Article Text

Statistics from Altmetric.com
- Jaks, Janus kinases
- IL, interleukin
- NK, natural killer
- SCID, severe combined immunodeficiency
- STAT, signal transducer and activator of transcription
- Th, T helper
- Janus kinases
- Jaks
- STATs
- signal transducer and activator of transcription
- immunosuppression
- cyokine signalling
While many effective immunosuppressive drugs exist and are in wide use, most target ubiquitously expressed molecules. Examples include steroids, ciclosporin A, tacrolimus, mycophenolate mofetil, and sirolimus. Consequently, these drugs have adverse effects unrelated to their immunosuppressive actions, and much effort has been directed towards identifying molecular targets with expression restricted to immune and inflammatory cells. In principle, such drugs could provide immunosuppression yet lack the toxicity associated with current therapies.
It is now well established that cytokines have critical roles in regulating immunity and inflammation; indeed targeting cytokines themselves has been an effective new strategy for immunosuppression.1 However, targeting intracellular cytokine signalling also now appears to be a viable new approach. Janus kinases (Jaks) and signal transducers and activators of transcription (STATs) have been demonstrated to be critical elements in signalling by certain families of cytokines. Importantly, the generation of a selective inhibitor of Jak3 appears to be feasible and effective. This report discusses the approach of targeting Jaks and STATs, with emphasis on Jak3 and a newly identified Jak3 antagonist.
CYTOKINES AND IMMUNOREGULATION
Cytokines comprise more than 50 secreted factors that regulate processes ranging from body growth, lactation, and adiposity to haematopoiesis but are especially important for regulating inflammatory and immune responses.2 That is, cytokines control both innate and adaptive immunity and are critical for lymphoid development and homoeostasis, as well as the ultimate differentiation of helper and memory T cells.
Interleukin (IL)-2, 4, 7, 9, 15, and 21 comprise one family of cytokines that uses a common receptor subunit termed the common γ chain, or γc (table 1).3–5 These cytokines are particularly important with respect to lymphoid development and function. For instance, IL-7 is critical for lymphocyte development and function and mice lacking IL-7R expression have arrested thymocyte development at the double negative (CD4–CD8–) stage.6–11 In addition, IL-7 is also important for the homoeostasis of mature peripheral lymphocytes and along with IL-15, IL-7 is important for generation of CD8+ memory cells.12–16 IL-15 is also essential for natural killer (NK) cell development.14,15,17,18 IL-4 is critical for the differentiation of T helper (Th) 2 cells, but also works in concert with IL-21 to regulate B cell function.19–22 IL-2, long recognised as a T cell growth factor, is also critical for maintenance of peripheral tolerance.23 Mice lacking either IL-2 or its receptor (IL-2Rα and β) have normal development of T cells but exhibit severe lymphoproliferative disease; whether this is due to cell death induced by impaired activation, lymphocyte selection, or the development of regulatory T cells has not been established.24–27
Cytokines that share the γc receptor subunit and their functions
CYTOKINE SIGNALLING AND PATHOGENESIS OF SEVERE COMBINED IMMUNODEFICIENCY (SCID)
All type I and II cytokine receptor subunits, including γc associate with Jaks (Jak1, Jak2, Jak3, and Tyk2), which are critical for initiating signalling (fig 1).24–28 While some Jaks are widely expressed and bind multiple cytokine receptors, Jak3 is predominantly expressed in haematopoietic cells and appears to interact uniquely with γc.29–34

Role of Janus kinases (Jaks) and signal transducers and activators of transcription (STATs) in cytokine signalling. Type I and Type II cytokine receptors associate with Jaks. Cytokine binding activates the Jaks, which phosphorylate the receptors allowing the recruitment of STATs. STATs are phosphorylated and accumulate in the nucleus where they regulate gene expression.
Severe combined immunodeficiency (SCID), a group of rare, inherited disorders in primary immunity, affects immune cell development and results in major defects in host defence.35–38 Infants with SCID develop opportunistic infections and have significant mortality during the first two years of life, unless they receive stem cell transplantation. Mutations of γc underlie X-linked SCID (SCIDX-1 or X-SCID) and account for roughly half of all known cases of SCID (fig 2).3,36,39 Deficiency of γc blocks signalling by IL-7, 15, 4, and 21, and thus it is not surprising that mutations of this chain have such dramatic consequences. The phenotype of this form of SCID is designated T−B+NK− SCID, and is indicative of the absence of T and NK cells. Although B cells are present, they are poorly functional, with defective B cell activation and abnormal class switching. Clinically, X-SCID patients have hypogammaglobulinaemia and do not mount specific antibody responses to immunisation. These humoral defects are explained in part by the lack of helper T cell function as well as intrinsic B cell abnormalities due to impaired cytokine signalling.

