


Abstract
We previously found a negative inotropic (NIR) and positive lusitropic response (LR) to C-type natriuretic peptide (CNP) in the failing heart ventricle. In this study, we investigated and compared the functional responses to the natriuretic peptides (NPs), brain (BNP) and C-type natriuretic peptide (CNP), and relate them to cGMP regulation and effects on downstream targets. Experiments were conducted in left ventricular muscle strips and ventricular cardiomyocytes from Wistar rats with heart failure 6 weeks after myocardial infarction. As opposed to CNP, BNP did not cause an NIR or LR, despite increasing cGMP levels. The BNP-induced cGMP elevation was mainly and markedly regulated by phosphodiesterase (PDE) 2 and was only marginally increased by PDE3 or PDE5 inhibition. Combined PDE2, -3, and -5 inhibition failed to reveal any functional responses to BNP, despite an extensive cGMP elevation. BNP decreased, whereas CNP increased, the amplitude of the Ca2+ transient. BNP did not increase phospholamban (PLB) or troponin I (TnI) phosphorylation, Ca2+ extrusion rate constant, or sarcoplasmatic reticulum Ca2+ load, whereas CNP did. Both BNP and CNP reduced the peak of the L-type Ca2+ current. Cyclic GMP elevations by BNP and CNP in cardiomyocytes were additive, and the presence of BNP did not alter the NIR to CNP or the CNP-induced PLB and TnI phosphorylation. However, a small increase in the LR to maximal CNP was observed in the presence of BNP. In conclusion, different responses to cGMP generated by BNP and CNP suggest different compartmentation of the cGMP signal and different roles of the two NPs in the failing heart.
Introduction
Circulating levels of natriuretic peptides (NPs) are increased in heart failure (Kalra et al., 2003; Abassi et al., 2004; Clerico and Emdin, 2004; Del Ry et al., 2005). The NPs activate particulate guanylyl cyclases, which generate cGMP. Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) activate the NP receptor (NPR)-A, whereas C-type natriuretic peptide (CNP) activates the NPR-B receptor. All three NPs bind to the clearance receptor NPR-C, which lacks guanylyl cyclase activity (Potter et al., 2009), but is suggested to have signaling properties on its own (Rose and Giles, 2008).
Studies show functional responses to NPs in the heart, but the effects are complex. We have recently shown a negative inotropic response (NIR) and a lusitropic response (LR) to CNP, with an increased response in failing compared with sham-operated rat hearts (Moltzau et al., 2013). The effects of ANP and BNP stimulation on contraction are controversial, as studies have reported NIR (Vaxelaire et al., 1989; Tajima et al., 1998; Zhang et al., 2005), positive inotropic responses (Lainchbury et al., 2000), or no effects on contractility (Brusq et al., 1999; Pierkes et al., 2002) in normal hearts. In failing hearts, ANP has shown no inotropic response (Lainchbury et al., 2000) or NIR (Ohte et al., 1999). In previous studies where we showed that CNP but not BNP increased cAMP-mediated inotropic responses through cGMP-dependent phosphodiesterase (PDE) 3 inhibition (Qvigstad et al., 2010; Afzal et al., 2011), we also found that CNP alone caused an NIR, whereas BNP did not in failing hearts (Qvigstad et al., 2010). Further, we found that concerted effects of phospholamban (PLB) Ser16 and troponin I (TnI) Ser23/24 phosphorylation most likely represent the main mechanisms explaining the cGMP–protein kinase G (PKG)–mediated NIR and LR to CNP (Moltzau et al., 2013). Whereas PDE2 was the major phosphodiesterase-hydrolyzing cGMP generated by CNP, we also found a role for both PDE2 and PDE3 in the regulation of CNP-induced functional responses (Moltzau et al., 2014). Thus, a major discrepancy between cGMP levels and functional responses was revealed.
In the present study, we further investigated and compared the functional responses to BNP (NPR-A agonist) and CNP (NPR-B agonist) in failing rat left ventricle. In recent years, there has been an interest in using natriuretic peptides in heart failure therapy (Boerrigter et al., 2009), although studies have shown limited, if any, effects of the BNP-analog nesiritide (O’Connor et al., 2011). However, the use of a chimeric natriuretic peptide, CD-NP, with dual selectivity for both NPR-A and NPR-B receptors, has also been suggested in the treatment of heart failure (Dickey et al., 2008). No studies have compared the signaling pathways after BNP and CNP stimulation in failing hearts. Thus, we wanted to clarify whether BNP exerted effects on downstream targets and the effects of cGMP (PLB, TnI, and Ca2+ transients) possibly involved in the CNP-induced NIR and LR, as well as whether simultaneous stimulation by both NPs could influence the effects seen under separate stimulation. Further, we investigated the effect of PDEs in the regulation and compartmentation of BNP-generated cGMP.
