


Abstract
N-Arachidonoylethanolamine (AEA) is a proposed endogenous ligand of the central cannabinoid receptor (CB1). Previous studies indicate that AEA is translocated across membranes via a process that has the characteristics of carrier-mediated facilitated diffusion. To date, studies of this mechanism have relied on [3H]AEA as a substrate for the carrier. We have synthesized an analog of AEA, SKM 4-45-1, that is nonfluorescent in the extracellular environment. When SKM 4-45-1 is exposed to intracellular esterases, it is de-esterified and becomes fluorescent. We have carried out studies to demonstrate that SKM 4-45-1 accumulation in cells occurs via the AEA carrier. SKM 4-45-1 is accumulated by both cerebellar granule cells and C6 glioma cells. Uptake of SKM 4-45-1 into C6 glioma is inhibited by AEA (IC50=53.8 ± 1.8 μM), arachidonoyl-3-aminopyridine amide (IC50=10.1 ± 1.4 μM), and arachidonoyl-4-hydroxyanilineamide (IC50=6.1 ± 1.3 μM), all of which also inhibit [3H]AEA accumulation. Conversely, [3H]AEA accumulation by cerebellar granule cells is inhibited by SKM 4-45-1 with an IC50 of 7.8 ± 1.3 μM. SKM 4-45-1 is neither a substrate nor inhibitor of fatty acid amide hydrolase, an enzyme that catabolizes AEA. SKM 4-45-1 does not bind the CB1 cannabinoid receptor at concentrations <10 μM. In summary, the cellular accumulation of SKM 4-45-1 occurs via the same pathway as AEA uptake and provides an alternative substrate for the study of this important cellular process.
N-Arachidonoylethanolamine (AEA) was isolated from porcine brain and has been postulated to be an endogenous ligand of the central cannabinoid receptor (CB1) (Devane et al., 1992). AEA administration to animals produces the physiological effects characteristic of the classical cannabinoids, including the tetrad of hypothermia, analgesia, catalepsy, and decreased locomotion (Fride and Mechoulam, 1993). Biochemical effects of AEA are similar to those of other CB1 agonists, including inhibition of adenylyl cyclase activity (Vogel et al., 1993) and inhibition of voltage-operated calcium channels (Mackie et al., 1993). These effects are blocked by the CB1 antagonistN-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride (Rinaldi-Carmona et al. 1994, 1995; Jung et al., 1997). AEA synthesis and release from neuronal cells in culture is dependent on increased intracellular calcium (DiMarzo et al., 1994), further support its role as a neurotransmitter or neuromodulator.
AEA is accumulated by neurons and the accumulation is rapid and temperature-sensitive. Previous work done in our laboratory has extended these studies and demonstrated that AEA accumulation by neurons occurs by facilitated diffusion, is bidirectional, and is inhibited by phloretin (Hillard et al., 1997). TheKm andVmax for AEA transport were determined previously to be 41 ± 15 μM and 0.61 ± 0.04 nmol/min/106 cells, respectively (Hillard et al., 1997). AEA uptake also is inhibited competitively by several AEA analogs, including benzylarachidonamide (Hillard et al., 1997) arachidonoyl-3-aminopyridine amide (A3AP) (Muthian et al., 1998), and arachidonoyl-4-hydroxyaniline amide (AM404) (Beltramo et al., 1997;Calignano et al., 1998) but not by arachidonic acid orN-palmitoylethanolamine (PEA) (DiMarzo et al., 1994). Cellular uptake of AEA has been demonstrated in a lymphoma cell line (Maccarone et al., 1998) and neuroblastoma cells (Deutsch and Chin, 1993; Maccarone et al., 1998). Although the biochemical evidence supports a carrier protein-mediated uptake of AEA, many questions remain regarding this process; including the identification of the carrier protein itself and whether AEA uptake is regulated by other signaling events in the cell.