Cytokine signalling and the pathogenesis of severe combined immunodeficiency (SCID). Cytokines that use γc, interleukin (IL)-2, 4, 7, 9, 15, and 21, are essential for many aspects of immune cell development and function (see table 1). Mutations of IL-7R, γc, and Janus kinase3 (Jak)3 disrupt the action of these cytokines and lead to SCID. Mutations of these genes account for roughly two thirds to three quarters of the cases of SCID. X-SCID, X-linked SCID, AR-SCID, autosomal recessive SCID.
The association of Jak3 with γc suggested the former as another candidate gene and Jak3 deficiency is now known to account for approximately 7–14% of SCID (see fig 2).40–45 It has an identical phenotype to X-SCID, T−B+NK−. Mutations of IL-7R also result in autosomal recessive SCID and underlie about 10% of SCID (see fig 2),9,37 but its phenotype, T–B+NK+ SCID, differs from X-SCID and Jak3 SCID. This is consistent with the finding that IL-7 is essential for the development of T but not NK cells. In aggregate, mutations of the IL-7R/γc/Jak3 axis account for nearly three-quarters of SCID.
DEVELOPMENT OF A SELECTIVE JAK3 ANTAGONIST
The observation that Jak3 is required for immune cell development and function implied that Jak3 might be desirable therapeutic target—that is, because Jak3 mutations result in SCID, purposefully interfering with Jak3 activity or function might generate a novel class of immunosuppressants. Moreover, following successful stem cell transplantation, patients with Jak3-SCID are healthy.39 Therefore a corollary is that because Jak3 deficiency results in immunodeficiency and not pleiotropic defects, a highly specific Jak3 inhibitor should also have very limited and precise effects. This contrasts sharply with the widely used immunosuppressive drugs (ciclosporin A, tacrolimus, and steroids), which are directed against ubiquitous targets and have diverse side effects. In principle, a potent and selective Jak3 inhibitor might be expected to have advantages over the current agents.
But is the generation of a selective protein tyrosine kinase inhibitor an achievable goal? Kinases catalyse the transfer of phosphate from ATP and many inhibitors interact with the kinase ATP-binding cleft. Given that hundreds, if not thousands of other proteins also bind ATP, it might be predicted that it would be difficult or even impossible to generate selective kinase inhibitors. Indeed, until recently, the idea of generating a selective kinase inhibitor has met with considerable scepticism. However, the successful generation of imanitib (Gleevec), which targets the kinase BCR-Abl, has firmly established that targeting protein kinases can be a sound approach.46 Currently, kinases are among the most popular targets for drug development.
Since the generation of selective kinase inhibitors appears to be feasible, are there specific challenges in generating a specific Jak3 antagonist? One critical issue is the extent to which other Jaks are inhibited. Inhibition of Jak2 would be particularly problematic in that experimental deficiency of this kinase results in embryonic lethality.47 Jak2 is essential for signalling by many haematopoietic cytokines including erythropoietin, thrombopoietin, macrophage-colony stimulating factor (M-CSF), and granulocyte macrophage-colony stimulating factor (GM-CSF). By inference, pharmacological inhibition of Jak2 in vivo would be expected to result in anaemia, thrombocytopenia, and leucopenia; obviously, such an inhibitor would hardly be an improvement over the existing immunosuppressant compounds. Similarly, a Jak3 inhibitor that inhibited Jak1 might also be a problem in that Jak1 is critical for signalling via interferon (IFN) and gp130 cytokines. Inhibition of Jak1 might lead to dramatically increased susceptibility to viral and bacterial pathogens. Moreover, Jak1 knockout mice die perinatally from an ill defined neurological abnormality,48 which could also be an issue with Jak1 inhibition.