We found that whereas both BNP and CNP caused a similar global cGMP elevation in the failing rat ventricle, they caused very different effects on downstream targets. These different effects on downstream targets and functional responses, despite similar effects on cGMP levels, suggest different cGMP compartmentation after BNP and CNP stimulation.
Materials and Methods
For more detailed methods, see the Supplemental Methods.
Materials.
Prazosin hydrochloride, atropine sulfate, and timolol maleate were purchased from Sigma-Aldrich (St. Louis, MO); cilostamide (Cil), sildenafil citrate (Sfil), and EHNA [erythro-9-(2-hydroxy-3-nonyl)adenine hydrochloride] were from Tocris Bioscience (Bristol, UK); BNP and CNP were from GenScript Corp (Piscataway, NJ); and isoflurane (Forene) was from Abbott Scandinavia (Solna, Sweden).
Heart Failure Animal Model.
The investigation conforms with the Guide for the Care and Use of Laboratory Animals by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and the Animal Research Reporting of In Vivo Experiments (ARRIVE) guidelines. Animals were cared for according to the Norwegian Animal Welfare Act, which conforms to the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (Council of Europe no. 123, Strasbourg, 1985) and was approved by the Norwegian Animal Research Authority. Male Wistar rats (300 g) were sedated in an anesthesia chamber (64% N2O, 32% O2, and 4% isoflurane) and subsequently ventilated on a Zoovent ventilator (Triumph Technical Services, Milton Keynes, UK) through endotracheal intubation. Anesthesia was maintained by 68% N2O, 29% O2, and 2–3% isoflurane, and deep surgical anesthesia was confirmed by abolished pain reflexes and no spontaneous breathing before the surgical procedure. Myocardial infarction was induced by left coronary artery ligation as described earlier (Sjaastad et al., 2000). Sham-operated rats (Sham) went through the same procedure except ligation of the left coronary artery. Six weeks later, rats with heart failure (HF) with a left atrial diameter >5 mm and increased lung weight (>2.0 g) in the presence of a large anterolateral infarction were included in the study. In addition, echocardiographic analysis was performed in a subset of animals during anesthesia (68% N2O, 29% O2, and 2–3% isoflurane through endotracheal intubation), which revealed marked left ventricular dilatation (left ventricular diameter in diastole, 9.9 ± 0.2 mm) and increased left ventricular end-diastolic blood pressure (26 ± 1 mm Hg). Before sacrifice, all animals were sedated in an anesthesia chamber (64% N2O, 32% O2, and 4% isoflurane) and further ventilated through endotracheal intubation, maintaining anesthesia with 68% N2O, 29% O2, and 2–3% isoflurane. Adequate and deep surgical anesthesia was obtained in the animal when spontaneous respiration and response to pain stimulation were reabolished. The chest was opened, and the heart was then rapidly excised, weighed, and rapidly perfused retrogradely for preparation of muscle strips or isolated cardiomyocytes. See Table 1 for detailed animal characteristics.
Animal characteristics and echocardiographic and hemodynamic data of heart failure hearts
Echocardiographic and hemodynamic characteristics were measured in a subset of animals.
Isolation of Cardiomyocytes.
Failing hearts were perfused (Langendorff setup) and digested enzymatically with trypsin and collagenase type II (90 U/ml final) (268 U/mg; Worthington Biochem. Corp., Lakewood, NJ) as described in the Supplemental Methods. Cardiomyocytes used in Ca2+ transient measurements were isolated by a slightly different protocol as previously described (Bøkenes et al., 2008). Isolated cardiomyocytes were used for experiments on the day they were prepared.
Isolated Muscle Strips.
HF left ventricular muscle strips were prepared and stimulated at 1 Hz. Contraction-relaxation cycles were recorded and analyzed as previously described (Sjaastad et al., 2003) with respect to maximal development of force [(dF/dt)max], time to peak force (TPF), time to 80% relaxation (TR80), and relaxation time (RT = TR80 − TPF). Inotropic responses were expressed as change of (dF/dt)max in percent of basal, and NIR defined as the decrease in (dF/dt)max. LR was expressed as decrease of RT in percent of basal. Isolated muscle strips were taken distant from the infarction area.
Ca2+ Transient Measurements.
Isolated cardiomyocytes were loaded for 10 minutes in fluo-4-AM and plated to coverslips with laminin. Whole-cell Ca2+ transients were recorded in field-stimulated cardiomyocytes at 1 Hz and 37°C using a photomultiplier (Photon Technology International, Edison, NJ) as previously described (Bøkenes et al., 2008). Only rod-shaped, striated cardiomyocytes with regular contractions were included. Baseline Ca2+ transients were recorded for at least 180 seconds before either BNP or CNP (300 nM) was added in two different sets of experiments. BNP- and CNP-induced alterations in Ca2+ transients were recorded until new steady state in Ca2+ transients was achieved, typically within 5–8 minutes. Background fluorescence was measured in each experiment. Ca2+ transient amplitudes (F/F0) were calculated as the ratio between maximal (F) and resting (F0) fluorescence. Ca2+ extrusion was expressed as 1/τ (s−1), where τ was calculated by monoexponential fitting of the decay of single Ca2+ transients. To measure sarcoplasmatic reticulum (SR) Ca2+ load, in a separate set of experiments, the field stimulation was stopped and caffeine (10 mM) was rapidly applied. SR Ca2+ load was interpreted as the ratio between peak and basal fluorescence in the caffeine-induced Ca2+ release. BNP and CNP responses were interpreted as the change in SR Ca2+ load by BNP or CNP in the superfusate.
