


Abstract
CP-195543 [(+)-2-(3-benzyl-4-hydroxy-chroman-7-yl)-4-trifluoromethyl-benzoic acid] is a structurally novel, selective and potent leukotriene B4 (LTB4) receptor antagonist. In vitro CP-195543 inhibited [3H]LTB4binding to high-affinity LTB4 receptors on human neutrophils (HN) and murine spleen membranes with IC50values of 6.8 nM (Ki = 4.9 nM) and 37.0 nM (Ki = 26.9 nM), respectively. CP-195543 inhibited human and mouse neutrophil chemotaxis mediated by LTB4 with IC50 values of 2.4 nM and 7.5 nM, respectively. Evidence of noncompetitive antagonist effects on the HN high-affinity LTB4 receptor was obtained by Scatchard analysis of [3H]LTB4 binding to and chemotaxis of HN to LTB4. Scatchard analyses of [3H]LTB4 binding to low-affinity receptors on HN indicated that CP-195543 acted as a competitive antagonist at this receptor, and inhibition of LTB4-mediated CD11b up-regulation on HN was inhibited competitively by CP-195543 (pA2 = 7.66). In whole blood, CP-195543 also blocked CD11b up-regulation on HN (pA2 = 7.12) and murine neutrophils (pA2 = 7.06) with a similar potency. LTB4-mediated CD11b up-regulation on human monocytes and eosinophils in whole blood were inhibited by CP-195543 with IC50 values of 270 nM and 420 nM, respectively. CP-195543 at 10 μM failed to inhibit HN chemotaxis and CD11b up-regulation mediated through alternative (i.e., complement fragment 5a, interleukin-8, platelet-activating factor) G-protein-coupled chemotactic factor receptors. In vivo, after oral administration, CP-195543 blocked LTB4-mediated neutrophil infiltration in guinea pig and murine skin with ED50 values of 0.1 mg/kg and 2.8 mg/kg p.o., respectively. When administered in osmotic pumps, CP-195543 reduced the clinical symptoms and attendant weight loss in an IL-1-exacerbated murine model of collagen-induced arthritis with half-maximal effects associated with plasma drug levels of 0.4 to 0.5 μg/ml. Collectively these data provide evidence of thein vitro potency and in vivo efficacy of a novel LTB4 antagonist and support its clinical evaluation in a variety of inflammatory diseases in man.
Although the involvement of LTB4 in a variety of human inflammatory diseases is suggested but not clinically proven (reviewed in Samuelsson, 1983; Ford-Hutchinson, 1990; Lewis et al., 1990; Henderson, 1994), the potential importance of LTB4, or alternate ligands which also bind to the LTB4 receptor (e.g.,12-(R)-hydroxyeicosatetraenoic acid), as a mediator, involved in the pathogenesis of several preclinical animal disease models, recently has been reinforced through the demonstration of efficacy in these models of the potent LTB4receptor antagonists SC-53228 and CP-105696 (Fretland et al., 1995a, b; Stevens et al., 1995; Griffiths et al., 1995; Turner et al., 1996a; Gladue et al., 1996). Moreover, the potential contribution of products of the 5-LO pathway in animal models of rheumatoid arthritis and experimental asthma recently has been confirmed independently in mice in which either the FLAP or 5-LO gene was deleted by homologous recombination (Griffiths et al., 1997; Irvin et al., 1997). CP-105696 exhibited high potency toward both the high- and low-affinity LTB4 receptors on HNs inin vitro ligand binding and bioassays performed in low protein. However, significantly higher concentrations of compound were required to achieve the same pharmacological effect when bioassays were performed in whole blood (Showell et al., 1995). Moreover, plasma drug levels of CP-105696, which were associated with efficacy in the various animal disease models, were in a concentration range predicted from the in vitro whole-blood assays (Showellet al., 1996; Griffiths et al., 1995; Turneret al., 1996a; Gladue et al., 1996). The loss of potency of CP-105696 in whole blood was attributed to the high level of protein binding which obtains for this compound. High plasma protein binding also is likely to contribute to the remarkably long plasma half-life for CP-105696, which was seen both in animals and in human phase I clinical studies (Liston et al., 1998). A goal of our program was therefore to discover a potent LTB4 receptor antagonist that maintained its potency in complex biological fluids and exhibited preclinical pharmacokinetics which would be predicted to have a more favorable pharmacokinetic profile in man. Herein we describe the discovery and pharmacological profile of CP-195543 (fig.1) which fulfills these criteria and is thus a suitable candidate for clinical evaluation.

Chemical structures of CP-195543 and CP-105696.
Materials and Methods
Human Neutrophil Isolation and Chemotaxis Assay
Neutrophils were isolated from anticoagulated blood (7.5% EDTA, 1 ml/50 ml blood), obtained with informed consent, of normal human donors as described (Ferrante and Thong, 1978). Isolated neutrophils were resuspended (2.5 × 106 cells/ml) in HBSS (supplemented with 10 mM HEPES) containing 0.7 mM Mg++, 1.6 mM Ca++, 0.035% sodium bicarbonate and 2 mg/ml recrystallized BSA (Sigma, St. Louis, MO) (HBSS-BSA) and adjusted to pH 7.25. The chemotaxis assay was performed in a 48-well chamber apparatus (Neuroprobe, Cabin John, MD) with cellulose nitrate filters (pore size, 3.0 μm) as described (Harvath et al., 1987). The total numbers of cells (observed at ×400 magnification) migrating from 20 μm from beneath the surface monolayer to the leading front (usually ∼100–120 μm/45 min at optimal chemotactic factor concentrations) in response to varying concentrations of chemotactic factors were summed and provided an index of the chemotactic response. Each experimental condition was performed in duplicate, and three fields were assessed for cell migration. Results were expressed as the percentage maximal response, where 100% was equal to the peak response seen in the presence of the most efficacious concentration of chemotactic factor. The number of cells migrating spontaneously (i.e., negative controls) were subtracted from all measurements before data transformation. In experiments in which CP-195543 or other test compounds were evaluated for their ability to block chemotaxis, equal concentrations were present in both compartments of the chemotaxis chamber.
