Article Text

Abstract
Exacerbations of COPD are thought to be caused by complex interactions between the host, bacteria, viruses, and environmental pollution. These factors increase the inflammatory burden in the lower airways, overwhelming the protective anti-inflammatory defences leading to tissue damage. Frequent exacerbations are associated with increased morbidity and mortality, a faster decline in lung function, and poorer health status, so prevention or optimal treatment of exacerbations is a global priority. In order to evolve new treatment strategies there has been great interest in the aetiology and pathophysiology of exacerbations, but progress has been hindered by the heterogeneous nature of these episodes, vague definitions of an exacerbation, and poor stratification of known confounding factors when interpreting results. We review how an exacerbation should be defined, its inflammatory basis, and the importance of exacerbations on disease progression. Important aetiologies, with their potential underlying mechanisms, are discussed and the significance of each aetiology is considered.
- AECOPD, acute exacerbation of chronic obstructive pulmonary disease
- CRP, C-reactive protein
- CXCL8, interleukin 8
- E selectin, endothelial selectin
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- GROα, growth-related oncogene α
- IL, interleukin
- LTB4, leukotriene B4
- NE, neutrophil elastase
- PEF, peak expiratory flow
- sICAM-1, soluble intracellular adhesion molecule 1
- SLPI, secretory leukoproteinase inhibitor
- TNFα, tumour necrosis factor α
- chronic obstructive pulmonary disease
- exacerbations
- aetiology
Statistics from Altmetric.com
- AECOPD, acute exacerbation of chronic obstructive pulmonary disease
- CRP, C-reactive protein
- CXCL8, interleukin 8
- E selectin, endothelial selectin
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- GROα, growth-related oncogene α
- IL, interleukin
- LTB4, leukotriene B4
- NE, neutrophil elastase
- PEF, peak expiratory flow
- sICAM-1, soluble intracellular adhesion molecule 1
- SLPI, secretory leukoproteinase inhibitor
- TNFα, tumour necrosis factor α
Chronic obstructive pulmonary disease (COPD) is a progressive chronic disease which is characterised by an inexorable decline in respiratory function, exercise capacity, and health status.1 This underlying state is interrupted by exacerbations of symptoms which vary in severity and frequency both between patients and during the course of one patient’s illness. These exacerbations are important, not only because of their impact on an individual’s life but also because of their long term effects on health status, morbidity, and mortality. In fact, exacerbation frequency is one of the most important determinants of health related quality of life.2 Exacerbations are a significant cause of hospital admission and readmission, and the burden placed on health resources is immense.3 Just as COPD as an entity is heterogeneous, so are exacerbations, and despite wide scientific interest in these episodes, there is still much debate regarding how exacerbations should be defined, their aetiology, and their prognostic significance.
DEFINITION
Given the heterogeneity and the complexity of COPD in the stable state, it is perhaps understandable that there is no standardised definition of an acute exacerbation of COPD (AECOPD) in research or in clinical practice. Exacerbation symptoms vary between individuals,4 the most common being breathlessness, wheeze, chest tightness, increased cough, and increased sputum volume and purulence.5 Non-specific symptoms are also common (including fatigue and general malaise),5 as are small but significant changes in objective measurements such as a transient reduction in forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), and peak expiratory flow (PEF).3 However, no single or group of symptoms or measurements are diagnostic for an exacerbation, so working definitions have arisen from research to assess the clinical condition of the patients and to evaluate their responses to treatment.
The most referenced definition remains Anthonisen et al6 which divided patients into three groups according to their symptomatology. Type 1 exacerbations were defined by the presence of increased breathlessness, sputum volume and sputum purulence; type 2 by the presence of two of these symptoms, and type 3 by the presence of one of these symptoms in addition to one of the following: an upper respiratory tract infection in the past 5 days, fever without other cause, increased wheezing or cough, or an increase in heart rate or respiratory rate by 20% compared with baseline readings. This definition was devised prospectively to analyse the effect of antibiotics on exacerbations and not to structure patient management. More recent studies have adapted these criteria with varying degrees of success.
In order to compare studies and to clarify management strategies, a consensus definition is required. The Aspen workshop7 defined AECOPD as “a sustained worsening of the patient’s condition from the stable state and beyond normal day to day variations, that is acute in onset and necessitates a change in regular medication in a patient with underlying COPD”. While a sound description of an exacerbation, when used alone, the definition is still unwieldy as it provides no clear definition of “sustained” (the authors explain that this should be for longer than 24 hours but that acute presentations should not be excluded) nor clear guidance on how “worsening of the patients condition” should be assessed. However, when used with validated daily diary cards, the definition becomes more structured with a means for differentiating between day to day symptom variability and true exacerbations.