Studies of the uptake carrier in the past have relied exclusively on the use of radiolabeled AEA. The aim of the present studies was to synthesize and characterize a fluorescent analog of AEA with which to study the uptake mechanism. Previous studies have indicated that binding of substrates to the AEA uptake carrier is sensitive to modifications of AEA in the arachidonate moiety (Hillard et al., 1997;Piomelli et al., 1999), whereas changes in the ethanolamide region were well tolerated (Muthian et al., 1998, Piomelli et al., 1999). Based on this information, we designed SKM 4-45-1, an analog of AEA that is conjugated to fluorescein diacetate via the primary alcohol. Previous studies have shown that fluorescein diacetate is nonfluorescent until it is de-esterified by esterases within cells (Rothman and Papermaster, 1966). Therefore, fluorescence develops after cellular accumulation and de-esterification.
With the studies reported herein, we describe the synthesis and characterization of SKM 4-45-1 as a substrate for the AEA carrier. SKM 4-45-1 is nonfluorescent until modification by cellular esterases, and it inhibits [3H]AEA uptake into cells. Conversely, SKM 4-45-1 uptake into C6 glioma cells in culture is inhibited by AEA and analogs of AEA known to inhibit AEA uptake. Therefore, our data suggests SKM 4-45-1 transport across cell membranes occurs via the AEA uptake carrier.
Materials and Methods
Preparation of Cerebellar Granule Cells (CGCs) and C6 Glioma Cells.
Cell experiments were carried out with CGCs or C6 glioma cells. CGCs were cultured from 6- to 8-day old Sprague-Dawley rat pups as described in Hillard et al. (1997). Cells were maintained in basal minimal Eagle's media (Life Technologies, Gaithesburg, MD) supplemented with 10% heat-inactivated fetal bovine serum, 5 mM glutamine, 25 mM KCl, 0.1 mg/ml gentamicin, and 0.01 mg/ml ampicillin. Cells were grown at 37°C in 5% CO2- supplemented room air. Cytosine arabinoside (10 μM) was added at 24 h to inhibit growth of proliferating cells. Cells were used in experiments after 7 to 8 days in culture. C6 glioma cells (America Type Tissue Culture, Rockville, MD) were grown in Dulbecco's minimum essential medium with 10% fetal bovine serum. Confluent C6 glioma cells were used in these studies.
SKM 4-45-1 Synthesis.
SKM 4-45-1 was synthesized from AEA (Cayman Chemicals, Ann Arbor, MI) and fluorescein-5-carbonyl azide diacetate (Molecular Probes, Eugene, OR). AEA in ethanol was dried under nitrogen and resuspended in anhydrous toluene. Excess fluorescein-5-carbonyl azide was introduced into the vial containing AEA and allowed to react at 80oC for 3 h. The reactants were dried under nitrogen and separated on a silica gel thin-layer chromatography plate with 60:40 ethyl acetate:hexane as the mobile phase. The product band was identified with UV illumination, and extracted with methylene chloride and ethyl acetate. We confirmed the structural identity of SKM 4-45-1 by 1H NMR: (CDCl3, 300 Mhz) d 8.10 (s, 1H), 7.74 (br, s, 1H), 7.37 (s, 1H), 7.15 (s, 1H) 7.17 (d, 2H J = 5.8 Hz), 7.07 (m, 4H), 5.92 (s, 1H), 5.37 (m, 8H), 4.33 (m, 2H), 3.62 (m, 2H), 2.80 (m, 6H), 2.34 (m, 6H), 2.24 (d, 2H J = 7.6 Hz), 2.09 (m, 4H), 1.75 (m,2H), 1.31 (m, 6H), 0.89 (t, 3H J = 6.7 Hz).
Enzymatic Modification of SKM 4-45-1 by Cell Lysates.
C6 glioma cells in culture were lysed with ice-cold TME buffer (50 mM Tris-HCl, 1 mM EDTA, and 3 mM MgCl2, pH 7.4) and centrifuged at 1000g for 10 min. Increasing amounts of the supernatant was incubated with 100 nM SKM 4-45-1 and the fluorescence was monitored at 490-nm excitation and 519-nm emission on a Perkin-Elmer luminescence spectrometer LS50.
Measurement of AEA Uptake by Cells.