L-Type Ca2+ Channel Current.
Voltage-clamp experiments were performed using an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA). The holding potential was −45 mV, and L-type Ca2+ currents were triggered by a 100-millisecond square voltage step from −45 to 0 mV at 0.125 Hz as described in the Supplemental Methods.
cGMP Assays and Western Blot.
Muscle strips were freeze-clamped 15 minutes after the addition of agonist and kept at −70°C until use in cGMP assay. Isolated cells were preincubated with PDE inhibitors for 15 minutes and stimulated for 10 minutes with agonist as indicated. cGMP was measured using the cGMP enzyme immunoassay kit (Cat no. 581021) from Cayman Chemical Company (Ann Arbor, MI). PLB Ser16 and TnI Ser23/24 phosphorylation were measured by Western blot analysis using antibodies for total and phosphorylated protein.
Statistics.
All results are presented as mean ± S.E.M. Statistical significance was calculated by paired or unpaired Student’s t test or one sample t test where appropriate. Bonferroni corrections were made when relevant. Two-way analysis of variance was performed where relevant. P < 0.05 was considered to reflect significant differences (*P < 0.05; **P < 0.01; ***P < 0.001).
Results
BNP Increases cGMP but Does Not Cause NIR or LR in Left Ventricular Muscle Strips Compared with CNP.
We have earlier shown a concentration-dependent increase of cGMP by CNP in HF left ventricular muscle strips (Moltzau et al., 2013). We now also show that BNP increased cGMP concentration-dependently in isolated HF muscle strips to a similar extent as CNP. The cGMP increase by BNP leveled off toward 300 nM BNP (Fig. 1A). Despite BNP-induced cGMP increase, the contractility and relaxation after 300 nM BNP were not significantly different from normal decay in the time-matched controls (Ctr) (Fig. 1, B and Ci). The response to BNP seemed to have two different components as cGMP increased further at a higher BNP concentration (1 µM), possibly caused by loss of selectivity between NPR-A and NPR-B above approximately 300 nM (Fig. 1A). However, this does not fit easily with the lack of NIR and LR at this concentration of BNP (Fig. 1C). CNP caused both an NIR and an LR (Fig. 1, B and Cii), which was significantly different compared with equal concentrations of BNP (P < 0.05 at all concentration by multiple two-way analysis of variance). As in HF, BNP (300 nM) did not cause significant NIR or LR in sham-operated rats compared with time-matched controls (Fig. 4B), whereas CNP caused functional responses in line with Moltzau et al. (2013) (data not shown). Thus, despite cGMP-increase by both BNP and CNP in failing hearts, BNP did not cause NIR or LR compared with CNP.

BNP increases cGMP, but it does not elicit functional responses, as opposed to CNP. (A) cGMP levels in HF left ventricular strips stimulated with increasing concentrations of BNP for 15 minutes. Extended BNP curve is extrapolated by GraphPad Prism (GraphPad Software, La Jolla, CA) based on cGMP values for BNP (Ctr, 50, 100, and 300 nM) theoretically selective for NPR-A over NPR-B (n = 6). The cGMP increase by BNP leveled off toward 300 nM BNP, but it increased further at a higher BNP concentration (1 µM). (B) Illustration of a contraction-relaxation cycle (signal averaged from about 25 cycles) when stimulating a representative HF left ventricular muscle strip with BNP (300 nM) or CNP (200 nM) compared with basal. (C) Maximal NIR [−(dF/dt)max, % of basal] and LR (−ΔRT, % of basal) were measured in HF left ventricular strips in the presence of increasing concentrations of BNP (i) or CNP (ii). Number of experiments is indicated in the figure. **P < 0.01; ***P < 0.001 versus Ctr.
cGMP Elevations by BNP and CNP Are Additive in HF Ventricular Cardiomyocytes.
BNP and CNP significantly increased the cGMP level in isolated ventricular cardiomyocytes compared with controls (Fig. 2A), indicating the presence of both NPR-A and NPR-B in the isolated ventricular cardiomyocytes from HF rats. When BNP (100 nM) and CNP (100 nM) were combined, the effects on the cGMP level were additive (no difference between BNP + CNP and calculated BNP + CNP in Fig. 2A), compatible with independently-induced cGMP-increase by the two NPs. Maximal receptor-selective concentration of BNP (300 nM) and CNP (1 µM) caused a cGMP increase of 2020 ± 380 fmol cGMP/mg protein (n = 6, P < 0.01 versus Ctr) and 1900 ± 220 fmol cGMP/mg protein (n = 6, P < 0.001 versus Ctr), respectively. At these concentrations, BNP and CNP together (BNP + CNP) showed a nominally, but not significantly, lower cGMP elevation than the sum of separate BNP and CNP stimulation (calculated BNP + CNP; Fig. 2B). Considering the different functional responses to BNP and CNP, our results are consistent with different cGMP compartments generated by the two NPs.