It may be that a concise and readily applicable definition cannot be reached until we have a sound understanding of the inflammatory process which underpins the clinical presentation. Once this is achieved, a biomarker may present itself as indicative of an exacerbation when raised to a significant degree and, indeed, may characterise the aetiology.
INFLAMMATORY CHANGE AND AECOPD
There is a substantial and cogent body of evidence that airway inflammation is prevalent in stable COPD and is fundamental to its pathogenesis. Neutrophil proteinases, especially neutrophil elastase (NE), have been shown to cause all components of COPD, including emphysema, mucous gland hyperplasia and mucus hyper secretion,8 and studies have shown that smokers have increased numbers of neutrophils in lung tissue,9 sputum,10,11 and bronchoalveolar lavage (BAL) fluid.12 Many inflammatory proteins have been found to be raised in stable COPD compared with healthy controls who smoke, including tumour necrosis factor α (TNFα), interleukin-1β (IL-1β), and the chemoattractants leukotriene B4 (LTB4), interleukin 8 (CXCL8), and growth-related oncogene α (GROα).13–15
There is evidence that inflammation is amplified during exacerbations, but studies have been small and have varied in their definitions of an exacerbation. Increased neutrophil counts have been found in the bronchial walls and in BAL fluid samples from patients during exacerbations of COPD,16,17 with increased neutrophil sequestration in the pulmonary microcirculation prior to migration into the airways.18 Increases in various inflammatory markers have also been found at COPD exacerbation compared with the stable state, including inflammatory cytokines, IL-6, CXCL8, endothelin-1, the neutrophil chemoattractant (LTB4), and NE (table 1).19–21
Inflammatory cells and proteins raised in exacerbations of COPD
In one recent study CXCL8, soluble intracellular adhesion molecule 1 (sICAM-1), and soluble endothelial selectin (E selectin) (all of which are required for neutrophil migration from the circulation into airspaces) were higher in patients with COPD who were hospitalised for an acute exacerbation than in healthy controls and those with stable COPD. Furthermore, serum levels of CXCL8 and sICAM declined significantly during treatment.22 The rise in inflammatory cells and proteins during an exacerbation and the subsequent reduction with symptom resolution suggests a causative link between excess inflammation and symptomatology, but this finding has not been reproduced in all studies (table 1). In another recent study, serum levels of TNFα, IL-6, CXCL8 and C-reactive protein (CRP) were measured in patients with severe COPD admitted with respiratory failure due to an exacerbation. Although levels of CRP and IL-6 were raised, there was no significant change during recovery.23 The disparity in results may be explained by the small numbers in each study and the wide heterogeneity of exacerbations since clear rises and falls in CRP have been noted by others at the start and resolution of the episode.24 However, it is of fundamental importance to understand the impact of each exacerbation on the disease process. There are conflicting data as to whether exacerbations are associated with a step-like decline in physiology or whether patients generally return to their pre-exacerbation status. If inflammatory markers do not return to pre-exacerbation levels, it is likely that the increased ongoing inflammation would influence the subsequent decline in lung function.
The high levels of inflammatory proteins and neutrophils within the lung are believed to initiate the pathology seen in COPD. Environmental pollution, cigarette smoke, and adherent pathogens activate respiratory epithelial cells and phagocytes. These cells initiate a complex inflammatory cascade, with secretion of pro-inflammatory proteins by epithelial and endothelial cells, leucocytes, and other cells in the connective tissue.1 Some of these proteins (including LTB4 and CXCL8) can stimulate neutrophil migration towards the site of injury by means of a chemoattractant gradient.32 To pass through connective tissue, neutrophils are believed to release enzymes which degrade components of the extracellular matrix; once released, these proteinases diffuse away from the neutrophil. Initially, the concentration of proteinases far exceeds the concentration of resident anti-proteinases (such as α1-antitrypsin), so proteolysis occurs unabated; however, once the concentration of proteinases equals the levels of anti-proteinases, they are irreversibly inactivated so tissue damage is limited.33 Close to the neutrophil, therefore, there is an area of obligate proteolysis which allows passage through the matrix while tissue is protected further away.