LTB4 Receptor Binding Assays
The [3H]LTB4 HN binding assay was performed according to a method adapted from Linet al. (1984). Neutrophils at a density of 2.5 × 106 cells/ml were incubated with 0.3 nM [3H]LTB4 in HBSS containing 0.7 mM Mg++, 1.6 mM Ca++, 10 mM HEPES and 0.035% sodium bicarbonate, adjusted to pH 7.25 in the absence and presence of varying concentrations of CP-195543 for 30 min at 4°C. The assay was performed in triplicate in microtiter plates (Costar, Cambridge, MA) with a total volume of 200 μl, and bound ligand was separated from free ligand with the beta plate apparatus (Pharmacia LKB, Piscataway, NJ). Specific [3H]LTB4binding represents the value obtained when nonspecific binding (value obtained in the presence of 1 μM unlabeled LTB4) is subtracted from total binding. For saturation binding experiments, concentrations of [3H]LTB4 ranging from 0.05 to 10 nM were used.
CD11b Up-regulation Assay
Whole-blood assay.
Human blood was collected as described previously. Whole blood was incubated with the indicated concentration of CP-195543 or CP-105696 for 5 min at 37°C. A 200-μl aliquot of blood ± antagonist was then added to 12 × 75 mm NUNC minisorp tubes containing 20 μl of LTB4 at 10 times the desired final concentration of LTB4. The samples were then incubated for 10 min at 37°C. The samples were placed in an ice-water bath and 1 ml of cold PBS containing 0.2% sodium azide and 2% heat-inactivated fetal bovine serum (PBS-wash) was added. The cells then were labeled and analyzed for CD11b expression as indicated below.
Isolated HN.
Neutrophils (1 × 107/ml in HBSS-BSA) were warmed for 5 min at 37°C. Then 100-μl aliquots of cells were added to tubes containing the indicated concentrations of antagonist and LTB4 (final volume, 0.5 ml) and were incubated for 10 min at 37°C. The samples were placed in an ice-water bath, and 1 ml of cold PBS-wash was added. The cells then were labeled and analyzed for CD11b expression. The final dimethyl sulfoxide concentration in all samples, including controls, was 0.01%.
Quantitation of CD11b expression.
The cells were pelleted by gentle centrifugation (200–250 × g) at 4°C. The supernatant was aspirated/decanted, and the cells were resuspended by gentle shaking. After a 10-min incubation with 10 μl blocking antibody (1 mg/ml heat-aggregated human IgG), the cells were incubated with saturating concentrations of fluorescein-conjugated anti-CD11b (Bear 1 clone, Caltag Laboratories, Burlingame, CA) for 20 to 30 min at 4°C. To identify monocytes, 20 μl of CD14PE (M5E2 clone, Pharmingen, San Diego, CA) also had been added to whole-blood samples. To each sample, 1 ml PBS-wash was added, and the cells were pelleted as before. Red blood cells were lysed with Becton Dickinson FACS lysing solution (San Jose, CA) in accordance with the manufacturer’s directions. After two washings with PBS-wash, the cells were resuspended in 0.25 to 0.5 ml PBS-wash.
Fluorescent analysis.
The degree of fluorescent staining was quantitated on a Becton Dickinson FACscan flow cytometer by CELL Quest software. Data were collected in the list mode. Neutrophil gates were defined by the FSC vs. SCC dot plots, eosinophils were identified by a combination of their light-scatter profile and autofluorescence and monocytes were identified by both light-scatter profile and the binding of CD14 PE. Data were quantified by histogram analysis and expressed as mean (neutrophils, eosinophils) or median channel (monocytes) fluorescence.
Mouse Neutrophil Isolation and Chemotaxis Assay
Neutrophils were isolated from pooled anticoagulated blood obtained from DBA/1LacJ mice by intracardiac puncture according to the procedure described by Rot (1991). Isolated cells (usually ∼30% neutrophils) were adjusted to a density of 2.5 × 106 neutrophils/ml in HBSS containing 1.6 mM Ca++ and 0.7 mM Mg++, 10 mM HEPES, 0.035% sodium bicarbonate and 1% BSA (pH 7.2). Chemotaxis was performed as described above for human neutrophils.
Mouse Mac-1 Up-regulation Assay
Whole blood obtained by intracardiac puncture from DBA/1LacJ mice was anticoagulated with EDTA (0.15%). The blood was preincubated with antagonist for 5 min at 37°C, after which a 100-μl aliquot of blood was added to 12 × 75 mm borosilicate tubes containing 10 μl of a 10X stock of final indicated LTB4concentration and incubated for 10 min at 37°C. The tubes were then removed and placed in an ice-water bath and ∼1 ml of cold PBS-wash containing 10 mM EDTA, pH 7.25. The cells then were pelleted at 200 × g and incubated with fluorescein isothiocyanate-conjugated rat anti-mouse Mac-1 mAb (M1/70.15 clone, Caltag). The red blood cells were lysed with FACS Lysing Solution and the remaining cells washed with PBS-wash at 4°C. Fluorescent labeling was quantified as described for HNs.