The ability of SKM 4-45-1 to inhibit the cellular uptake of AEA was carried out according to methods described previously (Hillard et al., 1997). CGCs (7–9 days in vitro) were washed with KRH buffer (118 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, and 10 mM HEPES, pH 7.4) and incubated with fresh KRH buffer containing 40 pM [3H]AEA (223 Ci/mmol; NEN, Boston, MA) in the presence of increasing concentrations of SKM 4-45-1. At 2 min, the buffer was removed and the cells were lysed and scraped into water; the radioactivity of both buffer and lysates was determined. Cellular accumulation of [3H]AEA was calculated as the fraction of the total radioactivity in the cells.
Measurement of SKM 4-45-1 Uptake by C6 Glioma Cells.
Confluent C6 glioma cells in culture were washed free of media and incubated in KRH buffer for 30 min. The buffer was removed and replaced with KRH buffer containing 100 nM SKM 4-45-1 and increasing concentrations of competitors in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide was 1%, which had no effect on AEA transport (unpublished data). Fluorescence was quantified with a Cytofluor II fluorescence microplate reader (excitation 490 nm, emission 525 nm).
Fluorescence Microscopy.
CGCs were washed free of media and incubated in KRH buffer for 30 min. The buffer was then replaced with fresh buffer containing 100 nM SKM 4-45-1 alone or in the presence of either 100 μM AEA or PEA. Twenty minutes after treatment, cells were exposed to a mercury fluorescence light source and emission was captured on a Sensys Photometrics charge-couple device camera; the same field also was imaged under white light. Images were obtained at 40× magnification on a Nikon diaphot microscope with 480-nm excitation and 530-nm emission filters.
[3H]CP55940-Binding Assay.
Radioligand binding to CB1 receptors was determined with rat forebrain membranes according to the methods described in Hillard et al. (1995a). Membranes (10 μg) were incubated with [3H]CP55940 (120 Ci/mmol; NEN), and varying concentrations of SKM 4-45-1 in 0.2 ml TME buffer containing 1 mg/ml BSA. Incubations were carried out in a Multiscreen filtration system with Durapore 1.2-μm filters (Millipore, Bedford, MA). Incubation was terminated at 1 h by filtration and washed three times with 150 μl of cold BSA-containing buffer. Bound radioactivity was determined by liquid scintillation counting.
Fatty Acid Amide Hydrolase (FAAH) Assay.
FAAH assays were carried out in rat brain membranes according to methods described previously (Edgemond et al., 1998). The activity of FAAH was determined from AEA [labeled with 14C in the ethanolamine portion of the molecule (100 mCi/mmol; NEN)]. Rat forebrain membranes (0.05 mg/ml) were incubated with SKM 4-45-1 and 160 nM [14C]AEA in 1 mg/ml BSA containing TME buffer for 2 min. The reaction was stopped with the addition of 2 ml of chloroform:methanol (1:2). One hour after standing at room temperature, 0.67 ml of chloroform and 0.6 ml of water were added and vortexed. Organic and aqueous phases were separated by centrifugation at 100 rpm for 10 min. The radioactivity in each phase was determined by liquid scintillation counting, and the percentage of conversion was calculated.
Results
Synthesis.
