goren et al., 2010).
An interesting but poorly investigated aspect of MAO-B inhibitors is their effect on the metabolism of neuromodulatory amines. Inhibition of MAO-B by sembragiline increased the brain levels of PEA, spermine, and spermidine. PEA modulates dopaminergic neurotransmission by reducing the activity of dopamine neurons at low concentrations and increasing dopamine release at high concentrations (Berry, 2007). Given that degeneration of dopaminergic projections and low dopamine levels are observed in AD, MAO-B inhibition by sembragiline could potentially restore dopaminergic transmission and improve behavioral symptoms of patients with AD. Polyamines, such as spermine and spermidine, modulate N-methyl-d-aspartate receptors and there have been reports of an age-dependent reduction of polyamine levels in the brains of animals and humans (Minois et al., 2011). Although the roles of polyamines in aging and disease are still unclear, reversal of polyamine depletion through inhibiting MAO-B could conceivably improve memory deficits in AD patients (Yoon et al., 2014). Finally, a recent paper showed that MAO-B may play a role in the synthesis of γ-aminobutyric acid (GABA) in astrocytes via the polyamine pathway, and it has been suggested that increased MAO-B expression in AD leads to excessive release of GABA from astrocytes in the hippocampus, which is held responsible for tonic inhibition leading to memory impairment (Jo et al., 2014). Therefore, sembragiline, by regulating levels of neuromodulatory amines and GABA, may have a beneficial effect on neuropsychiatric and cognitive symptoms in AD.
Although mice expressing mutant forms of amyloid-precursor protein and/or presenilins are the mainstay for research in AD, they do not recap all the features of AD, and their use in target validation and drug discovery is somewhat limited to therapeutic agents directly affecting amyloidosis. In this context, it is worth noting that an AD mouse model, PS2APP (Richards et al., 2003), although showing astrogliosis around plaques, did not show any increase in MAO-B. Little or no data on MAO-B expression have been reported in other commonly used amyloid transgenic animal models. This makes these animals poorly suitable to investigate the role of MAO-B in AD. Conversely, the inducible astrocytic MAO-B mouse model (Mallajosyula et al., 2008; Siddiqui et al., 2011) is a useful tool to study neurodegenerative disorders where increased expression of MAO-B occurs, such as in AD. Also, as mentioned above, these mice express MAO-B only in glial cells at levels comparable to those observed in human AD brains. In this model, sembragiline showed neuroprotective effects as well as the ability to reduce ROS formation and to reduce reactive astrogliosis. The ability of sembragiline and other MAO-B inhibitors (Mallajosyula et al., 2008) to reduce reactive astroglyosis suggests a role for MAO-B in the processes that regulate activation of astroglia. This animal model has primarily been used to study the relevance of MAO-B for dopaminergic neurodegeneration in Parkinson’s disease. However, the occurrence of MAO-B-induced astrogliosis in regions other than in the striatum, e.g., the hippocampus and cortex, indicates that reactive gliosis and potentially events leading to neuronal damage may also be relevant for degenerative processes affecting other types of neurons. Further investigation of neuronal death and synaptic dysfunction in these regions, which are primarily affected in AD, may provide further insight into the effect of MAO-B overexpression.
A large body of evidence supports the involvement of glial cells in AD. Although their precise contribution to AD development has yet to be fully clarified (Wyss-Coray, 2006; Crotti and Ransohoff, 2016), glia dysfunction, also commonly referred to as “neuroinflammation,” likely represents an early event in AD development. Genome-wide association studies have shown an association between variants of immune biomolecules (e.g., TREM2) and AD (Cuyvers and Sleegers, 2016). Although the focus in the past has been on microglia in AD, a number of recent reports indicate a major toxic role for astrocytes in neurodegenerative disorders. Astrocytes have been shown to be involved in synapse pruning in the developing and adult brain (Chung et al., 2013), a process that may be deregulated in neurologic disorders (Chung et al., 2015). In addition, a subset of astroglia cells appears to be activated by microglial cells and to mediate neurotoxicity (Liddelow et al., 2017); this astrocyte subset is abundant in various human neurodegenerative diseases, including AD. It is worth mentioning that the term “neuroinflammation” may be misleading in AD; gene expression studies have shown that AD or other neurodegenerative disorders such as Parkinson’s disease do not cluster with “bona fide” inflammatory diseases, such as multiple sclerosis (Filiou et al., 2014). Thus, rather than generically framing research in terms of neuroinflammation, AD-specific molecular phenotypes among glia might bear druggable targets. Because MAO-B seems to represent one of these targets, it would be of relevance to expand research, possibly beyond its proposed role as ROS source, to other potential functions of MAO-B overexpression in astroglia pathophysiology.