Mouse Spleen Membrane Preparation and LTB4Receptor Binding Assay
Spleens were dissected from 40 to 50 DBA/1LacJ mice and kept in 50 mM Tris-HCl buffer (pH = 7.3, 4°C). After removal of connective tissue, the total weight of the spleens was determined. The pooled tissue was minced and homogenized twice by Polytron (Brinkmann Instruments, Westbury, NY) PCU-2 at setting 7 for 10 s in 50 mM Tris (4°C) at 3 ml/g tissue. The homogenate was centrifuged at 1000 × g for 15 min (4°C), and the resultant supernatant was centrifuged at 40,000 × g for 20 min (4°C) with washing of the intermediate pellet with 50 mM Tris. The final pellet was resuspended at a protein concentration of 5 mg/ml in 50 mM Tris pH = 7.3 with 10 mM MgCl2 and stored at −70°C. The protein concentration was determined by the Bradford method (Bradford, 1976) with BSA as standard.
The [3H]LTB4 mouse spleen binding assay was performed according to a method adapted from Chenget al. (1986). The [3H]LTB4 binding assays were carried out in a volume of 150 μl containing 50 μg membrane preparation, 0.7 nM [3H]LTB4, 50 mM Tris pH = 7.3, 10 mM MgCl2 and 10% methanol in the presence or absence of various concentrations of unlabeled LTB4 or CP-195543 for 30 min at 4°C. The assay was performed in triplicate in microtiter plates (Costar, Cambridge, MA) and filtered through Whatman GF/B glass fibre filters with a beta plate apparatus (Pharmacia LKB, Piscataway, NJ) to separate free from bound [3H]LTB4. Specific [3H]LTB4 binding represents the value obtained when nonspecific binding (value obtained in the presence of 5 μM unlabeled LTB4) is subtracted from total binding.
Guinea Pig and Mouse Skin Neutrophil Infiltration Assays
Male Hartley guinea pigs were shaved and fasted 1 day before conducting the experiment. On the day of the experiment, the guinea pigs were separated into groups of five and color coded, and their daily weights were recorded. One group was designated as a control group and was given doses of vehicle orally. Other groups were given doses orally of CP-195543 (1 ml/100 g b.wt.) in a suspension of 0.5% carboxymethylcellulose. One hour after oral dosing, the shaved dorsal surface of each guinea pig was challenged intradermally with 0.1 ml of LTB4 (100 ng), at duplicate sites, by a 1-cc syringe with a 30-gauge needle. The duplicate sites were arranged alternately (right anterior outside corresponding with the left posterior inside). One set of duplicate sites was injected with vehicle (0.25% BSA in saline) for background measurement. Each site was marked with a green dot by a Sharpee pen. Four hours after intradermal challenge, the guinea pigs were sacrificed in a CO2 desiccator and the skin was removed. Each injected site was punched out with a 13-mm gasket punch and placed in a tube containing 8 ml of 0.5% hexadecyltrimethylammonium bromide. The samples then were homogenized with a Polytron homogenizer and subjected to two free-thaw cycles (−70°C and 37°C). The samples then were poured into 15-ml centrifuge tubes and centrifuged at 3500 rpm for 30 min at room temperature. The supernatant was diluted 1:5 with 0.5% hexadecyltrimethylammonium bromide/50 mM K2PO4 buffer in 1.5-ml microfuge tubes and spun at full speed in an Eppendorf centrifuge for 10 min. Supernatants were assayed for MPO, a neutrophil marker, as follows. The diluted sample (50 μl) was mixed with 150 μl of substrate (0.2 mg/ml o-diansidine, 0.22 μl/ml H2O2 in 50 mM KPO4 buffer, pH 6.0) in a 96-well plate for 15 min at 37°C. The substrate solution and plates containing the samples were prewarmed before the reaction. The reaction was terminated by adding 100 μl of 0.4 M glycine, and the absorbance of each sample was measured in a microtiter plate reader at 450 nm. The number of neutrophils in each sample was quantified by including a standard curve.
Male DBA/1LacJ mice were anesthetized with methoxyflurane and injected intradermally with LTB4 (100 ng in 20 μl of saline containing 1 mg/ml BSA), and neutrophil infiltration was measured after 4 hr by the MPO content of the skin. Known numbers of MNs (harvested from the peritoneal cavity after injection of oyster glycogen) were included in each assay as a standard curve, and data are expressed as numbers of neutrophil equivalents per site. Drug was dosed orally as a suspension in 0.5% methyl cellulose 1 hr before the injection of LTB4.
Murine Collagen-Induced Arthritis
Male DBA/1LacJ (9–13 weeks old) mice were immunized at the base of the tail with 100 μg of chicken type II collagen in Freund’s complete adjuvant on days 0 and 21. The severity of the symptoms of arthritis was assessed by inspection of the paws (0 = normal paw; 1 = swelling and/or redness of one top or finger joint; 2 = two or more joints involved; 3 = severe arthritis in the entire paw; maximum score for each animal = 12). In this series of experiments, administration of IL-1α was used to cause a severe flare of the arthritis; 0.3 μg of recombinant murine IL-1α, diluted in PBS containing 1 mg of BSA per ml was administered s.c. on day 25 or 26. This protocol caused all the control animals to exhibit a severe form of the disease. Changes in body weight during the course of the experiment also were monitored. Treatment with CP-195543 commenced 1 day before the first injection of IL-1α. Each experiment was repeated four times with groups of seven to nine mice.
Histopathology
Knee joints were decalcified in Kristensen’s solution, embedded in Paraplast plus, sectioned and stained with Safranin O; approximately matched sections were examined by light microscopy on a Nikon FXA microscope.