The combination of BNP and CNP is additive on cGMP levels, but BNP affects only lusitropic response to maximal CNP. (A and B) Cyclic GMP levels in isolated HF ventricular cardiomyocytes stimulated for 10 minutes with either BNP or CNP or both (BNP + CNP). Calculated BNP + CNP: sum of separate BNP and CNP stimulation. (A) 100 nM BNP, 100 nM CNP (n = 5), (B) 300 nM BNP, 1 µM CNP (n = 5–6). (C and D) Concentration-response curves of inotropic response (C, normalized) and lusitropic response (D, % of basal) to CNP in the absence and presence of BNP (300 nM). HF left ventricular muscle strips were preincubated for 20 minutes with BNP before the first dose of CNP was added. n = 8. NS, not significant, *P < 0.05; **P < 0.01; ***P < 0.001.
BNP Does Not Influence the NIR to CNP but Increases the LR to Maximal CNP in Left Ventricular Muscle Strips.
To elucidate whether the lack of NIR and LR to BNP was due to an NPR-A–dependent inhibitory component attenuating the possible effects on contractility, the functional responses to CNP were studied in left ventricular muscle strips preincubated with BNP (300 nM). Neither the −logEC50 to CNP of the NIR nor the NIR to maximal CNP was affected by BNP preincubation (Fig. 2C). On the contrary, the LR to maximal CNP was significantly increased in the presence of BNP (ΔLRmax = 4.1% ± 1.6%, n = 8, P < 0.05), but BNP stimulation did not affect the −logEC50 of CNP (Fig. 2D) for the LR. BNP at 300 nM did not, however, cause any additional NIR or LR to maximal CNP when added to the muscles after CNP preincubation (data not shown).
cGMP Elevation by BNP Is Markedly and Mainly Regulated by PDE2 in Ventricular Cardiomyocytes.
To elucidate the regulation of cGMP generated by NPR-A, experiments with BNP (100 nM) were performed in ventricular HF cardiomyocytes in the presence of PDE2 (EHNA, 10 µM), PDE3 (Cil, 1 µM), and PDE5 (Sfil, 0.1 µM) inhibition, separately or in combination. A series of experiments showed no significant cGMP increase in the presence of PDE inhibitors (EHNA, Cil, Sfil, or the different combinations) alone [i.e., Ctr (167 ± 56 fmol cGMP/mg protein, n = 8) versus EHNA, Cil, and Sfil (278 ± 67 fmol cGMP/mg protein, n = 8), nonsignificant (NS)]. BNP in the presence of the PDE2 inhibitor EHNA caused an increase (P < 0.05) compared with BNP alone (Fig. 3). BNP in the presence of Cil, Sfil, or in combination did not cause a significant increase in cGMP compared with BNP alone (all NS). BNP in the presence of EHNA and Cil, EHNA and Sfil, and EHNA, Cil, and Sfil caused no significant cGMP elevation beyond what was seen with BNP in the presence of EHNA alone. Our results suggest that cGMP increase by BNP is markedly and mainly regulated by PDE2.

BNP causes a large cGMP increase in the presence of a PDE2 inhibitor. cGMP levels in isolated HF ventricular cardiomyocytes after BNP (100 nM) stimulation in the absence (Ctr) and the presence of individual PDE2 (EHNA, 10 µM), PDE3 (Cil, 1 µM), or PDE5 (Sfil, 0.1 µM) inhibition or with combined PDE3 and -5; PDE2 and -3; PDE2 and -5; or PDE2, -3, and -5 inhibition. Number of experiments is indicated in the figure. *P < 0.05; ***P < 0.001.
PDE2, PDE3, and PDE5 Inhibition Did Not Reveal Any NIR or LR to BNP in Left Ventricular Muscle Strips.
Despite their similar ability to increase cGMP, only CNP, and not BNP, modulated contractility. Thus, we tested whether PDEs could constitute a barrier between the cGMP generated by BNP and the contractile machinery. We preincubated left ventricular muscle strips with the PDE inhibitors EHNA, Cil, and Sfil and challenged the muscle strips with BNP (300 nM). However, there was no significant difference in HF or Sham rats or between the effect of BNP in the presence of combined PDE2, -3, and -5 inhibition (NS versus BNP alone) and BNP alone (NS versus Ctr; Fig. 4, A and B).