In conclusion, sembragiline is a potent and selective MAO-B inhibitor with a favorable preclinical safety profile. Sembragiline was able to modulate neuromodulatory monoamines, reduce oxidative stress, and decrease astrocyte activation. Safety of sembragiline treatment has also been demonstrated preclinically and by numerous clinical studies where the drug was well tolerated without the appearance of adverse effects. Sembragiline has completed a Phase II study in moderate AD (Nave et al., 2017). Although 1-year treatment with sembragiline was well tolerated with no difference between treated groups and placebo in treatment emergent adverse events, including laboratory parameters, the study missed the preselected primary cognitive endpoint (Nave et al., 2017). However, a beneficial effect of sembragiline on secondary endpoints was observed for neuropsychiatric symptoms and for activity of daily living after 52 weeks of treatment. Together, although this Phase II study suggests that sembragiline administered in addition to standard-of-care therapy (i.e., acetylcholinesterase inhibitors, memantine) in patients with moderate AD may not enhance cognitive benefit, it may provide therapeutic benefit in AD patients with behavioral and neuropsychiatric symptoms. These symptoms are among the most complex, stressful, and costly aspects of care (Kales et al., 2015) and are poorly served by available treatments.
Acknowledgments
The authors thank Francoise Kahn, Patricia Glaentzlin, Valerie Goetschy, Robert Schubenel, Raquel Herrador, Jasmin Graehlert, Marie-Therese Miss, and Marie Haman (F. Hoffmann-La Roche Ltd., Basel, Switzerland) for technical support. The authors also thank Athena Lemesiou, Megan Allen, and Joanna F. McCarter of MediTech Media for providing medical writing support, which was funded by F. Hoffmann-La Roche in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Authorship Contributions
Participated in research design: Borroni, Bohrmann, Prinssen, Loetscher, Chinta, Ellenbroek, Paehler, Andersen, Wyler, and Cesura.
Conducted experiments: Borroni, Grueninger, Chinta, Rajagopalan, Rane, Siddiqui, and Messer.
Contributed new reagents or analytic tools: Wyler.
Performed data analysis: Borroni, Grueninger, Prinssen, Chinta, Ellenbroek, Paehler, Andersen, and Cesura.
Wrote or contributed to the writing of the manuscript: Borroni, Bohrmann, Grueninger, Prinssen, Nave, Loetscher, Ellenbroek, Messer, Paehler, and Cesura.
Footnotes
- Received March 24, 2017.
- Accepted May 23, 2017.
↵1 Current affiliation: Biological and Pharmaceutical Sciences, College of Pharmacy, Touro University California.
↵2 Current affiliation: Victoria University of Wellington, Wellington, New Zealand.
↵3 Current affiliation: Novartis Institutes for BioMedical Research, Basel, Switzerland.
This study was funded by F. Hoffmann-La Roche.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- Aβ
- amyloid-β
- AD
- Alzheimer’s disease
- ANOVA
- analysis of variance
- DCF
- dichlorofluorescein diacetate
- GABA
- γ-aminobutyric acid
- GFAP
- anti-glial fibrillary acidic protein
- HRP
- horseradish peroxidase
- l-5-HTP
- l-5-hydroxytryptophan
- MAO-B
- monoamine oxidase B
- NPS
- neuropsychiatric symptoms
- PBS
- phosphate-buffered saline
- PEA
- 2-phenylethylamine
- PK
- pharmacokinetics
- ROS
- reactive oxygen species
- SN
- substantia nigra
- SSRI
- selective serotonin reuptake inhibitors
- TH
- tyrosine hydroxylase
- Copyright © 2017 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}