An orally available, selective Jak3 antagonist has now been developed (fig 3).49 The drug, designated CP-690 550 has nanomolar potency against Jak3 in vitro and is approximately 30 and 100 times less potent for Jak2 and Jak1, respectively. Importantly, the drug has been found to be efficacious in preventing transplant rejection in two animal models, a murine heterotopic heart transplant model and a non-human primate renal transplant model. In both cases, the drug significantly prolonged graft survival. While many compounds are effective in murine models of organ transplantation, their utility in primate models is a better predictor of efficacy in humans. In the primate model, CP-690 550 was more effective as single agent than ciclosporin A, the most widely used drug for transplant rejection, establishing that Jak3 blockade alone can successfully block alloimmune responses.49 CP-690 550 was found to potently block cytokine signalling and cytokine induced gene expression without affecting T cell receptor signalling. This is important because one could imagine that the combination of a calcineurin inhibitor, which would block signals emanating from the T cell receptor, with CP-690 550 might have synergistic effects.
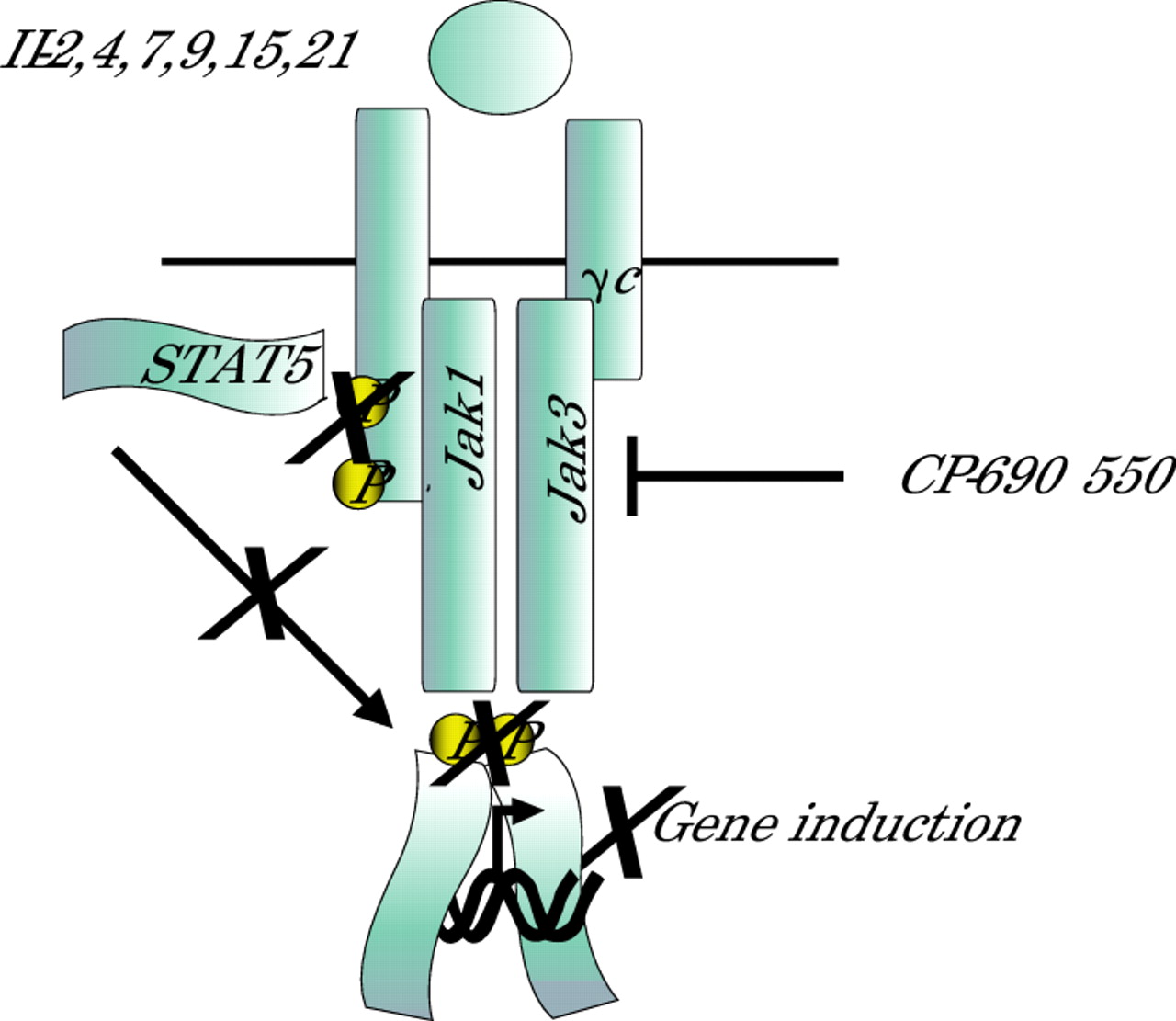
A selective Jak3 antagonist inhibits Janus kinase (Jak) activity, blocking early cytokine signalling and resultant signal transducer and activator of transcription (STAT) activation. The drug, CP-690 550, abrogates cytokine dependent gene regulation and subsequent lymphocyte activation. In this manner it blocks transplant rejection. STATs also have critical functions in mediating cytokine signalling and in principle, targeting STATs would also be useful to generate novel immunosuppressants.