Data Analysis
Results are expressed as the means ± S.E. from at least three independent experiments, unless otherwise indicated. IC50 and ED50 values were calculated by plotting the mean of the percent control of each compound-treated group on a log scale, fitting the linear portion of the graph to a linear regression curve and calculating the xvalue at y = 50. Results of the in vivoexperiments were analyzed by Student’s t test with a Bonferroni correction factor for multiple comparisons. Binding site densities and respective affinities for [3H]LTB4 were calculated by nonlinear regression by Prism Software (one-site model) (Graph Pad, San Diego, CA). Comparisons between groups were made by analysis of variance and Dunnett’s post hoc test (RSI, BNN Software, Cambridge, MA). pA2 values were calculated according to the method of Schild (1947).Ki was calculated according to the method of Cheng and Prusoff (1973).
Materials
CP-195543 was synthesized as described (Koch, 1993) and CP-105696 was synthesized as described (Koch et al., 1994). LTB4 and PAF were obtained from BioMol Research Labs (Plymouth Meeting, PA), [3H]LTB4 (specific activity, 150–200 Ci/mmol) from DuPont/NEN (Boston, MA), IL-8 from Peprotech (Rocky Hill, NJ) and complement fragment 5a from Sigma (St. Louis, MO).
Results
Effects of CP-195543 on LTB4 binding to high-affinity receptors on HN and MSM.
The ability of CP-195543 to compete for [3H]LTB4binding to the high-affinity receptor on HN is shown in figure2A. An IC50 value of 6.8 ± 0.75 nM (Ki = 4.9 ± 0.5 nM) (n = 9) was obtained for CP-195543. IC50 values obtained in the same assay for CP-105696 (n = 9) and LTB4(n = 9) were 6.7 ± 0.83 nM (Ki = 4.8 ± 0.59 nM) and 1.3 ± 0.18 nM, respectively. The ability of CP-195543 (n = 3) to compete for [3H]LTB4binding to the high-affinity receptor on MSM is shown in figure 2B. An IC50 value of 37.0 ± 15.0 nM (Ki = 26.9 ± 10.7 nM) was obtained for CP-195543. IC50 values obtained in the same assay for CP-105696 (n = 3) and LTB4 (n = 3) were 22.0 ± 10.7 nM (Ki = 15.7 ± 7.6 nM) and 4.0 ± 2.1 nM, respectively. These results indicate that CP-195543 interacts with the high-affinity receptor on HN with a potency similar to that obtained previously with CP-105696. In addition, in an animal species, mice, suitable for in vivo testing, CP-195543 maintains potency in the nanomolar range. Scatchard analyses of [3H]LTB4 binding to high-affinity receptor on HN in the presence and absence of CP-195543 provided clear evidence of noncompetitive interactions at this high-affinity binding site (n = 5, table1).

Inhibition of [3H]LTB4binding to HN (A) and MSM (B) by CP-195543, CP-105696 and LTB4. HN and MSM were incubated with 0.3 nM and 0.7 nM [3H]LTB4, respectively, for 30 min at 4°C in the presence and absence of a range of concentrations of CP-195543, CP-105696 or LTB4, and specific binding was determined. Data are means ± S.E. from nine and three experiments, respectively, each performed in triplicate. Zero percent specific binding refers to the level of [3H]LTB4binding obtained in the presence of 1.0 μM unlabeled LTB4.
Analysis of saturation binding of [3H]LTB4 to high- and low-affinity receptors in the absence and presence of CP-195543
Effect of CP-195543 on [3H]LTB4 binding to low-affinity receptors on HN.
Scatchard analyses of [3H]LTB4 binding to low-affinity receptors on HN in the absence and presence of CP-195543 were performed. Evidence of competitive interactions at this low-affinity binding site was obtained (n = 3, table1).
Effect of CP-195543 on LTB4-mediated HN and MN chemotaxis.
With fixed concentrations of LTB4 (HN = 5 nM, MN = 5 nM) in the lower chamber of the chemotaxis apparatus, HN and MN chemotaxis was inhibited by CP-195543 with IC50 values of 2.4 ± 1.6 nM (n = 4) and 7.5 ± 4.0 nM (n = 3), respectively (fig.3). When HN chemotaxis was assessed across a full range of concentrations of LTB4, the effect of CP-195543 could not be overcome by increasing concentrations of LTB4. This provides evidence that a noncompetitive form of inhibition is occurring at the LTB4 receptor which mediates neutrophil chemotaxis (fig. 4).

Inhibition of LTB4-mediated HN and MN chemotaxis by CP-195543. LTB4 was used at a fixed concentration of 5 nM (HN) and 5 nM (MN) in the lower chamber, and CP-195543 was present at varying concentrations (0.1 nM –10 μM) in both the upper and lower chambers. Chambers were incubated for 60 min at 37°C, and then the filters were removed and processed and counted as described under “Materials and Methods” (Showell et al., 1995). Data are the means ± S.E. from three independent experiments.

Effect of CP-195543 on the concentration-dependent curve of LTB4-mediated HN chemotaxis. Chemotaxis was induced by a range of concentrations of LTB4 in the absence and presence of CP-195543 at the concentrations indicated. Data are expressed as the percentage of maximal chemotaxis occurring to LTB4 in the absence of CP-195543 and are means ± S.E. from three independent experiments.
Effect of CP-195543 on LTB4-mediated up-regulation of the adhesion molecule CD11b on isolated HN and on HN in whole blood.
