Article Text

Statistics from Altmetric.com
Editor—Retinitis pigmentosa (RP, MIM 268000) is the term applied to a clinically and genetically heterogeneous group of retinal degenerations primarily affecting the rod photoreceptors. RP is characterised by progressive loss of vision, initially manifesting as night blindness and reduction in the peripheral visual field, and later involving loss of central vision.1 Ophthalmoscopic examination typically shows pigmentary disturbances of the mid-peripheral retina. RP may be inherited as an autosomal recessive, autosomal dominant, digenic, or X linked trait. Autosomal recessive RP (ARRP) accounts for around 20% of all cases of RP, while sporadic RP, which is presumed to be recessive in most cases, accounts for a further 50%.2
Mutations causing autosomal recessive RP (ARRP) have been found in the genes encoding rhodopsin,3 the α and β subunits of rod phosphodiesterase,4 5 the α subunit of the cyclic GMP gated channel protein,6 and the genesRPE65,7 8 RLBP1,9 ABCR,10 andTULP1.11 12 In addition, genetic linkage studies have identified ARRP loci at 1q31-q32.1,13 14 2q31-q33,156cen-q15,16 and 16p12.1-p12.3.17 (MIM numbers for the loci identified by these studies are 180380, 180071, 180072, 123825, 180069, 180090, 601708, 600132, 600105, and 602594 respectively. No MIM number has yet been assigned to the 2q31-q33 and 6cen-q15 loci.) Of these four loci, only linkage to the 1q31-q32.1 locus (RP12) and the 6cen-q15 locus has been reported in more than one pedigree.
The first report of linkage of ARRP to 1q31-q32.1 was in a large inbred Dutch family with ARRP in which most patients exhibited para-arteriolar preservation of the retinal pigment epithelium (PPRPE).13This was followed by linkage of a second, consanguineous, pedigree from Pakistan.14 In both the Dutch and the Pakistani families there was evidence that only the branches of the family with PPRPE were linked to 1q31-q32.1, while other branches of each family had classical RP and were unlinked to 1q. In both cases the authors attributed this finding to non-allelic heterogeneity. The occurrence of two very similar eye conditions in one family would presumably be because of the high prevalence of recessive disorders in inbred populations. Recently the critical interval for the RP12 locus was reduced to a 3 cM region between the markers D1S412 and AFM207wb12.18
The human gene for a retinally expressed regulator of G protein signalling (RGS16/RGS-r) has been mapped to 1q25-1q31.19 Since it has been shown that RGS16/RGS-r can interact with the α subunit of transducin, a heterotrimeric G protein that is an integral part of the rod phototransduction cascade, theRGS16/RGS-r gene is an excellent functional candidate for ARRP.20 21 We sought to determine: (1) how important the 1q31-q32.1 locus is as a cause of ARRP by performing linkage analysis and homozygosity mapping in a panel of ARRP families from Europe and Asia, and (2) whether mutations inRGS16/RGS-r are responsible for ARRP at this locus (RP12). In order to perform linkage analysis and homozygosity mapping, 14 large/consanguineous families with ARRP from the UK (two families), Pakistan (nine families), and Spain (three families) were ascertained. In addition, PMK214, the family originally studied by Leutelt et al,14 was included for mutation screening of the candidate geneRGS16/RGS-r. Clinical examination of all affected subjects showed typical features of retinitis pigmentosa (pigmentary retinopathy, associated with symptoms of night blindness and loss of visual fields). Informed consent was obtained by local clinicians.
To identify the locus responsible for the disease in each family we performed a linkage or homozygosity analysis. The initial set of markers chosen to be analysed were those corresponding to the loci of genes responsible for ARRP, and those known to flank the ARRP loci for which no gene has as yet been identified (table 1). When evidence of linkage was obtained for the 1q31-q32.1 locus, further polymorphic markers from this region were analysed to determine whether recombinant subjects within the family would permit further refinement of the locus. Marker order was determined from the Généthon sex averaged genetic map.22 Primers were obtained from the MapPairs set (Research Genetics, Huntsville, AL), or synthesised commercially according to data from GDB (Johns Hopkins University:http://gdbwww.gdb.org/).