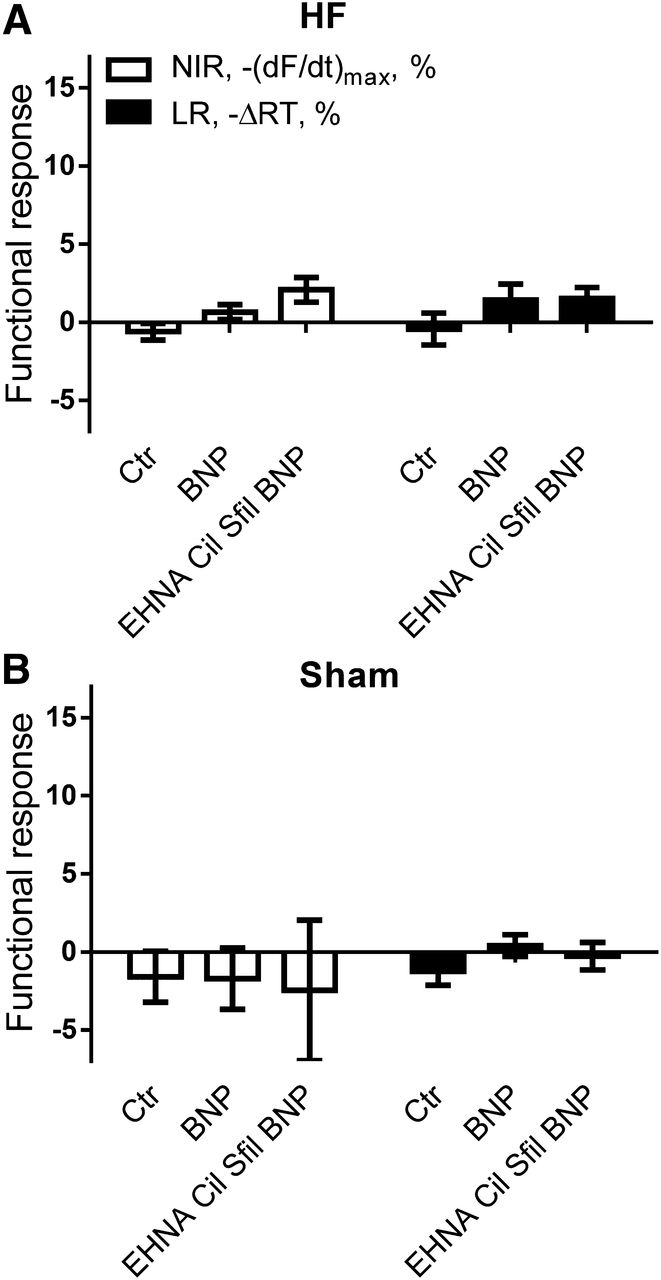
Combined PDE2, PDE3, and PDE5 inhibition does not reveal an NIR or an LR to BNP. Maximal NIR [−(dF/dt)max, %] and LR (−ΔRT, %) in HF (A) and Sham (B) left ventricular muscle strips stimulated with BNP (300 nM) in the absence (Ctr) and presence of PDE2, 3, and 5 inhibition. EHNA: 10 µM; Cil: 1 µM; Sfil: 0.1 µM. (A) n = 4–6; (B) n = 6–8.
Different Influence of BNP Compared with CNP on PLB Ser16 and TnI Ser23/24 Phosphorylation in HF Ventricular Cardiomyocytes.
As both BNP and CNP increase cGMP and BNP failed to cause an NIR or LR even in the presence of PDE2, -3, and -5 inhibition in left ventricular muscle strips, we wanted to investigate further whether BNP and CNP differentially affected downstream targets and the effects of cGMP in the failing heart. We measured PLB Ser16 and TnI Ser23/24 phosphorylation in the presence of BNP and/or CNP in isolated HF ventricular cardiomyocytes. BNP (300 nM) did not cause any significant increase neither on PLB Ser16 nor TnI Ser23/24 phosphorylation. Confirming our previous results (Moltzau et al., 2013), CNP (1 µM) caused a more than 20-fold increase in PLB Ser16 phosphorylation and about a 2-fold increase in TnI Ser23/24 phosphorylation compared with controls (Fig. 5, A and B). The combination of BNP and CNP (BNP + CNP) did not cause any further increase in PLB Ser16 or TnI Ser23/24 phosphorylation compared with CNP alone. However, in the presence of PDE2, -3, and -5 inhibition (EHNA, Cil, Sfil), BNP caused a nominal but nonsignificant increase in PLB Ser16 phosphorylation (Fig. 5A).

BNP does not increase PLB and TnI phosphorylation, whereas CNP does. (A and B) Phosphorylation of PLB at Ser16 (A) and TnI at Ser23/24 (B) in isolated HF ventricular cardiomyocytes stimulated with BNP (300 nM) or/and CNP (1 µM). BNP stimulation was also done in the presence of PDE2, -3, and -5 inhibition (EHNA, Cil, Sfil) (n = 4); EHNA: 10 µM; Cil: 1 µM; Sfil: 0.1 µM. This figure shows representative Western blots of the expression of total (PLB/TnI) and phosphorylated (p-PLB/p-TnI) protein. The excised bands show monomeric PLB at about 5 kDa and TnI at 28kDa.