With use of 5 nM LTB4 with isolated HN, and 20 nM LTB4, with HN in whole blood, we observed half-maximal inhibition of CD11b up-regulation by CP-195543 at 280 ± 60 nM (n = 4) and 660 ± 60 nM (n = 4), respectively (fig.5). For comparative purposes, the results obtained with CP-105696 also are shown. Whereas half-maximal inhibition on isolated HN occurred with an IC50 = 410 ± 60 nM (n = 3), with HN in whole blood it required concentrations of CP-105696 that were significantly (>2 log) higher with an IC50 = 54.7 μM ± 9.3 μM (n = 5) (fig. 5). Thus, in contrast to CP-105696, the intrinsic antagonist potency of CP-195543 on LTB4-mediated CD11b up-regulation on neutrophils is maintained in whole blood. When CD11b up-regulation was assessed across a full range of concentrations of LTB4, the addition of increasing concentrations of CP-195543 caused parallel rightward shifts in the LTB4-concentration curves, which indicates that competitive interactions are occurring at the LTB4 receptor that mediates CD11b up-regulation (fig. 6A). The pA2 value for CP-195543 obtained with isolated cells was 7.66 ± 0.1 (n = 7). In confirmation of these results and the findings above with use of a single concentration of LTB4, CP-195543, when evaluated in whole blood for effects on CD11b up-regulation across a full range of concentrations of LTB4, behaved as a competitive inhibitor and also retained potency similar to that seen with isolated cells (fig. 6B). The pA2 value for CP-195543 with HN in whole blood was 7.12 ± 0.12 (n = 7).
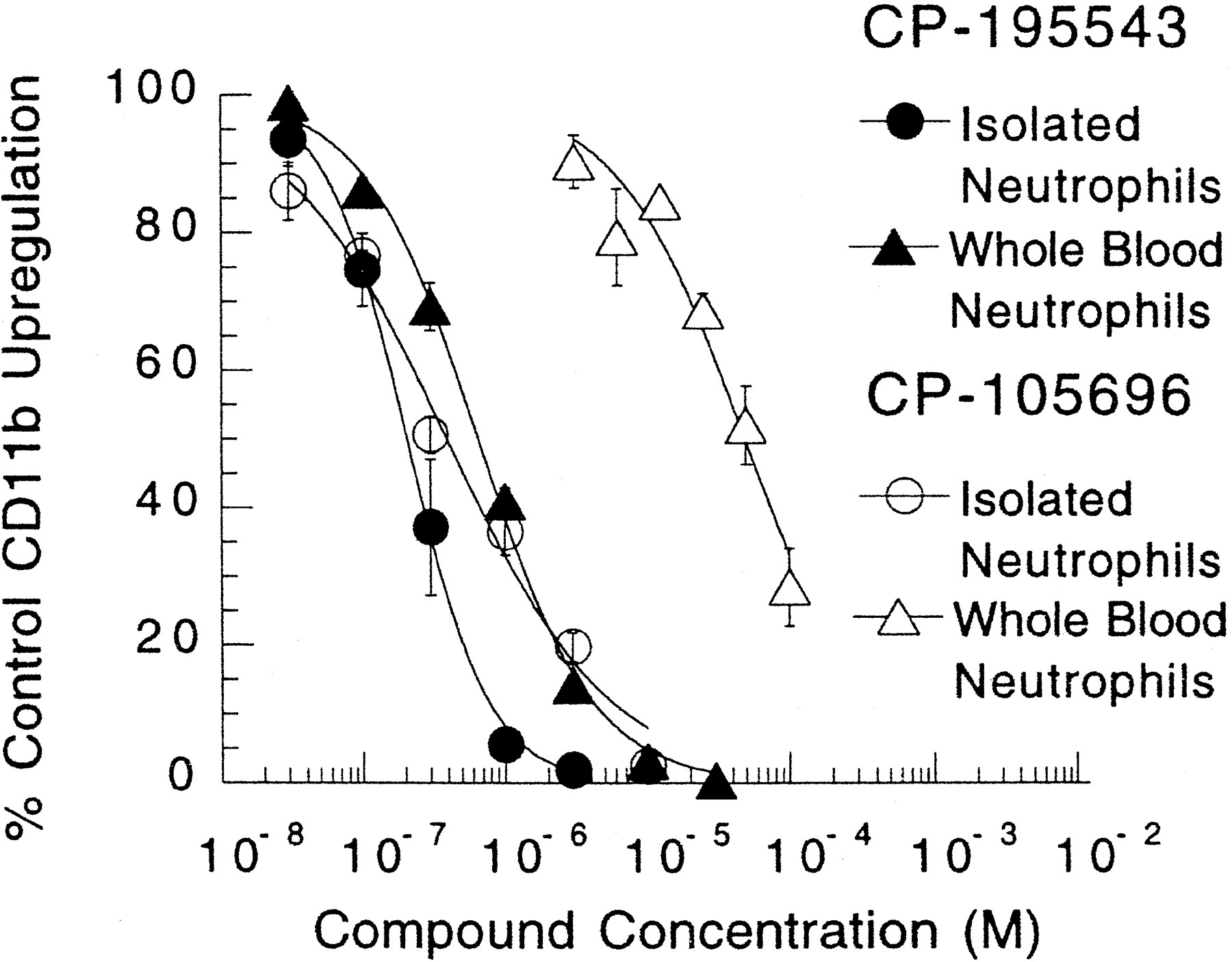
Inhibition of LTB4-mediated CD11b up-regulations in isolated HN and HN in whole blood by CP-195543 and CP-105696. LTB4 was used at a fixed concentration of 5 nM for isolated HN and 20 nM for HN in whole blood. CP-195543 was present at varying concentrations (30 nM –30 μM) in isolated cells and whole blood; CP-105696 was present at varying concentrations in isolated cells (30 nM–30 μM) and whole blood (3–100 μM). Isolated HN and HN in whole blood were incubated for 10 min at 37°C, and then samples were processed for FACS analysis as described under “Materials and Methods” (Showell et al., 1995). Data are expressed as the percentage of the response occurring in the absence of drug and are the means ± S.E. from three to six independent experiments.