In patients with α1-antitrypsin deficiency, lung levels of the inhibitor are low. There is an imbalance between proteinases and anti-proteinases and normal inactivation of proteinases is reduced and the area of obligate proteolysis is increased, leading to widespread tissue damage that is thought to be crucial to disease progression.34,35 In COPD there is a similar imbalance between proteinases and anti-proteinases—at least during exacerbations—with more pro-inflammatory proteins, the recruitment and activation of inflammatory cells, and the potential for more tissue damage during cell emigration, degranulation, and secretion in situ. This is reflected in the detection of active enzymes in the secretions during such episodes.25
Although the role of other cells and proteinases has been implicated in the distal lung, the neutrophil remains the cell central to many of the features and effects of exacerbations in the more proximal airways. The neutrophil is a green cell due to the presence of myeloperoxidase (MPO) in its azurophilic granules,36 and the sputum appears green (purulent) when it contains high numbers of neutrophils.37 It has been shown that neutrophil recruitment is increased during lower respiratory tract bacterial infections,25 which can be identified by the presence of purulent sputum using a simple graded colour chart.38 This provides one distinct identifying feature of some episodes that is clearly associated with the presence of excess elastase activity.24
IMPORTANCE OF EXACERBATIONS
Acute exacerbations are an important cause of mortality and morbidity in patients with COPD.39 In 1996 a study of survival following hospital admission for acute exacerbations reported in-hospital mortality rates of 11% and 1 year mortality rates of 43%.40 A more recent study reported in-hospital mortality at 8% with a 1 year mortality rate of 23%,41 confirming the prognostic importance of these episodes.
Exacerbations have a marked effect on the quality of life of patients. Frequent exacerbations (more than two per year) have been associated with more dyspnoea and reduced exercise capacity,42 a greater decline in health status,2,43 and a greater likelihood of becoming housebound44 than in patients with fewer acute episodes.
It is understandable that repeated exacerbations should be associated with a faster decline in lung function. The urinary excretion of desmosine and isodesmosine (byproducts of lung elastin degradation) are raised in exacerbations compared with the stable state,45 and higher levels of these proteins have been associated with a faster decline in FEV1 in COPD.46 In a 15 year study, increased sputum neutrophil counts (as found during bacterial exacerbations) have been correlated with a rapid decline in FEV111 and, in another study, the extent of emphysema (quantified by computed tomography) correlated with the number of previous exacerbations.47
It may be that patients who experience frequent AECOPD have increased bronchial inflammation in the stable state compared with those with fewer episodes. Bhowmik et al19 found a relationship between stable state CXCL8 and IL-6 concentrations in induced sputum and exacerbation frequency. However, this study did not account for important confounding factors such as the presence or absence of bronchiectasis, chronic bronchitis, treatment with inhaled steroids, degree of pulmonary impairment, smoking status, or bacterial colonisation (all of which influence inflammation48). Gompertz et al49 accounted for the above factors and found no difference in pro-inflammatory mediators between frequent and less frequent exacerbators. The only difference noted was the concentration of the anti-proteinase secretory leukoproteinase inhibitor (SLPI) which was reduced in the sputum of patients with frequent exacerbations. The authors speculated that reduced activity of SLPI (which has been shown to have antibacterial and antiviral functions in vitro50) may predispose towards infective exacerbations, which has been supported by studies of recurrent mucosal infections outside the lung.51
The underlying aetiology of the exacerbation may also affect FEV1 decline, as Wilkinson et al described the greatest reduction in FEV1 (56 ml in 1 year) in patients with a higher airway bacterial load, and this decline was greater still in patients with a change in bacteria as opposed to a single colonising species.52
CAUSES OF ACUTE EXACERBATIONS OF COPD
Published data suggest that 50–70% of exacerbations are due to respiratory infections53 (including bacteria, atypical organisms and respiratory viruses), 10% are due to environmental pollution (depending on season and geographical placement),54 and up to 30% are of unknown aetiology.40
Role of bacterial infections
Studies using bronchoscopic sampling of the lower airways have found a relationship between bacteria and exacerbations with approximately 30% of sputum cultures and 50% of bronchial secretion cultures associated with the presence of a potential pathogenic bacteria.55,56 In severe exacerbations requiring ventilatory support, this number is even higher (over 70%).57,58 Commonly isolated organisms include Haemophilus influenzae (11% of all exacerbating patients), Streptococcus pneumoniae (10%), Moraxella catarrhalis (10%), Haemophilus parainfluenzae (10%), and Pseudomonas aeruginosa (4%), with Gram negative bacteria occurring more rarely (data taken from four studies55–58). It appears that infections with Pseudomonas spp, Stenotrophomonas spp, and Gram negative bacteria occur in more severe exacerbations, affecting the most debilitated patients.58,59 This may reflect previous antibiotic pressure, but more research is required to link patient characteristics with likely pathogens (table 2).