Known loci for ARRP including some of the polymorphic markers used in this study
Non-radioactive PCR was performed with 300 ng of genomic DNA, 10 pmol of each primer, 200 μmol/l dNTPs, 1.5 mmol/l MgCl2, and 1 unit of Taq DNA polymerase. A three stage PCR consisting of 35 cycles of 94°C, 50-62°C, and 72°C, each for one minute, was used. The amplified products were then separated by electrophoresis on 6-8% non-denaturing polyacrylamide gels (Protogel, National Diagnostics) and stained with ethidium bromide. Two point lod scores were calculated using the LINKAGE package.23
Primers for heteroduplex analysis and direct sequencing of the five exons and the intron-exon boundaries of theRGS16/RGS-r gene were designed using the published genomic sequence19 (Genbank AF009356) as follows: exon 1F - GCCTGCCACCATCCTGCCTAC; 1R - CTCTCT CTCAAGCCTCTAGC (208 bp); exon 2F - CACCAC CCAGAGCTCTTCTGG; 2R - GTTGCCAAGGTCCA CTTAGCG (312 bp); exon3F - CAGCAATCACCAAGCAAGCC; 3R - CAGGCTCTCAGCAGTCCTGG (281 bp); exon 4F - GATCCCCTAGTGTGCCAGCCTC; 4R CTCTACACCTAGCCCTTCCTCC; (429 bp); exon 5F CACTTCCTGATGCAGCATCCG; 5R CTCTTCCCG GCTGGCTTCC (372 bp). Polymerase chain reaction PCR was performed in a 50 μl reaction with 1 μg of genomic DNA, 20 pmol of each primer, 200 μmol/l dNTPs, 0.75-1.5 mmol/l MgCl2, and 1 unit ofTaq DNA polymerase. A three stage PCR consisting of 40 cycles of 94°C, 55-60°C, and 72°C, each for one minute, was used. The resulting product was allowed to cool slowly to room temperature to maximise the formation of heteroduplexes.24
All five exons of RGS16/RGS-r were directly sequenced in two or more members of each potentially 1q linked family using an automated sequencer (ABI Biosystems model 373). PCR products were purified and then reamplified using the FS kit (Perkin Elmer). Variants were sequenced in both the forward and reverse directions to confirm the location of sequence changes.
AciI restriction digests were performed directly on 20 μl of exon 4 PCR product by overnight incubation at 37°C. The resultant bands were photographed after electrophoresis on a 2% agarose gel and stained with ethidium bromide.
Three of the 14 families studied showed evidence of linkage to the 1q31-q32.1 ARRP locus and were excluded by linkage analysis from all of the other ARRP loci (fig 1). Two of these pedigrees were of Pakistani origin (18RP and RP112) and one was Spanish (RP23/91). Analysis of additional polymorphic markers on 1q in these families did not permit further refinement of the RP12 locus.