Importantly, we found that the drug did not cause granulocytopenia or thrombocytopenia; at the highest doses, mild anaemia was noted, indicating that Jak2 antagonism is likely not to be an overwhelming concern problem with CP-690 550. There was a trend in the reduction of CD8+ T cells, but no significant decline in total T lymphocytes. Animals treated with CP-690 550 did show a modest decline in NK cells, presumably due to inhibition of IL-15 signalling; whether this will be clinically relevant remains to be determined.
A theoretical issue with the use of a Jak3 antagonist relates to inhibition of IL-2 signalling. Because IL-2 deficiency is associated with autoimmunity, it is conceivable that a Jak3 inhibitor could have effects on peripheral tolerance. However, there was no evidence of this in the recent study.49 In addition, monoclonal antibodies against IL-2Rα (CD25, basiliximab and daclizumab) are used for transplant rejection.50 However, there is no evidence at this time that these monoclonal antibodies are associated with a breakdown in peripheral tolerance. Furthermore, it is likely that a Jak3 inhibitor, by antagonising all of the γc cytokine receptors, would be more immunosuppressive than an IL-2R antagonist. It therefore seems unlikely that treatment of patients with a selective Jak3 inhibitor would result in autoimmunity and breakdown in peripheral tolerance. Obviously though, this possibility will need to be borne in mind in clinical trials.
In addition to transplant rejection, clearly CP-690 550 has potential utility in a number of other clinical settings. The issue of adverse effects is especially important given that autoimmune disorders occur more frequently in young women in their childbearing years and that treatment is often lifelong. Inhibition of Jak3 might be useful for the treatment of psoriasis, psoriatic arthritis, graft versus host disease, multiple sclerosis, inflammatory bowel disease, systemic lupus erythematosus, rheumatoid arthritis, as well as other autoimmune diseases. IL-4 and IL-9 promote allergic responses, so a Jak3 inhibitor might also be useful in these disorders.51–53
Another setting in which the utility of a Jak3 inhibitor will need to be considered is haematological malignancy. Some leukaemias and lymphomas are associated with constitutive activation of Jak3; as such, one might anticipate that inhibition of Jak3 might be an effective agent for these tumours.54,55
TARGETING OTHER JAKS
The discrete but essential function of Jak3 makes this kinase a logical pharmacological target. The successful generation of a selective inhibitor (CP-690 550) establishes the feasibility of generating other selective Jak inhibitors. However, the first question, based on physiological function, is whether antagonism of other Jaks would be useful and for what settings (table 2).
The four members of the Janus kinase (Jak) family and the major cytokines reported to activate them
Tyk2 is known to be involved in signalling by type I IFNs, but the phenotype associated with its deficiency is relatively modest.56–58 Tyk2−/− mice have residual signalling by type I IFNs and limited susceptibility to viral infections. In contrast, IL-12 signalling is more significantly impaired in these mice. Furthermore, mice with a mutation in Tyk2, have marked resistance to the development of collagen induced arthritis.59,60 Therefore, it might be anticipated that targeting Tyk2 would be a useful strategy for the treatment of Th 1 mediated disorders such as arthritis. It should be noted that IL-23 also uses IL-12Rβ and activates Tyk2, but the effect of Tyk2 deficiency on IL-23 responses has not been examined.61,62
As noted above, experimental deficiency of Jak1 or Jak2 is lethal, and therefore one might conclude that targeting these kinases would not be a useful strategy. However, one could imagine that in the treatment of cancers or leukaemia, a greater level of toxicity may be acceptable, assuming that the drug is efficacious. It is possible that drugs with effects on Jak2 and Jak1 might be useful in these settings, despite their likely toxicities.