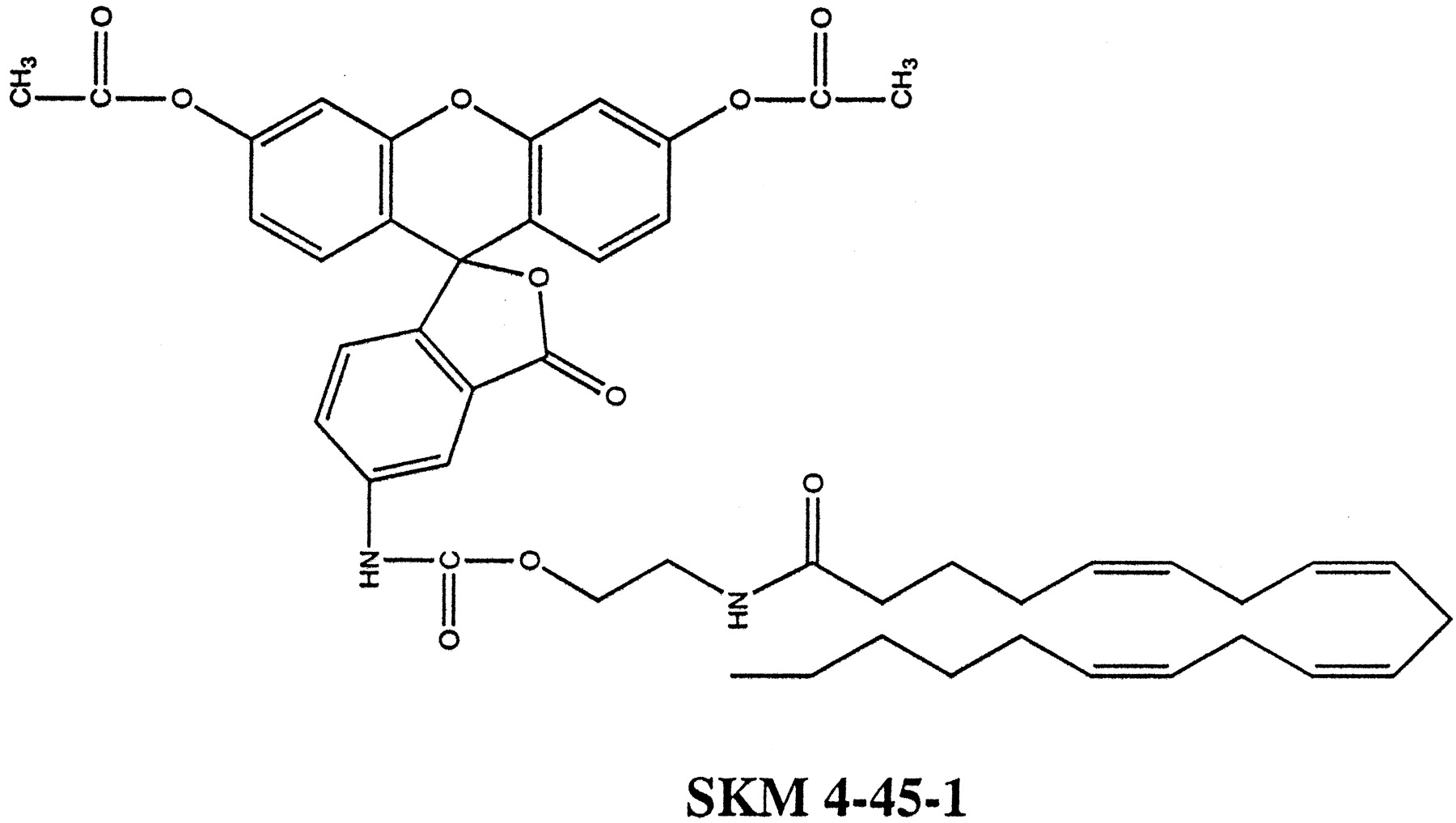
SKM 4-45-1 was synthesized from AEA and fluorescein-5-carbonyl azide diacetate under anhydrous conditions at 80°C. The heat activation of carbonyl azide converts it to an isocyanate, which readily reacts with alcohols to form stable urethanes (Takadate et al., 1984). Shown in Fig. 1is the structure of SKM 4-45-1, the resulting product of AEA reaction with fluorescein-5-carbonyl azide diacetate.

SKM 4-45-1 was synthesized under anhydrous conditions from AEA and fluorescein-5-carbonyl azide and its structure verified by NMR.
Esterase Modification and Fluorescence.
Fluorescein diacetates have fluorescence only after esterase activation (Rothman and Papermaster, 1966; Jones and Senft, 1985). Therefore, we tested the fluorescence activation of SKM 4-45-1 by C6 glioma lysates. SKM 4-45-1 in buffer alone did not show any fluorescence (Fig.2). However, when SKM 4-45-1 (100 nM) was incubated with cell lysates, fluorescence intensity increased in proportion to the volume of cell lysate added. In all incubations, the fluorescence intensity was time-dependent.

SKM 4-45-1 fluorescence is dependent on hydrolysis by cytosolic esterases. Increasing amounts of C6 glioma cell lysates in TME buffer (0–150 μl) were incubated with 100 nM SKM 4-45-1 and fluorescence measured. Data are reported as time course of relative fluorescence intensity.