Causes of COPD exacerbations
Following bacterial exacerbations, patients develop new strain-specific antibodies as part of their immune response. Sethi et al60 studied this phenomenon and described antibodies to surface exposed epitopes on H influenzae after exacerbations. These antibodies were extremely strain-specific, showing bactericidal activity for only 11 of 90 heterologous strains. Furthermore, a recent study found Moraxella catarrhalis in approximately 10% of exacerbations with organism clearance occurring after 30 days, and again there was evidence of strain-specific immunity.61 The development of a new immune response supports the hypothesis that bacteria cause exacerbations, and the specificity of this response provides an explanation for recurrent exacerbations (with infection by the same bacterial species but a different genetic strain).
There are a number of mechanisms by which bacteria could affect symptoms in COPD. S pneumoniae, H influenzae and P aeruginosa have been shown to induce mucus hypersecretion in vitro.62 The latter two organisms have also been shown to reduce ciliary beat frequency,63 and H influenzae can cause epithelial damage during adherence.64 Bacteria enhance pulmonary inflammation in COPD, which could potentially cause increased tissue damage due to leucocyte recruitment and proteinase secretion. The isolation of potentially pathogenic bacteria in BAL fluid from COPD patients during bronchoscopy is associated with a higher neutrophil count and higher TNFα levels in BAL fluid than in culture negative patients.65 The bacterial load is proportional to pro-inflammatory mediators irrespective of the pathogen present,37 although some bacteria are associated with a greater response than others. In vitro studies have demonstrated higher levels of IL-6, CXCL8, and TNFα release following infection with H influenzae,66 and studies in man have shown higher concentrations of TNFα in the sputum of patients with positive cultures for this organism.67 White et al27 showed that eradication of bacteria was associated with a corresponding resolution of inflammation (including MPO, used as a surrogate marker for neutrophils, and LTB4), while a continued bacterial presence was associated with a lesser reduction. This provides further support for the hypothesis that bacteria cause exacerbations and that the size of the load determines the degree of inflammation.
If bacteria are the cause of at least 50% of exacerbations, one might expect a significant clinical response to antibiotics; however, many controlled trials of antibiotic therapy show little or no benefit with treatment.68,69 This may be explained by small patient numbers, poor identification of patients who were likely to have bacterial exacerbations of COPD before starting treatment, choice of antibiotic, and the end points measured. As stated previously, high neutrophil counts and increasing concentrations of MPO in sputum cause increasing sputum purulence.69 MPO concentrations are proportional to bacterial load, with increasing likelihood of bacterial isolation in culture as sputum darkens. When only purulent exacerbations are treated with antibiotics, a clear clinical and biological improvement is seen.24
Although some studies have shown that the likelihood of isolating potentially pathogenic bacteria is increased during exacerbations (with increased bacterial numbers compared with the stable state),24,55–57,70 others (using sputum cultures) have not,71,72 and these negative findings have questioned the role of bacteria during acute episodes. However, these older studies must be interpreted with caution.
Firstly, there is some evidence to suggest that standard sputum cultures may miss the presence of bacteria in the lung both during the stable state and during exacerbations. An American study demonstrated the presence of intracellular H influenzae in 87% of bronchial biopsy samples from patients with COPD during an acute exacerbation, compared with 33% of patients with stable COPD and none in healthy controls. However, standard microbiological sampling techniques identified bacteria in only 7% of the same acutely unwell patients.73 A more recent study found positive sputum cultures in only 21% of patients with COPD persistently colonised with the same strain of H influenzae, with strain-specific H influenzae genes seen in both positive and negative sputum cultures.74 Negative sputum cultures may be explained by the presence of low concentrations of bacteria during colonisation (less the 103 colony forming units/ml sputum will yield negative cultures) or the sensitivity of culture techniques to certain bacteria.
Secondly, many older studies were unable to differentiate between strains of bacteria as molecular techniques were unavailable at the time. Authors described the presence of the same bacteria during stable periods and exacerbations, thereby discounting their role,71,72 but would have been unable to appreciate differences in genotype which indicate the presence of a different strain of the same species75 or the overall bacterial load.
Bacterial colonisation
Approximately 20–30% of patients with COPD have positive sputum bacterial cultures when clinically stable,72,76,77 and the presence of bacteria has been shown to be related to the degree of airflow obstruction and current cigarette smoking status.78 However, there remains controversy as to whether bacterial colonisation causes a deterioration in COPD or whether colonisation is simply a marker of disease severity when mucociliary clearance is likely to be impaired as a result of epithelial damage. It is also unclear whether asymptomatic colonisation leads to exacerbations caused by the same bacterial strain, or whether colonisation predisposes towards new bacterial growth. The presence of bacteria already suggests a breach of host defences with associated epithelial cell damage, mucus hypersecretion, reduced ciliary beat frequency, increased submucosal vascular leakage, and inflammatory cell infiltration. It therefore seems likely that patients with chronic colonisation would have a greater inflammatory burden, more frequent exacerbations, and a faster decline in lung function. Indeed, this has been shown in other inflammatory respiratory conditions such as cystic fibrosis and bronchiectasis,79,80 and a number of studies have aimed to clarify this point in COPD.