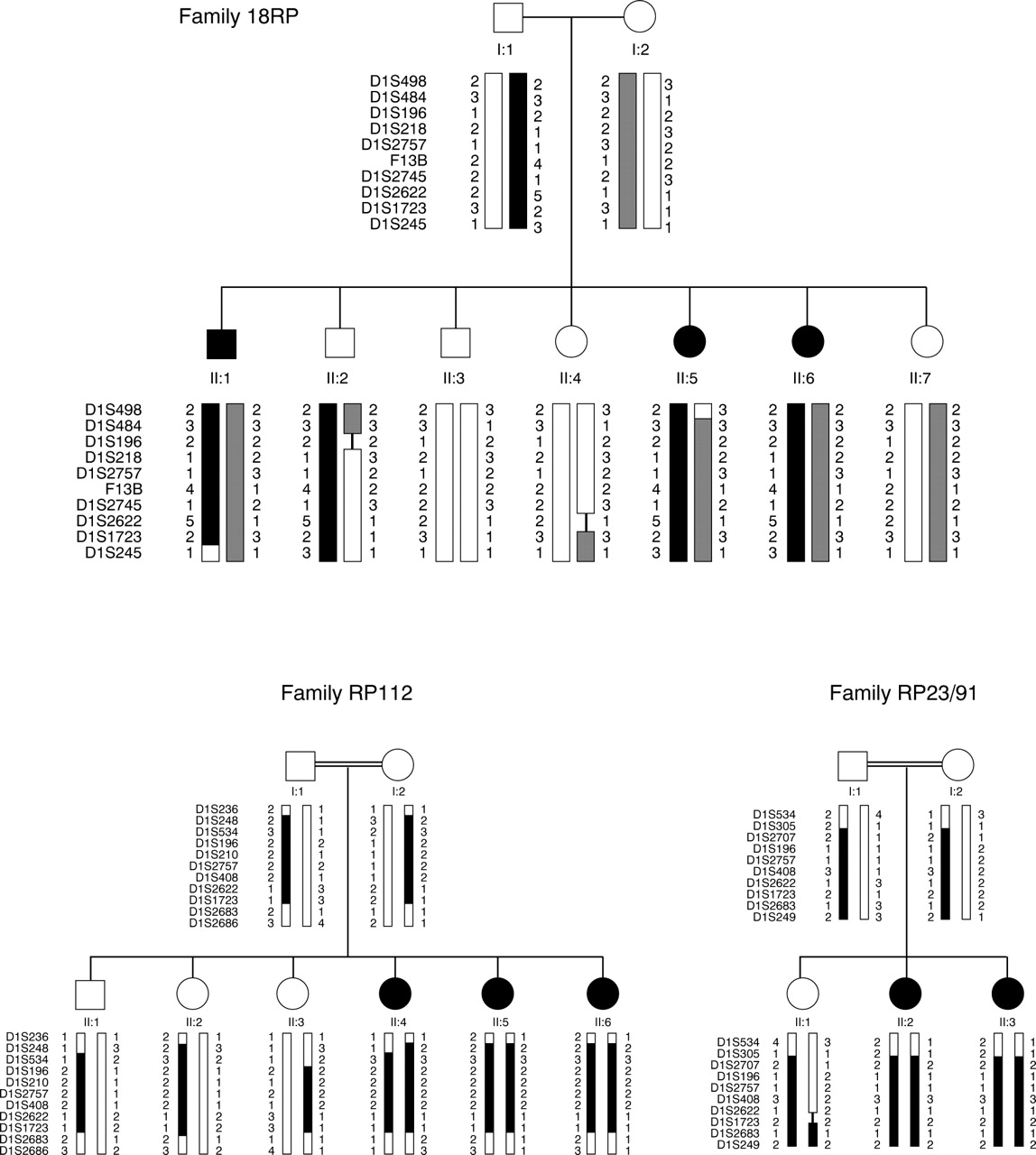{kind=link}
Pedigrees of families exhibiting evidence of linkage to 1q31-q32.1 showing haplotypes for polymorphic markers in this region. Maximum lod scores for each family were: 18RP Zmax=1.70 (at θ=0.00) for the marker F13B, RP112 Zmax=2.48 at D1S2622, and RP23/91 Zmax=1.63 at D1S408. The RP12 locus lies between markers D1S2757 and D1S2622. Black or hatched bars situated between the marker alleles indicate the affected haplotype.
Clinical examination of the small Spanish RP23/91 family showed para-arteriolar preservation of the retinal pigment epithelium (PPRPE) as previously described in RP12 linked families.13 14 A relative lack of pigmentation adjacent to retinal arterioles was also seen in the Pakistani RP112 family. This feature could not be conclusively shown in the 18RP pedigree, possibly because the three affected subjects are only 22, 14, and 13 years old respectively. The fundus photograph showing PPRPE in the original paper of van Soestet al 13 shows heavy retinal pigmentation and was taken from a 41 year old patient.
In order to show that the three families of Pakistani origin were not distantly related and that RP12 is an important ARRP locus in that population, we compared the disease haplotypes for markers within and immediately surrounding the RP12 locus in each family (table 2). The maternal disease haplotype in family 18RP and the homozygous disease haplotype for PMK214 was identical for three adjacent markers (F13B, D1S2745, and D1S1660) within the RP12 locus. A founder effect could not be excluded in these two families. However, the paternal disease haplotype in 18RP and that seen in RP112 were entirely different, indicating that at least three different RP12 alleles may exist in these families.