Cellular Uptake of SKM 4-45-1.
SKM 4-45-1 is taken up into CGCs (Fig. 3). Shown is a photograph of an experiment where CGCs were incubated with 100 nM SKM 4-45-1 for 20 min. SKM 4-45-1 uptake occurs in CGCs as confirmed by fluorescence. Parallel experiments were carried out in the presence of either 100 μM AEA or PEA. As expected, AEA inhibited SKM 4-45-1 uptake, whereas PEA had no effect.

Accumulation of SKM 4-45-1 by CGCs. CGCs were incubated with 100 nM SKM 4-45-1 alone (top) and in the presence of 100 μM AEA (middle) or PEA (bottom). Twenty minutes after incubation, cells were exposed to a mercury fluorescence source and emission was photographed for 1 min. Experiments were carried out on a Nikon Diaphot microscope with a 40× magnification and a Sensys Photometrics charge-coupled device camera; excitation wavelength was 485 nm and emission wavelength was 530 nm.
SKM 4-45-1 Accumulation Occurs Via AEA Uptake Carrier.
AEA is accumulated by CGCs by an uptake mechanism that has the characteristics of facilitated diffusion (Hillard et al., 1997). Several approaches were used to determine whether SKM 4-45-1 was accumulated via the AEA transmembrane carrier.
First, SKM 4-45-1 inhibits [3H]AEA accumulation in CGCs with an IC50 of 7.8 ± 1.3 μM (Fig. 4). This compares favorably with other inhibitors of AEA accumulation in CGCs, including AM404 (IC50 = 3.4 ± 1.2 μM) and A3AP (IC50 = 4.8 ± 1.1 μM) (Muthian et al., 1998).

Effect of SKM 4-45-1, AM404, and A3AP on [3H]AEA uptake into CGCs. CGCs (7–9 days in vitro) were incubated with KRH buffer containing 40 pM [3H]AEA in the presence of increasing concentrations of SKM 4-45-1, AM404, and A3AP. At 2 min, the buffer was removed and the cells were scraped into fresh buffer. Both the buffer and scraped cells were counted for radioactivity, and the percentage of accumulation was calculated as (dpm in cells)/(dpm in cells + dpm in buffer) × 100. Nonspecific association was eliminated by subtracting the uptake measured in the presence of 100 μM AEA. Shown is the mean of four experiments; vertical lines represent S.E.
AEA inhibits the accumulation of SKM 4-45-1 (100 nM) by C6 glioma cells with an IC50 value of 53.8 ± 1.8 μM (Fig.5). A3AP and AM404 also inhibit SKM 4-45-1 accumulation (100 nM) into C6 glioma cells with IC50 values of 10.1 ± 1.4 and 6.1 ± 1.3 μM, respectively (Fig. 5). A comparison of the IC50 values of the inhibitors used against 40 pM [3H]AEA and 100 nM SKM 4-45-1 accumulation is shown in Table 1. Collectively, these data suggest that SKM 4-45-1 accumulation occurs via the AEA transmembrane carrier, SKM 4-45-1 inhibits [3H]AEA accumulation in CGCs, and its uptake is sensitive to inhibitors of AEA uptake.

SKM 4-45-1 uptake is inhibited by AEA, A3AP, and AM404. C6 Glioma cells in culture were incubated for 30 min at room temperature with SKM 4-45-1 and competitors as indicated. Fluorescence was quantified with a Cytofluor II fluorescence microplate reader (excitation 490 nm, emission 525 nm), and data are reported as relative fluorescence intensity versus increasing concentrations of treatments. Shown is the mean of three experiments done in triplicates; vertical lines represent S.E.
IC50 values for inhibitors of [3H]AEA and SKM 4-45-1 accumulation in CGCs
CB1 Binding and FAAH Activity.
AEA binds to the CB1 cannabinoid receptor in the brain with high affinity (Devane et al., 1992) and it is catabolized by FAAH (Deutsch and Chin, 1993; Cravatt et al., 1996). The possibility that SKM 4-45-1 is also a ligand for the CB1 receptor and either a competitor or substrate for FAAH was explored. At concentrations <3 μM, SKM 4-45-1 has no effect on the binding of [3H]CP55940, a high-affinity cannabinoid ligand, to rat brain membranes (Fig.6). In contrast, AEA displaced [3H]CP55940 binding with aKD of 144 nM.