Bogaert et al studied 269 patients with mild to severe COPD, collecting four sputum samples over a 10 month period in cases of stable disease and collecting at least one sample during an exacerbation. 34% of patients had positive sputum cultures for one potential pathogen while stable and 49% had positive sputum cultures during exacerbations. The most common colonising organisms were H influenzae (19%), S pneumoniae (13%), and M catarrhalis (9%). Interestingly, only colonisation with a monoculture of S pneumoniae predisposed towards frequent exacerbations once potential confounding factors were taken into account.59
Patel et al70 found higher rates of colonisation in a smaller study of 29 patients with moderate to severe COPD; 51% of patients were colonised by one or more possible pathogens, with H influenzae present in 53% of patients with positive cultures, S pneumoniae in 33%, and H parainfluenzae, M catarrhalis, and P aeruginosa in 20%.
Hill et al37 showed that colonisation was associated with increased inflammation (with increased neutrophil counts, and increased CXCL8, LTB4 and MPO) and that bacterial load was proportional to the inflammatory response seen. Bacterial loads of <106 colony forming units (cfu) were not associated with neutrophilic inflammation (and represented low level, stable colonisation), while loads of >106 cfu were associated with increased neutrophil influx to pro-migratory inflammatory proteins.
Colonisation does not appear to be static as changes in bacterial strain, species, and load have been seen with sequential sampling in individual patients. In a study of 30 stable COPD patients with positive sputum cultures, a change in colonising bacterial type was associated with a greater decline in FEV1 and higher levels of inflammatory proteins than persistent colonisation with the same bacteria; the higher the bacterial load, the more pronounced the decline in lung function and the increase in inflammation.52 The authors suggest that higher bacterial loads would stimulate the release of more pro-migratory cytokines leading to more neutrophils and therefore more proteinase induced lung damage. In the presence of a new type of bacteria, there may be an enhanced inflammatory reaction leading to a faster decline in lung function via the same pathway.
In support of this, Patel and colleagues70 found a positive correlation between lower airway colonisation, exacerbation frequency and CXCL8 concentration in the sputum. Colonisation may also affect the severity of an exacerbation as the presence of H influenzae when stable was associated with more symptom reporting and increased sputum purulence during the exacerbations. Also, although results did not reach significance, there was a notable trend between colonisation and the duration of an exacerbation.
In a recent study of COPD, a comparison was made between colonising and exacerbation strains of H influenzae. Colonising strains were defined by the presence of a bacterial strain with no clinical signs of exacerbation and no new serum antibody response to the bacterial strain. Exacerbating strains were defined by the presence of a bacterial strain at symptom onset and new bactericidal antibodies to the bacterial strain 1 month later.81 Using a mouse model and cell cultures, the exacerbating strains of H influenzae caused more neutrophil recruitment, adhered to epithelial cells in higher numbers, and (in cell cultures) induced more CXCL8 release from epithelial cells than colonising strains. Similar findings have been reported by Sethi et al82 who found that more exacerbations were associated with a new strain or type of bacteria than with a previously colonising strain. However, a study by Bresser et al67 described contradictory results, with 67% of exacerbations being due to a previously colonising bacteria strain and 86% of new strain acquisitions occurring without an exacerbation. The difference in results is not unexpected because the interactions between host and pathogen are complex, strains of bacterial pathogens differ in virulence with a degree of subtlety that may not be easily detected, and there is great interpatient variability in the intensity and duration of the inflammatory response.
Despite continued debate, there is compelling evidence that bacteria do cause exacerbations of COPD, with greater bacterial loads seen in bronchoscopic “protected specimen brush” sampling compared with the stable state,55 more airway inflammation25 with resolution of inflammation if bacteria are eradicated,27 and a new immune response seen following infection.60,61 However, it is still unclear whether exacerbations are due to colonising strains that undergo mutation or to new bacterial strains or bacterial species. Nevertheless, there is evidence that colonised patients experience more exacerbations and have more inflammation and perhaps a faster deterioration in lung function.