Effect of CP-195543 on the concentration-dependent curves of LTB4-mediated up-regulation of CD11b on isolated HN (A) and HN in whole blood (B). CD11b up-regulation was induced by a range of concentrations of LTB4 in the absence and presence of CP-195543 at the concentrations indicated. Data are expressed as the percentage of maximal response to LTB4 in the absence of CP-195543 and are the means ± S.E. from seven experiments. The pA2 (see inserts) values were determined according to the method of Schild (1947).
Effect of CP-195543 on LTB4-mediated up-regulation of the adhesion molecule Mac-1 on MN in whole blood.
With LTB4 at a fixed concentration (100 nM), the observed half-maximal inhibition of Mac-1 up-regulation on MN in whole blood by CP-195543 was 700 ± 170 nM (n = 3) (fig.7A). When Mac-1 up-regulation on MN in whole blood was assessed across a full range of concentrations of LTB4, as seen with HN, the addition of increasing concentrations of CP-195543 caused parallel rightward shifts in the LTB4-concentration curves, providing evidence of competitive interactions at the LTB4 receptor on MN which mediates Mac-1 up-regulation (fig. 7B). The pA2 value for CP-195543 with MN in whole blood was 7.06 ± 0.05 (n = 3). These results suggest that CP-195543 exhibits a similar level of potency on the murine LTB4 receptor that mediates Mac-1 up-regulation when compared with its human counterpart.

Inhibition of LTB4-mediated Mac-1 expression on MN in whole blood. (A) LTB4 was used at a fixed concentration of 100 nM and CP-195543 was present at varying concentrations. (B) CD11b was induced by a range of concentrations of LTB4 in the absence and presence of CP-195543 at the concentrations indicated. Data are expressed as the percentage of the maximal response to LTB4 occurring in the absence of CP-195543 and are the means ± S.E. from three independent experiments. The pA2 (see insert, B) value was determined according to the method of Schild (1947).
Effect of CP-195543 on LTB4-mediated CD11b up-regulation on HE and HM in whole blood.
Previously we used a Ca++ mobilization assay in isolated HM to assess the antagonist potency of compounds on monocyte LTB4 receptors (Showell et al., 1995). This assay is not compatible with complex biological fluids such as whole blood, however. We subsequently discovered that LTB4-mediated CD11b up-regulation on monocytes in whole blood with FACS is a reliable and quantifiable end point, suitable for evaluating LTB4 antagonists. Moreover, this technique is also suitable for assessing effects of antagonists on eosinophils (Conklyn et al., 1996). Shown in figure 8 are the results obtained with CP-195543 by this assay. Half-maximal effects of LTB4 on CD11b up-regulation on HM and HE in whole blood occurred at 5.9 nM and 16.9 nM, respectively. Maximal effects were seen at ∼100 nM and ∼100 nM, respectively (see insert, fig.8). With use of 20 nM LTB4 with HM and 100 nM LTB4 with HE, we observed half-maximal inhibition of CD11b up-regulation by CP-195543 at 270 ± 110 nM (n = 6) and 420 ± 40 nM (n = 6), respectively. Shown for comparative purposes are the results we obtained with CP-105696. For CP-105696 the respective IC50 values for HM and HE were 11.9 ± 1.7 μM (n = 4) and 12.5 ± 1.5 μM (n = 4). Clearly significantly higher concentrations of CP-105696 are required to achieve the same level of inhibition compared with CP-195543. The CP-195543 inhibitory concentration-response curves obtained for HM and HE are similar to those seen with HN in whole blood; in addition, in experiments where a full range of concentrations of LTB4 were evaluated, evidence for competitive antagonism was found (results not shown). These data provide evidence of a functional and pharmacological overlap of LTB4 receptors that mediate CD11b up-regulation on HN, HM and HE.

Inhibition of LTB4-mediated CD11b up-regulation in HM and HE in whole blood by CP-195543 and CP-105696. LTB4 was used at a fixed concentrations of 20 nM (HM) and 100 nM (HE). CP-195543 was present at a range of concentrations (30 nM–30 μM) and CP-105696 (30 nM–100 μM). HN and HE in whole blood were incubated for 10 min at 37°C and then samples were processed for FACS analysis as described under “Materials and Methods” (Conklynet al., 1996). Data are expressed as the percentage of the response occurring in the absence of antagonist and are means ± S.E. from four to six independent experiments. Insert shows LTB4 concentration-response curves.
Effect of CP-195543 on HN chemotaxis and CD11b up-regulation mediated by other chemotactic factors.
A panel of chemotactic factors, namely complement fragment 5a, IL-8 and PAF which mediate their effects through independent G-protein-coupled receptors, were examined in chemotaxis and CD11b up-regulation assay in the absence and presence of CP-195543 at 10 μM. As previously found for CP-105696, CP-195543 had no effect on these responses (results not shown).
Effect of CP-195543 on neutrophil infiltration into guinea pig and mouse skin mediated by intradermal injection of LTB4.
Injection of LTB4 into guinea pig skin previously was demonstrated to induce time- and concentration-dependent infiltration of neutrophils (Pettipher et al., 1993; Showell et al., 1995). With this model we evaluated CP-195543 after oral dosing for its ability to block cell infiltration mediated by LTB4 by examining the excised skin sites for the presence of MPO. When a range of amounts of LTB4(10–100 ng/site) was injected into the skin, half-maximal inhibition of neutrophil infiltration occurred at 0.1 ± 0.02 mg/kg p.o. (n = 3) and was independent of the amount of LTB4 injected (fig.9A). These data confirm that CP-195543 also acts noncompetitively in vivo on the LTB4 receptor that mediates neutrophil recruitment. In a species, mice, used for disease models,e.g., collagen-induced arthritis, we also evaluated the ability of CP-195543 to block LTB4 (100 ng/site)-mediated neutrophil infiltration. The results are shown in figure 9B. Half-maximal inhibition of neutrophil infiltration occurred at 2.8 ± 0.6 mg/kg (n = 4).