Different Regulation of Ca2+ Transients and SR Ca2+ Load, but Not L-Type Ca2+ Current, by BNP and CNP in Failing Ventricular Cardiomyocytes.
We next tested the functional effects of BNP and CNP on Ca2+ transients in isolated HF ventricular cardiomyocytes. BNP (300 nM) decreased the Ca2+ transient magnitude (P < 0.05; Fig. 6C) gradually over a period of 5–8 minutes before reaching a steady state (Fig. 6A). The Ca2+ extrusion rate was not altered by BNP (Fig. 6D). In contrast, the addition of CNP (300 nM) increased the Ca2+ transient magnitude (F/F0) (in a biphasic manner with a first increase within approximately 60 seconds before a second steady state, P < 0.01; Fig. 6C). At steady state after CNP stimulation, the Ca2+ extrusion rate constant was increased (Fig. 6D). The Ca2+ transient response was significantly different between BNP and CNP stimulation in isolated cardiomyocytes, consistent with a compartmented effect of BNP and CNP on the Ca2+-handling proteins. To evaluate more fully the compartmented effects produced by CNP and BNP, we next examined two main determinants of Ca2+ transients in cardiomyocytes, namely, L-type Ca2+ channel currents and SR Ca2+ load. As shown in Fig. 6, E and F, the peak amplitude of the L-type Ca2+ channel current was reduced by both BNP (P < 0.05 versus Ctr) and CNP (P < 0.01 versus Ctr), in contrast to the SR Ca2+ load, which was increased by CNP and unaltered by BNP.

Ca2+ transients, Ca2+ extrusion rate, and SR Ca2+ load, but not L-type Ca2+ current, are differently regulated by BNP and CNP in failing ventricular cardiomyocytes. (A) Representative recordings of Ca2+ transients before and after the addition of BNP (left) or CNP (right) to the superfusate. (B) Representative single Ca2+ transients (average of five recordings) in baseline and after BNP (left) and CNP (right) stimulation. (C and D) Change of the Ca2+ transients (C) and the Ca2+ extrusion rate constant (1/τ) (D) after stimulation with BNP (n = 7) or CNP (n = 8). (E) Change of SR Ca2+ load as a measure of the increase of the Ca2+ content in the SR by BNP (n = 14) or CNP (n = 6). (F) Change of the peak of the L-type Ca2+ current after stimulation with BNP (n = 5) or CNP (n = 6). BNP, CNP: 300 nM. *P < 0.05; **P < 0.01; ***P < 0.001 BNP versus CNP.
Discussion
The main and novel finding in this report is the different ability of BNP and CNP to regulate myocardial contractility and Ca2+ transients, despite comparable increases in cGMP levels in failing hearts. We did not find an NIR or an LR to BNP in either HF or Sham, as opposed to CNP. Further experiments in failing hearts showed that cGMP elevations by BNP and CNP were additive. Further, BNP did not increase phosphorylation of PLB Ser16 and TnI Ser23/24 or the rate of Ca2+ extrusion from cytosol or SR Ca2+ load, whereas CNP did. However, BNP decreased, whereas CNP increased, the peak of the Ca2+ transient. Both NPs reduced the peak L-type Ca2+ channel current. In the presence of BNP, CNP-induced LR was increased, but not the NIR. The total cGMP level generated by BNP was markedly and mainly regulated by PDE2. Inhibition of PDE2, -3, and -5 nominally increased the phosphorylation of PLB Ser16 to BNP. Despite this finding, an NIR or LR to BNP was not revealed in the presence of PDE2, -3, and -5 inhibition. Thus, our results suggest that CNP and BNP signal in different compartments in failing rat left ventricle.
Role of PDEs in Compartmentation of the cGMP Signal after BNP Stimulation.
We earlier had found that the NIR and LR to CNP are regulated by PDEs (Moltzau et al., 2014). A possible explanation for the lack of NIR and LR to BNP could be PDE barriers protecting the contractile machinery from NPR-A–generated cGMP. Even though PDE2 inhibition more than doubled BNP-stimulated cGMP levels and combined PDE2, -3, and -5 inhibition revealed a nominal, although nonsignificant, increase in PLB Ser16 phosphorylation, no NIR or LR to BNP was revealed. Thus, PDE barriers are not a sufficient explanation for the inability of BNP stimulation to reach the same functional compartment as CNP stimulation, unless PDEs other than the ones tested could be involved. The cGMP increase by both BNP (this study) and CNP (Moltzau et al., 2014) is markedly and mainly regulated by PDE2, in line with earlier studies (Castro et al., 2006). Still, the effect of PDE2 inhibition on the cGMP elevation was significantly higher for CNP than BNP (P < 0.05), despite comparable cGMP elevation with the two NPs alone. This may also be consistent with different compartmentation of cGMP generated from the two receptors, with apparently less PDE2 activity in the NPR-A compartment than in the NPR-B compartment.