TARGETING STATS
Jaks are activated following cytokine stimulation, and their substrates include cytokine receptors, which when phosphorylated, bind signalling molecules, including members of the STAT family (see fig 1). Latent cytosolic transcription factors, STATs accumulate in the nucleus, bind DNA and regulate gene expression following activation.24,27 There are seven STATs and their specific and critical roles are evidenced by gene targeted mice (summarised in table 3).27,28
Summary of the major cytokines that activate various signal transducers and activators of transcription (STATs) and the phenotypes of STAT deficient mice
At present, it has been simpler to identify small molecules that interfere with Jaks than STATs, as the former are enzymes. However, because of their critical and selective functions, STATs remain attractive targets. Because they do not have enzymatic activity, one must block STAT expression, recruitment to cytokine receptors, dimerisation or DNA binding. Cytokine recruitment and dimerisation are mediated by phosphotyrosine-SH2 interactions and so peptidomimetics have been designed to disrupt these interactions.63 However, phosphotyrosine-SH2 interactions are important for many different receptors and the successful generation of peptidomimetics has been elusive.
Assuming a successful strategy can be devised, which STATs would be useful to target? In terms of immunoregulation, two STATs that might be useful targets are STAT4 and STAT6.20,21,28,64 These STATs are critically important for the differentiation of helper T cells. IL-4 activates STAT6, promoting Th 2 cell differentiation and allergic response, whereas IL-12 activates STAT4 and drives differentiation of naive T cells to Th 1 cells. These cells produce IFNγ, which is not only critical for host defence against intracellular pathogens but also contributes to many autoimmune diseases. Constitutive activation of STAT3 and STAT5 has been noted in a significant proportion of tumours and increasing attention is being paid to targeting these STATs in cancer.63,65,66 Interestingly, statins have also been reported to inhibit Th 1 mediated disease and block STAT4 activation; the exact mechanism(s) underlying these effects remains to be determined.67,68
NEGATIVE REGULATORS OF CYTOKINE SIGNALLING
A number of mechanisms downregulate cytokine signalling including tyrosine phosphatases, protein inhibitors of activated STATs (PIAS) family members, and suppressors of cytokine signalling (SOCS) (fig 4).69 SOCS proteins are classic feedback inhibitors of signalling; by virtue of their SH2 domains, they inhibit Jak activity, block recruitment of signalling molecules to cytokine receptors and promote degradation of cytokine receptors and/or Jaks. In principle, mimics or inducers of SOCS and PIAS proteins would be immunosuppressive in that such agents would be expected to attenuate effects of cytokines. However, at this point it is not clear how such drugs would be designed. Similarly, activators of the tyrosine phosphatases that target Jaks and STATs would in principle do the same; again, it is not clear if this is truly a feasible approach.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Negative regulation of cytokine signalling. Cytokine signalling is attenuated by tyrosine phosphatases (PTPases), which dephosphorylate Janus kinases (Jaks), cytokine receptors and signal transducers and activators of transcription (STATs); several PTPases have been implicated in these functions. In addition, a family of protein inhibitors of STATs (PIAS) has also been shown to antagonise the action of these transcription factors. Finally, a family of cytokine inducible regulators of the Jak-STAT pathway, suppressors of cytokine signalling (SOCS proteins) has also been identified. In principle, activators, inducers, or mimics of the negative regulators of cytokine might also be useful as novel immunosuppressants.
SUMMARY AND CONCLUSIONS
Studies in humans with mutations of Jak3 and its associated receptor subunits have predicted that selective Jak3 antagonists could represent a new class of immunosuppressants. In contrast to existing drugs, Jak3 has limited tissue expression and discrete functions. A highly selective inhibitor would therefore not be expected to result in toxicities seen with existing immunosuppressants. A selective Jak3 antagonist, CP-690 550, has now been developed and is effective in models of transplant rejection including studies in non-human primates. Importantly, it is also not associated with unacceptable adverse effects indicative of substantial Jak2 inhibition. As the drug moves towards clinical trials in humans, it will be important to determine other clinical settings ranging from autoimmunity and allergy to cancer in which this new agent might be useful. Given the importance of cytokines in regulating all phases of the immune response, the successful generation of a selective Jak inhibitor demonstrates the feasibility of targeting Jaks and raises the question as to whether targeting other Jaks might also be useful. In principle, targeting STATs and the negative regulators of cytokine signalling could be effective and these molecules will surely continue to receive considerable attention as therapeutic targets.