Role of viral infections
Viral infections of the respiratory tract are a common cause of morbidity and it is unclear whether patients with COPD are more susceptible to these infections than healthy individuals. Some studies have isolated viruses more frequently in COPD patients83,84 while others have not.85,86 Certainly, exacerbations have been associated with a prodrome of coryzal symptoms and are more frequent in winter months when viral infections are more common in the community.87 Many studies have provided only indirect evidence of a viral aetiology of AECOPD as they have relied on the presence of serological conversion as a marker of infection,83,88 although some recent studies have used more robust techniques including viral culture85 and polymerase chain reaction (PCR) to identify viral RNA or DNA sequences.89 However, there is evidence that at least a proportion of episodes are associated with viral isolation. Cell culture and serological studies suggest that viral infections are the likely cause of approximately 20% of exacerbations,90–92 while PCR studies have suggested that up to 40% of acute respiratory infections in COPD are associated with viruses.93
Serological evidence of infection with influenza virus has been detected in 5–28% of patients following an exacerbation, but is present in only 6% of patients who have not had an exacerbation. The prevalent strain of influenza has some effect on exacerbation frequency and severity. A recent American study carried out over four consecutive winters noted that, when the dominant strain of influenza was H1N1 (one not associated with substantial morbidity and mortality in the elderly), hospital admission for influenza related chest infections were low.94,95 In contrast, when H3N2 influenza was prevalent (a more virulent strain), admissions for chest infections and related deaths were significantly raised,94,96 suggesting a cause and effect.
Rhinovirus has been detected in up to 23% of patients during exacerbations but in <1% of stable patients.89 Serological studies have indicated that respiratory syncytial virus (RSV) infection has been associated with up to 6% of exacerbations of COPD, although Falsey et al97 reported that 11.4% of hospital admissions for COPD could be accounted for by the presence of RSV when diagnosed using serology, reverse transcriptase-PCR, and viral culture. As with other viruses, colonisation with RSV in the stable clinical state has been demonstrated in some studies,90,98 although the significance of these findings is unknown.
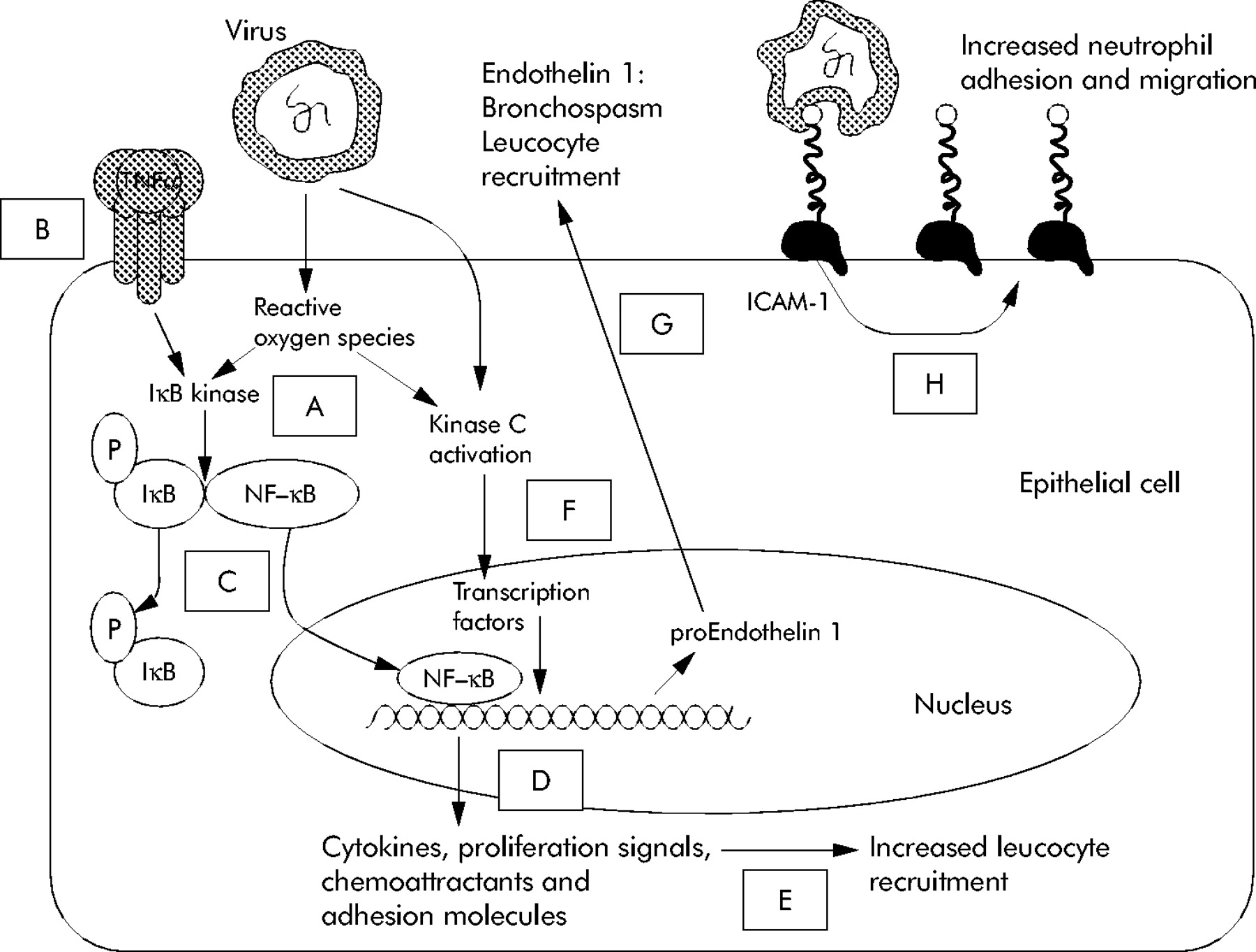
Parainfluenza viruses and adenoviruses have been associated with a lower percentage of exacerbations. Recent studies have suggested that picornaviruses, coronoviruses,93 and the newly identified human metapneumovirus99 may also be important causes of acute episodes. However, the role of these pathogens in COPD remains unclear as a positive PCR result without evidence of an acute immune response to the virus is at variance with conventional understanding of an infection. Nevertheless, viruses can affect many aspects of the respiratory tract that can cause damage and inflammation associated with exacerbations (fig 1).
{kind=link}
Key inflammatory effects of viral infection. Viruses cause increased neutrophil recruitment into airways by several mechanisms. Viral attachment and subsequent epithelial cell damage causes release of reactive oxygen species (ROS) as shown in point A. ROS and activation of inflammatory receptors (such as tumour necrosis factor α (TNFα) binding to its receptor) as shown in point B causes dephosphorylation of nuclear factor kappa B (NF-κB) (shown in point C) which in turn causes the transcription of inflammatory proteins (D), increasing white cell recruitment to lung tissue (E). Viral attachment and ROS also activate protein kinase C (F) which stimulates further transcription of inflammatory proteins including the precursor of endothelin 1 which is a potent bronchoconstrictor (G). Some viruses (including rhinoviruses) bind to ICAM-1, increasing its expression, which provides a target for neutrophil attachment and migration (H).