Compartmentation of the Signaling Pathways after BNP and CNP Stimulation.
BNP and CNP exerted different effects on downstream targets (PLB and TnI) and effects (Ca2+ transients, NIR, and LR) of cGMP, despite similar cGMP levels. In line with this, others have found effects of CNP on contractility and PLB phosphorylation in normal hearts (Pierkes et al., 2002; Frantz et al., 2013), but not of ANP, which stimulates the same receptor as BNP (NPR-A). We previously found that PLB Ser16 and TnI Ser23/24 phosphorylation most likely represent the main mechanisms for the cGMP-PKG–mediated NIR and LR to CNP in failing hearts (Moltzau et al., 2013). Whether the NIR to BNP seen in some studies is induced by the same mechanisms as CNP is not clear. Previous studies have suggested a role for the Na+/H+ exchanger in the ANP-induced NIR (Tajima et al., 1998) and a common PKG-mediated mechanism for the NIR induced by ANP, BNP and CNP (Zhang et al., 2005). A key finding in our study is that whereas CNP increases PLB phosphorylation and causes associated increase in Ca2+ transients, SR Ca2+ load and Ca2+ extrusion in cardiomyocytes, BNP increases neither. The slightly decreased amplitude of the Ca2+ transient seen with BNP in our study is in accordance with the corresponding finding of reduced L-type Ca2+ channel current, which is in line with reports of reduced L-type Ca2+ channel currents by NPs (Tohse et al., 1995; Rose and Giles, 2008; Sodi et al., 2008) and could in theory contribute to a NIR. The lack of an NIR, however, indicates either that the effect is too small or that it is opposed by other, unknown effects. Thus, we interpret our results showing different effects by BNP and CNP on NIR and LR, PLB, and TnI phosphorylation, Ca2+ amplitude, and Ca2+ extrusion rate despite similar effects on cGMP levels to indicate clearly that the two NPs exert their signaling in different compartments. Furthermore, our data indicate that the different functional response to BNP and CNP on Ca2+ transients and contractility is not due to compartmented regulation of L-type Ca2+ channels, as both NPs reduced the L-type Ca2+ channel current. As CNP, and not BNP, was able to increase SR Ca2+ load, we suggest that different functional response on cellular Ca2+ transients in failing cardiomyocytes is due to compartmented regulation of SR Ca2+ cycling by CNP and BNP.
Lack of Correlation between Functional Responses and cGMP Increase.
We recently showed that CNP, but not BNP, increased β1-adrenergic receptor (Qvigstad et al., 2010; Afzal et al., 2011) and 5-HT4 serotonin receptor–induced inotropic responses (Afzal et al., 2011) through cGMP-inhibition of PDE3. This study and other studies with ANP and CNP (Pierkes et al., 2002; Frantz et al., 2013) support our present results that BNP and CNP signal in different functional compartments. The discrepancy between BNP and CNP on NIR and LR, despite both increasing cGMP, is analogous to findings in the cAMP pathway, where PGE1 and isoprenaline both increased cAMP, but only isoprenaline showed contractile effects (Kaumann and Birnbaumer, 1974; Hayes et al., 1979). The reason for the differential functional compartmentation by BNP and CNP could be different localization of the receptors on the cell membrane combined with restricted diffusion of cGMP. Several studies address the role of A-kinase anchoring proteins in compartmentation of cAMP-mediated signals (Carnegie et al., 2009), but a similar family of PKG-anchoring proteins is not identified (Francis et al., 2010). However, cGMP-dependent kinase interacting proteins can localize PKG to certain regions of the cell (Francis et al., 2010). Physical barriers restricting diffusion of Na+ and Ca2+ near the plasma membrane in cardiomyocytes have been suggested (Lederer et al., 1990; Aronsen et al., 2013) and could also apply to the diffusion of cyclic nucleotides. Further studies are needed to determine whether there are different binding proteins for NPR-A and NPR-B, like PKG-anchoring proteins or cGMP-dependent kinase interacting proteins; whether there are structural barriers separating the signaling from the two receptors; or whether other mechanisms are involved in the compartmentation of NPR-A and NPR-B signaling.
BNP Increased the CNP-Induced Lusitropic Response.