Effect of SKM 4-45-1 and AEA on the binding of [3H]CP55940 to the CB1 receptor. Rat forebrain membranes (10 μg protein/incubate) were prepared as outlined and incubated with SKM 4-45-1 or AEA and 500 to 600 pM [3H]CP55940 for 1 h at room temperature. Data are from a single experiment done in quadruplicate; vertical lines represent S.E.
FAAH activity was assessed in rat brain membranes with [14C]AEA in the presence of AEA or SKM 4-45-1 (Fig. 7). As expected, AEA inhibited the catabolism of [14C]AEA with an IC50 value of 300 nM. However, SKM 4-45-1 had no effect on the catabolism of [14C]AEA at or below 10 μM. These data indicate that SKM 4-45-1 is neither a competitor nor a substrate for FAAH.

Effect of SKM 4-45-1 and AEA on FAAH activity in rat brain membranes. Rat forebrain membranes were prepared as outlined and were incubated (0.05 mg/ml final concentration) for 2 min at 37°C with 160 nM [14C]AEA labeled in the ethanolamide moiety of the molecule. Release of [14C]ethanolamine was measured.
Discussion
These results demonstrate that SKM 4-45-1 is a substrate for the AEA transmembrane carrier and its presence in cells can be detected by fluorescence. We have demonstrated that SKM 4-45-1 is nonfluorescent extracellularly; its fluorescence is dependent on cellular uptake and nonspecific esterase cleavage of the diacetates. SKM 4-45-1 inhibits the cellular accumulation of [3H]AEA and conversely, the uptake of SKM 4-45-1 into C6 glioma cells is inhibited by AEA. In addition, SKM 4-45-1 uptake into C6 glioma is inhibited by analogs of AEA with IC50 values similar to the IC50 values for their inhibition of [3H]AEA uptake. Similar to [3H]AEA accumulation, SKM 4-45-1 uptake into CGCs is inhibited by AEA but not by PEA. Moreover, SKM 4-45-1 does not bind the cannabinoid CB1 receptor, and it is not a substrate or competitor for FAAH. Therefore, our data demonstrate that SKM 4-45-1 is a substrate for the AEA carrier and should be a useful molecule for the investigations of the AEA uptake mechanism.
Our previous study indicated that AEA transport in CGCs is via a protein-mediated facilitated diffusion that is bidirectional (Hillard et al., 1997). The bidirectionality of AEA transport raises the interesting possibility of the presence of endogenous intracellular AEA that could affect SKM 4-45-1 transport into cells. However, we have been unable to measure endogenous AEA in cells due to its low amounts; therefore, we do not expect endogenous AEA to influence the transport of SKM 4-45-1 into cells under our experimental conditions. Furthermore, we have previously determined that AEA is concentrated inside cells beyond its concentration gradient (unpublished data), therefore, any undetectable presence of endogenous AEA may be a negligible factor with regard to SKM 4-45-1 transport into cells. The mechanism for this intracellular accumulation of AEA is yet unknown.
In the last several years, many laboratories have provided evidence for AEA as an endogenous neurotransmitter or neuromodulator. AEA is a natural constituent of brain that is biologically active (Devane et al., 1992) and is synthesized and released from neurons in a calcium-dependent manner (DiMarzo et al., 1994). Administration of AEA to animals produces physiological effects that are characteristic of the cannabinoids (Fride and Mechoulam, 1993). The biochemical effects of AEA administration include inhibition of adenylyl cyclase activity (Felder et al., 1993; Vogel et al., 1993) and the inhibition of the opening of voltage-operated calcium channels (Mackie et al., 1993). However, an important criterion that must be met by any neuromodulator/neurotransmitter is that there must exist a mechanism to terminate its activity. There are two documented mechanisms of AEA inactivation, a microsomal enzyme (FAAH) that catabolizes AEA to arachidonic acid and ethanolamine (Deutsch and Chin, 1993; Hillard et al., 1995b; Cravatt et al., 1996), and an uptake mechanism that removes AEA from the extracellular environment (DiMarzo et al., 1994;Hillard et al., 1997). Because FAAH is an intracellular, membrane-bound enzyme (Desarnaud et al., 1995; Hillard et al., 1995b; Ueda et al., 1995), it is likely that these two mechanisms of AEA inactivation act in series, such that AEA from the extracellular milieu is taken up into the cells to be catabolized by intracellular FAAH.