Inhibition of LTB4-mediated neutrophil infiltration in guinea pig skin (A) and mouse skin (B). LTB4 (10–100 ng/site, guinea pigs and 100 ng/site, mice) was injected intradermally, and 4 hr later skin sites were excised from sacrificed animals and assessed for MPO content. Various doses of CP-195543 were administered p.o. 1 hr before injection of LTB4. Data are expressed as means ± S.E. from three to four independent experiments.
Effect of CP-195543 on zymosan-induced abdominal stretching and prostaglandin production in mice.
At oral doses 10-fold greater than the ED50 obtained for inhibition of LTB4-mediated neutrophil infiltration in mouse skin, CP-195543 failed to inhibit either abdominal stretching or 6-keto prostaglandin F1α production induced by intraperitoneal administration of zymosan (results not shown). These data provide evidence that CP-195543 is not an inhibitor of cyclooxygenase.
Effect of CP-195543 on the development of IL-1-exacerbated collagen-induced arthritis in mice.
Previous studies showed that the LTB4 receptor antagonist CP-105696 was an effective inhibitor of the spontaneous development of murine collagen-induced arthritis as well as the protocol involving IL-1 exacerbation (Griffiths et al., 1995). We therefore evaluated CP-195543 in this same preclinical model of human rheumatoid arthritis. Because the plasma half-life of CP-195543 in mice is short (t½ = 1.3–1.6 hr), and thus impractical for oral dosing in a chronic protocol, the compound was administered in osmotic pumps. Sustained plasma levels of drug were achieved during the course of the study (8 days) by this mode of administration. From preliminary pharmacokinetic studies a range of doses were chosen for administration via osmotic pumps which maintained targeted plasma levels from 0.25 to 3.0 μg/ml. Using this dosing regimen, half-maximal inhibition of IL-1-exacerbated, collagen-induced arthritis reflected by either clinical disease scores (fig. 10A) or weight loss (fig. 10B) occurred at CP-195543 plasma concentrations of approximately 0.4 to 0.5 μg/ml. At plasma concentrations of 2 to 3 μg/ml, CP-195543 caused complete suppression of disease and eliminated weight loss (fig. 10, A and B). Histological analysis of knee joints indicated that in collagen-immunized mice treated with osmotic pumps containing vehicle control, there was destruction of articular cartilage, erosion of bone and a massive influx of inflammatory cells in the subsynovial connective tissue. In contrast, in immunized mice treated with CP-195543 where clinical signs and weight loss approached control values (i.e., associated with plasma drug levels of 2–3 μg/ml), histological analysis of joints was indistinguishable from control, sham-immunized mice (results not shown). Collectively, these data provide evidence of the ability of CP-195543 to antagonize directly the effects of the exogenous administration of LTB4 in vivo and inhibit the development of an inflammatory disease, murine collagen-induced arthritis, in which either LTB4 or alternate ligands which may also act via interactions with LTB4receptors are formed endogenously.

Suppression of IL-1 exacerbated collagen-induced arthritis by varying doses (targeted plasma concentration range, 0.25–3.0 μg/ml) of CP-195543 administered in osmotic pumps with a prophylactic dosing regimen. Panel A shows the effect of CP-195543 on disease scores and compared with vehicle control, and panel B shows the effect of CP-195543 on weight loss compared with vehicle control. Data points are the means ± S.E. from four independent experiments.
Discussion
Herein we show that CP-195543 is an intrinsically potent and specific antagonist of LTB4 receptors on human cells. As exemplified by its ability to block LTB4-mediated CD11b up-regulation on neutrophils in whole blood, CP-195543 maintained potency in a high-protein environment. Experimental evidence from in vitro ligand binding assays suggested that CP-195543 is a noncompetitive antagonist of the high-affinity LTB4 receptor on human neutrophils. The high-affinity LTB4 receptor on neutrophils mediates chemotaxis and in an in vitro HN chemotaxis assay, CP-195543 blocked this functional response noncompetitively. Moreover, in confirmation of this noncompetitive pharmacological effect on the high-affinity receptor, in vivo, LTB4-dependent neutrophil recruitment in guinea pig skin also was antagonized by CP-195543 in a noncompetitive manner. In contrast to the high-affinity LTB4 receptor, both through the use of ligand binding assays and bioassays on human neutrophils, CP-195543 was shown to be a competitive antagonist of the low-affinity LTB4 receptor. In whole human blood CP-195543 competitively antagonized LTB4-mediated CD11b up-regulation on human neutrophils and, in addition, blocked LTB4-mediated CD11b up-regulation on eosinophils and monocytes with similar potencies. In mouse blood CP-195543 antagonized LTB4-mediated up-regulation of Mac-1 on MN at concentrations similar to that found for HN. When administeredvia osmotic pumps CP-195543 blocked the development of IL-1-exacerbated collagen-induced arthritis in mice. Plasma drug levels that were associated with efficacy in this disease model caused an approximately ≥1 log shift in the LTB4concentration-response curve for Mac-1 up-regulation on MN in vitro. CP-195543 did not inhibit prostaglandin synthesis in mice at doses 10-fold greater than those which blocked LTB4-mediated cell infiltration, providing evidence of a lack of cyclooxygenase inhibitory activity for this compound.