Comparison of the disease haplotypes in the three families of Pakistani origin for polymorphic markers in the critical region on 1q31
These four families then underwent mutation screening of the five exons of the RGS16/RGS-r gene by heteroduplex analysis and direct sequencing. Heteroduplexes were observed with the exon 4 product in the parents from consanguineous families RP112, PMK214, and RP23/91, but not in their affected children. No heteroduplexes were observed in members of 18RP.
Direct sequencing of the exon 4 and exon 5 products showed polymorphisms in introns 3 (−10 a to g) and 4 (−35 c to t) respectively. The intron 3 a>g change, which creates a recognition site for the restriction enzyme AciI, was found to be present in the heterozygous state in the parents from families RP112, PMK214, and RP23/91 in whom a heteroduplex band had been observed, and in the homozygous state in their affected children.AciI restriction digest analysis was, therefore, used to show the segregation of this polymorphism in the large linked family PMK214. All seven affected members of this consanguineous pedigree were found to be homozygous for the intron 3 (−10 a to g) polymorphism. This indicates thatRGS16/RGS-r cannot be excluded from the critical genetic interval for RP12 in this family. In addition, 10 Pakistani controls were subjected to AciI restriction digest analysis, two of whom were found to be heterozygous for the intron 3 polymorphism. No sequence variants within the coding regions of RGS16/RGS-r were observed in any subject.
While there are nine published loci for autosomal dominant RP (ADRP),rhodopsin mutations may account for up to 25% of cases.25 In contrast, none of the 11 published genes/loci for ARRP has been estimated to account for more than 5% of recessive RP. In this study, two out of nine Pakistani and one out of five European families showed evidence of linkage to RP12 and exclusion from the other loci. While this result is based upon a small sample it suggests that mutations in the gene situated at this locus may be responsible for a significant proportion of ARRP, at least in the Pakistani population.
In rod photoreceptors, the GTP bound α subunit of the heterotrimeric G protein transducin (Tα) activates the effector enzyme, cGMP phosphodiesterase (PDE), by binding to its inhibitory gamma subunit (PDEγ). Hydrolysis of GTP by the intrinsic GTPase activity of Tα releases bound PDEγ and results in inactivation of PDEαβ during the phase of recovery from photoactivation. In vitro experiments with purified Tα subunits have shown that the intrinsic GTPase activity of Tα is inadequate to account for the rapid hydrolysis of GTP that is observed in vivo.26 It has been postulated, therefore, that a GTPase activating protein (GAP) that is capable of greatly accelerating this process must exist in rod outer segments.
RGS16/RGS-r, which is highly expressed in the retina, has been proposed to fill the role of the Tα GAP.19 It has been shown in vitro that the rate of transducin GTPase activity in the presence of RGS16/RGS-r is sufficient to correlate with the in vivo recovery kinetics of the visual cascade, and that in addition RGS16/RGS-r facilitates signal termination by competing with and thereby displacing PDEγ from Tα.20 21 This biochemical evidence suggests an important role for RGS16/RGS-r in retinal photoreceptors.
Since the human gene for RGS16/RGS-rhas been mapped to 1q25-1q31,19 and since in the present study it could not be excluded from the RP12 locus by analysis of the intron 3 polymorphism in the linked family PMK214, this gene must remain as a potential candidate for the ARRP linked to this locus. We failed to find any sequence changes in the coding region of the gene in our four families, though it is possible that disease causing mutations may lie in the promoter region of the gene, which was not screened.
This study must, however, reduce the prospect ofRGS16/RGS-r mutations being found in other 1q linked families and lead to a renewed search for candidates within the RP12 critical interval.
Acknowledgments
We would like to thank the family members for taking part in this study. Drs David A R Bessant and Annette M Payne are supported by the Medical Research Council of the UK (grant No G9301094). We would also like to thank the Wellcome Trust for an Equipment Grant for the sequencing facility (No 039283/2/93/Z/MW/JF).