Transport of polar molecules across cell membranes have been well studied and characterized. Lipid molecule transport however, has been more difficult to study. It has been hypothesized that lipid transport into cells occurs by simple diffusion: adsorption to cell membrane, transmembrane movement, and desorption into the cytosol (Hamilton, 1998). Recent studies however, have identified and cloned protein carriers for lipids, including carriers for the prostaglandins (Kanai et al., 1995) and long-chain fatty acids (LCFAs) (Schaffer and Lodish, 1994). Therefore, protein carriers exist and function to transport lipid molecules across cell membranes. The availability of a fluorescent substrate for the AEA carrier may aid in its molecular characterization and identification.
The purpose of our studies was to design a tool with which to study the AEA uptake mechanism. Similar tools have been used by others to study various transport processes, including the characterization of the kinetics of nucleotide transport into synaptic vesicles with a fluorescent analog of ATP (Gualix et al., 1999) and excretion transport with a fluorescently labeled version of the anthelmintic drug ivermectin (Fricker et al., 1999). In another study, Schaffer and Lodish (1994) used a fluorescent analog of LCFAs to measure uptake and ultimately clone the LCFA transporter by an elegant functional assay. Therefore, there are several potential uses for a fluorescent substrate in the study of transport mechanisms.
In this report, we describe the synthesis and characterization of an analog of AEA, SKM 4-45-1, that is transported into the cell by the same mechanism that transports AEA. Furthermore, SKM 4-45-1 is specific for the AEA transporter and does not interact with either the cannabinoid CB1 receptor or FAAH at micromolar concentrations. Therefore, SKM 4-45-1 may be used to study AEA transport in ways similar to the examples described above. For example, SKM 4-45-1 could be used to identify cells that transport AEA and potentially identify intracellular sites of AEA sequestration, if they exist. Furthermore, similar to the study of Schaffer and Lodish (1994), SKM 4-45-1 also may prove advantageous in the identification and cloning of the AEA transporter. In addition, because SKM 4-45-1 is only fluorescent after activation by intracellular esterases, it also could potentially be used to study the bidirectionality of the AEA transporter based on the accumulation of extracellular fluorescence. One limitation of SKM 4-45-1, however, is that the manifestation of intracellular fluorescence occurs as a result of two kinetic processes, uptake and intracellular de-esterification. Therefore, the utility of SKM 4-45-1 in kinetic studies of AEA transport is limited. In conclusion, SKM 4-45-1 is a fluorescent analog of AEA that interacts selectively with the AEA transporter and hence, will be a useful tool for the study of AEA movement across cell membranes.
Acknowledgments
We thank Dr. Sukumar Manna, Marcie Greenberg, Peter Schmidt, and Daniel Horvatin for technical contributions to these studies. We also thank Dr. Isabel Nogueron for helpful discussions.
Footnotes
-
Send reprint requests to: Cecilia J. Hillard, Ph.D., Department of Pharmacology and Toxicology, Medical College of Wisconsin, 8701 Watertown Plank Rd., Milwaukee, WI 53226. E-mail:chillard{at}mcw.edu
-
↵1 This work was supported by National Institute on Drug Abuse Grant DA09155.
- Abbreviations:
- AEA
- N-arachidonoylethanolamine
- CB1
- central cannabinoid receptor
- A3AP
- arachidonoyl-3-aminopyridine amide
- AM404
- arachidonoyl-4-hydroxyanilineamide
- PEA
- N-palmitoylethanolamine
- CGC
- cerebellar granule cell
- FAAH
- fatty acid amide hydrolase
- LCFA
- long- chain fatty acid
- Received November 8, 1999.
- Accepted January 11, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}