Previously we showed that CP-105696 was a potent inhibitor of LTB4 receptors on leukocytes from a variety of animal species and man, however, this activity was expressed robustly under assay conditions of low protein. In assays in high protein,e.g., whole blood and isolated neutrophils reconstituted into plasma, ≥2 log higher concentrations of CP-105696 were required for the same degree of inhibition (Showell et al., 1995; Showell HJ and Conklyn MJ, unpublished observations). Nevertheless, follow-up studies in a series of disease models in animals showed that CP-105696 was efficacious (Griffiths et al., 1995; Turneret al., 1996a; Gladue et al., 1996). At efficacious doses of CP-105696 given p.o., plasma levels of CP-105696 were found to be in a concentration range that predicted at least a 1 log shift in the LTB4-concentration response curves for CD11b/Mac-1 up-regulation from in vitrowhole-blood studies. Such drug levels were approached in Phase I human clinical studies; however, development of this compound was halted owing to its extraordinarily long plasma half-life (∼400 hr) (Listonet al., 1998). Based on the promising profile of CP-105696 we therefore set out to discover a second generation compound with similar intrinsic potency which maintained its potency in an environment of high protein. CP-195543 meets these criteria and as such represents a suitable candidate for clinical evaluation.
To date only limited clinical data are available in the public domain on LTB4 antagonists. LY-293111 was reported to block neutrophil infiltration subsequent to antigen challenge in atopic asthmatics; however, no evidence of efficacy in patients with active asthma was reported (Evans et al., 1996). Several other compounds were reported to be potent antagonists of LTB4 receptors as reflected by activity in a variety of in vitro binding and bioassays; however, no efficacy data from clinical trials are available on these compounds (reviewed in Djuric et al., 1992; Cohen and Yagaloff, 1994;Fretland and Penning, 1996; Sawyer, 1996). A limited number of published clinical studies in inflammatory diseases (e.g.,rheumatoid arthritis, psoriasis, inflammatory bowel disease) with either 5-LO inhibitors or FLAP antagonists, in which significant inhibition of LTB4 was predicted to have occurred, failed to show robust evidence of efficacy, and conclusions were offered that these diseases therefore do not depend on either 5-LO activity or LTB4 (de Jong et al., 1991; Weinblatt et al., 1992; Collawn et al., 1992; Ford-Hutchinson, 1993; Roberts et al., 1997; Hawkeyet al., 1997). Although this hypothesis may be correct, alternate hypotheses also can be offered; for example, in addition to LTB4 other ligands, e.g.,12(R)-hydroxyeicosatetraenoic acid and isoleukotrienes that bind to LTB4 receptors and do not derive from the 5-LO pathway, may be present in inflammatory lesions (Hammarströmet al., 1975; Woolard, 1986; Djuric and Fretland, 1989;Harrison and Murphy, 1995). Also, formation of LTB4 levels, sufficient for biological activity (≤1 nM), may be occurring in tissue even in the presence of 5-LO inhibitors/FLAP antagonists, and this is not reflected by eitherex vivo whole-blood or urine LT measurements. In support of this latter possibility, data from Turner et al. (1996a)show that CP-105696 blocked airway hyperreactivity in a monkey model of asthma. In contrast, in the same model, ZD2138, a potent 5-LO inhibitor, did not prevent the development of airway hyperreactivity even though 5-LO product formation was blocked ex vivo in whole blood (Turner et al., 1996b). Further, in clinical trials ZD2138, while blocking LTB4 production in blood ex vivo, showed diminished activity when compared with LTD4 antagonists in an antigen-challenge protocol in asthmatics (Nasser et al., 1994; Taylor et al., 1991; Findlay et al., 1992; Rasmussen et al., 1992).
As discussed above, CP-195543 affords a level of potency as an LTB4 antagonist that, providing adequate systemic exposure can be achieved in humans, will be a suitable pharmacological agent to test the validity of the hypothesis that LTB4 and (or) other ligands that bind to LTB4 receptors are important mediators of clinical inflammatory conditions.
Acknowledgments
The chemistry contributions of Dr. Kelvin Cooper, Michael Biggers, Bob Chambers, Mukesh Ramchandani and Joe Dibrino to the CP-195543 program are gratefully acknowledged. The authors are indebted to Doreen Gale for her help in typing this manuscript. Drs. Leland Loose, Steve Williams and Thasia Woodworth are thanked for their helpful comments.
Footnotes
-
Send reprint requests to: Henry J. Showell, Department of Cancer, Immunology and Infectious Diseases, Central Research Division, Pfizer Inc., Groton, CT 06340.
- Abbreviations:
- LTB4
- leukotriene B4, 5(S),12(R)-dihydroxy,6-cis,8,10-trans,14-cis-eicosatetraenoic acid
- 5-LO
- 5-lipoxygenase
- FLAP
- 5lipoxygenase-activating protein
- HN
- human neutrophil
- MN
- murine neutrophil
- MSM
- murine spleen membrane
- HM
- human monocyte
- HE
- human eosinophil
- IL-8
- interleukin 8
- MPO
- myeloperoxidase
- HEPES
- N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- EDTA
- ethylenediaminetetraacetic acid
- BSA
- bovine serum albumin
- HBSS
- Hanks’ balanced salt solution
- PBS
- phosphate-buffered saline
- FACS
- fluorescence-activated cell sorter
- Received October 21, 1997.
- Accepted February 5, 1998.
- The American Society for Pharmacology and Experimental Therapeutics
References
JPET articles become freely available 12 months after publication, and remain freely available for 5 years.Non-open access articles that fall outside this five year window are available only to institutional subscribers and current ASPET members, or through the article purchase feature at the bottom of the page.
|




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}