In summary, the association between exacerbations of COPD and the identification of viruses and an appropriate immune response has not been consistent. Overall, the data suggest that viruses do play a role in some episodes, and the variation in frequency of viral identification may be influenced by epidemics present at the time of data collection48 as well as the sensitivity of the methods.
Lower respiratory tract viral infections damage the airway epithelium causing loss of ciliated cells, increased plasma exudation, and increased mucus production.100 Rhinovirus attaches to the airway epithelium through the intercellular adhesion molecule (ICAM)-1, inducing further ICAM-1 expression and thereby promoting neutrophilic recruitment and inflammation.101 Respiratory viruses may also increase the expression of RANTES leading to increased eosinophil recruitment into the airways with associated release of platelet activating factor, major basic protein, cysteinyl leukotrienes, and eosinophil peroxidase.28 Indeed, the association of eosinophilia with some exacerbations is well documented,102,103 although it has mainly been seen in patients with milder disease.104 In vitro, rhinovirus infection induces expression of CXCL10, a ligand for activated T lymphocytes and natural killer cells, and in vivo levels of CXCL10 correlate with symptom severity and viral titres in airway secretions.105 Rhinovirus infection also increases production of reactive oxidant species and activates nuclear factor-κB, which is important in the regulation of CXCL8 production.106
Viral and chlamydial infections have been associated with increased endothelin-1 concentrations in spontaneous and induced sputum.20 Endothelin-1 is a potent bronchoconstrictor produced by bronchial epithelial cells which increases production of IL-6. IL-6 is a pro-inflammatory cytokine which activates lymphocytes and also enhances endothelin-1 production, suggesting a positive feedback loop between these two proteins.107 Respiratory viruses can also increase the release of acetylcholine by activating M2 muscarinic receptors, also (potentially) leading to bronchoconstriction.108
Finally, the effects of viral infection may predispose to secondary bacterial infections of the respiratory tract. There appears to be an association between viral and simultaneous or sequential bacterial infection of the airways. Increased isolation of H influenzae and S pneumoniae has been obtained from sputum in patients with positive viral seroconversion, suggesting that viral infection preceded bacterial infection.109
Atypical organisms
Mycoplasma pneumoniae and Chlamydia spp are intracellular bacteria that share some characteristics of viruses. They are usually diagnosed retrospectively by serological testing and have been implicated in AECOPD. One recent study suggested that up to14% of exacerbations were associated with mycoplasma.110 Recent animal studies and in vitro work have highlighted a potential mechanism for mycoplasma to induce features of exacerbations as mycoplasma derived lipoproteins activate Toll Like Receptor 2, increasing mucin production.111
Meloni et al112 described serological and molecular evidence of current Chlamydia pneumoniae infection in 8.9% of patients during exacerbations of COPD, while Beaty et al113 found evidence of acute infection in 5% of exacerbations. In vitro work suggests that this organism can also promote features of AECOPD by increasing the secretion of IL-1β and decreasing the secretion of its anti-inflammatory counterpart, IL-1 receptor antagonist, thereby potentially increasing inflammatory cell sequestration into the airways.114
NON-INFECTIOUS AETIOLOGIES OF AECOPD