The lack of NIR and LR to BNP could imply the generation of a component induced by BNP inhibiting the signaling pathway, in line with others showing that lack of NPR-A increased CNP effects on contractility and PLB phosphorylation in normal mouse hearts, possibly correlated to increased PKGI expression (Pierkes et al., 2002). However, the lack of NIR and LR to BNP in our study is not likely explained by such a component, as BNP did not reduce CNP-induced NIR or LR. On the contrary, we found that BNP increased the LR to maximal CNP, even though BNP did not affect CNP-induced phosphorylation of PLB Ser16 and TnI Ser23/24. However, the effect of BNP on the CNP-induced LR might be explained by the effect of BNP on other cGMP-mediated functions. Such an effect of BNP must then be present before stimulation by CNP as we showed no additional LR when BNP was given to the muscles after CNP preincubation. However, we cannot rule out that the BNP effect on LR is mediated by cGMP-independent effects through NPR-C (Rose and Giles, 2008), but the effect is not easily explained by possible Gi signaling.
Therapeutic Use of Natriuretic Peptides.
Since the initial promising results of the BNP analog nesiritide (Colucci et al., 2000), there has been great interest in using NPs in therapy for heart failure (Boerrigter et al., 2009). Even though the BNP analog nesiritide did not give a survival benefit (O’Connor et al., 2011), studies are still evaluating CD-NP, a combined NPR-A and NPR-B agonist (Hobbs, 2014). In our experiments, BNP did not have any contractile effect, whereas CNP did. Cyclic GMP–mediated effects through PKG are suggested to be beneficial in HF (Boerrigter et al., 2009). However, the increased sarcoplasmatic reticulum Ca2+-ATPase 2 (SERCA2) activity by CNP, potentially causing futile Ca2+ cycling (Moltzau et al., 2013), might be an energy-demanding process. Although disputable, the increased SERCA2 activity by CNP might also be a protective mechanism as increased SERCA2 activity is suggested as therapy in HF (Gwathmey et al., 2011). The effects of CNP in the heart are complex, as we previously showed increased β1-adrenergic receptor–induced, cAMP-mediated inotropic responses in the presence of CNP (Qvigstad et al., 2010), a potentially harmful effect. Thus, when evaluating new drugs that target NP signaling, direct effects on the heart should be taken into consideration (Levy, 2013; Bach et al., 2014).
Conclusion.
Our results show extensive functional evidence of differential compartmentation of BNP- and CNP-induced cGMP with different influences on downstream targets and effects (Fig. 7). We conclude that BNP and CNP signal in different compartments, which again indicates different roles for the two NPs in the failing heart.

Illustration of cGMP compartmentation in cardiomyocytes after BNP and CNP stimulation. 1) Separate BNP and CNP stimulation: BNP and CNP increase cGMP in different compartments where CNP causes functional responses, whereas BNP does not, even when cGMP degradation by PDEs is abolished. 2) CNP stimulation in the presence of BNP stimulation. BNP increases the CNP-induced LR. Dotted lines represent hypothetical signaling pathways of BNP through NPR-A and NPR-C.
Acknowledgments
The authors thank Iwona Gutowska Schiander, Cam H. T. Nguyen, and Marie Dahl for excellent technical assistance, and Ståle Nygård for statistical analysis.
Authorship Contributions
Participated in research design: Moltzau, Aronsen, Meier, Skogestad, Skomedal, Osnes, Levy, Qvigstad.
Conducted experiments: Moltzau, Aronsen, Meier, Skogestad, Ørstavik, Lothe, Qvigstad.
Performed data analysis: Moltzau, Aronsen, Skogestad, Lothe, Sjaastad, Qvigstad.
Wrote or contributed to the writing of the manuscript: Moltzau, Aronsen, Meier, Skogestad, Ørstavik, Lothe, Sjaastad, Skomedal, Osnes, Levy, Qvigstad.
Footnotes
- Received March 24, 2014.
- Accepted July 8, 2014.
This work was supported by The Norwegian Council on Cardiovascular Disease; The Research Council of Norway; South-Eastern Norway Regional Health Authority; The Kristian Gerhard Jebsen Foundation; Anders Jahre’s Foundation for the Promotion of Science; The Family Blix Foundation; The Simon Fougner Hartmann Foundation; University of Oslo; and the COST Action BM1005 (European Network on Gasotransmitters).
This work, with some modifications, was part of a Ph.D. thesis: Moltzau LR (2012) Effects of Natriuretic Peptides on Contractility and cGMP Compartmentation in Failing Rat Myocardium. Ph.D. thesis, Faculty of Medicine, University of Oslo, Oslo, Norway.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- ANP
- atrial natriuretic peptide
- BNP
- brain natriuretic peptide
- Cil
- cilostamide
- CNP
- C-type natriuretic peptide
- EHNA
- erythro-9-(2-hydroxy-3-nonyl)adenine hydrochloride
- HF
- heart failure
- LR
- lusitropic response
- NIR
- negative inotropic response
- NP
- natriuretic peptide
- NPR
- natriuretic peptide receptor
- NS
- nonsignificant
- PDE
- phosphodiesterase
- PKG
- protein kinase G
- PLB
- phospholamban
- SERCA2
- sarcoplasmatic reticulum Ca2+-ATPase 2
- Sfil
- sildenafil
- SR
- sarcoplasmatic reticulum
- TnI
- troponin I
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}