Despite the importance of infections, other factors may provoke exacerbations. In a study of 1016 patients with severe COPD, infection was the proven cause in only 51% of exacerbations. Heart failure was thought to account for 26% of episodes of increased symptoms, but about 30% of exacerbations had no identified aetiology.40
Environmental pollution
Epidemiological research has identified more exacerbations during periods of increased pollution.115,116 Increases in black smoke particulate matter, sulphur dioxide (SO2), ozone (O3), and nitrogen dioxide (NO2) are associated with increases in respiratory symptoms, admissions for exacerbations, and COPD associated mortality.117 These adverse effects have been most clearly described with high levels of ozone resulting in an increased relative risk of hospital admission with AECOPD118–120 and with particulate air pollution comprising 50% organic and 50% inorganic matter of ⩽10 μm diameter (PM10).121,122 These studies have concluded that up to 9% of admissions with AECOPD may be due to atmospheric pollution, especially during the summer months.54
Pollutants have been shown to be pro-inflammatory. In animal models, SO2 and PM10 have been shown to increase mucus secretion in airways leading to increased airway resistance and bronchial plugging.123 Intratracheal instillation of PM10 into the lungs of rats causes an influx of neutrophils, a subsequent rise in pro-inflammatory mediators, and increased epithelial permeability,124 supporting this concept.
Exposure to diesel exhaust fumes for 1 hour has been shown to induce a marked neutrophil influx into the proximal airways of healthy volunteers with a corresponding increase in CXCL8, GROα, and IL-5 levels in bronchial washings.125 There was also an increase in the expression of transcription factors (such as NFκB, c-jun and c-fos) in bronchial biopsies,126 consistent with oxidative stress triggering the increased synthesis of pro-inflammatory cytokines. Exposure to ozone is associated with increased nasal inflammation in non-atopic children,127 and SO2 and NO2 enhance the response to inhaled allergens.128
Mucus hypersecretion and ciliary damage are features of COPD1 and would be expected to impair clearance of inhaled particles or soluble gases. This may make the patient more susceptible to the pro-inflammatory effects of inhaled pollutants120 which, in turn, would lead to more mucus secretion, airway oedema, and neutrophil recruitment and activation, all of which are features of AECOPD.
Miscellaneous
Exacerbations and a decline in FEV1 have also been associated with a fall in outdoor or indoor room temperature, suggesting a cause and effect. However, coryzal symptoms are also more common during colder periods, and the increase in exacerbation frequency could be accounted for by viruses rather than the physical effects of temperature.129
Despite some patient concerns, there is no evidence that influenza vaccination is associated with AECOPD. Indeed, two recent studies reported no ill effects (with no changes in dyspnoea, exercise capacity, lung function or exacerbation frequency) following vaccination, irrespective of the severity of the disease.130,131
CONCLUSIONS
There have been significant advances in our understanding of the aetiology and pathogenesis of acute exacerbations of COPD. Exacerbations appear to be related to heightened airway inflammation which leads to the composite of symptoms seen during these acute episodes. The evidence suggests that bacteria, viruses, and environmental pollution are the most important causes of exacerbations, and that purulent sputum is the key sign which differentiates between the former and latter two aetiologies. Frequent exacerbations appear to be associated with worsening health outcomes, and efforts should focus on preventing these episodes or prompt effective treatment of each episode. However, treatment options are limited, and further research is needed to clarify the mechanisms that commence and sustain exacerbations to identify new therapeutic targets.
REFERENCES
Footnotes
-
Funding: none.
-
Competing interests: none declared.
Linked Articles
- Airwaves