


Abstract
Crucial as molecular sensors for many vital physiological processes, seven-transmembrane domain G protein-coupled receptors (GPCRs) comprise the largest family of proteins targeted by drug discovery. Together with structures of the prototypical GPCR rhodopsin, solved structures of other liganded GPCRs promise to provide insights into the structural basis of the superfamily's biochemical functions and assist in the development of new therapeutic modalities and drugs. One of the greatest technical and theoretical challenges to elucidating and exploiting structure-function relationships in these systems is the emerging concept of GPCR conformational flexibility and its cause-effect relationship for receptor-receptor and receptor-effector interactions. Such conformational changes can be subtle and triggered by relatively small binding energy effects, leading to full or partial efficacy in the activation or inactivation of the receptor system at large. Pharmacological dogma generally dictates that these changes manifest themselves through kinetic modulation of the receptor's G protein partners. Atomic resolution information derived from increasingly available receptor structures provides an entrée to the understanding of these events and practically applying it to drug design. Supported by structure-activity relationship information arising from empirical screening, a unified structural model of GPCR activation/inactivation promises to both accelerate drug discovery in this field and improve our fundamental understanding of structure-based drug design in general. This review discusses fundamental problems that persist in drug design and GPCR structural determination.
I. Introduction
The biological and medical importance of G protein-coupled receptors (GPCRs1) is well established and extensively documented. The breadth of GPCR distribution across nearly all of the body's organs and tissues and the cellular role GPCRs play as signal transducers make GPCRs key regulatory elements in a broad range of normal and pathological processes. Thus, GPCRs have been and will continue to be an important focus for drug discovery (Drews, 2000; Ma and Zemmel, 2002).
Over the past decade, the pursuit of GPCRs as targets for drug discovery campaigns has benefited greatly from the development and adoption of high-throughput approaches to their pharmacological assay and medicinal chemistry. Availability of these tools in conjunction with a genomically complete GPCR target palette has effectively enabled researchers to rapidly screen GPCRs of specific therapeutic interest and quickly elaborate upon potential leads during the ensuing drug development process, thus sparking a renaissance in GPCR pharmacology. The emergence of new types of ligand-receptor-effector relationships, including positive and negative allosterism, inverse agonism, multimeric receptor pharmacology, and ligand biased signaling (discussed in section II) has widened our perspective beyond simple, two-state (on/off) receptor models and suggests entirely new mechanistic avenues for therapeutic intervention. Such an expanded scope of options is both enticing and vexing from a drug discovery point of view, a paradox only magnified by our limited structural insights into the molecular mechanics of the GPCR superfamily.
For nearly 30 years we have probed GPCRs through laborious mutagenesis and assay procedures in an attempt to distinguish the residues that are functionally important and then fitting our findings into hypothetical models of their structure. Despite the significant toil devoted to these efforts and the consolidated lists of residues cataloged as important for binding and/or function, the utility of the findings were inevitably limited by their individualized nature. The goal of achieving a highly resolved and practical understanding of specifically “druggable” receptor sites remains elusive, and the iterative structure-function process has proven too slow and laborious to proactively guide drug discovery. New developments in the area of X-ray crystallography suggest that the structural veil has now lifted and that we are on the threshold of a new era for GPCR drug discovery. Reports have been rapidly emerging about the successful crystallization and structural determination by X-ray diffraction methods of GPCRs, including the β2-adrenergic (Cherezov et al., 2007; Hanson et al., 2008; Rasmussen et al., 2011; Rosenbaum et al., 2011), β1-adrenergic (Warne et al., 2008, 2011), A2a-adenosine (Jaakola et al., 2008; Xu et al., 2011), chemokine C-X-C chemokine receptor type 4 (CXCR4) (Wu et al., 2010), and dopamine D3 receptors (Chien et al., 2010). Demonstration of high-resolution structures for multiple receptors and the accelerated rate at which they are appearing suggests that we have bypassed some of the roadblocks historically associated with crystallizing members of this integral membrane protein family. Application of such methods to the GPCR superfamily promises to illuminate at atomic resolution just how these important membrane proteins work and, in so doing, significantly change the tactics of our empirical drug discovery process.
Given the likelihood that structures for more GPCRs will be forthcoming, rather than attempt to dissect differences in the currently available structures, we will describe the commonalities of successful strategies from a technical perspective and indicate how they can be implemented to best benefit future GPCR drug discovery efforts.
II. Current Challenges and Opportunities in G Protein-Coupled Receptor Drug Discovery
A. Overview of the Current G Protein-Coupled Receptor drug Discovery Process
1. Therapeutic Relevance.
The medicinal importance of GPCRs can be partially appreciated by considering their location and function within the cell. The physical location and disposition of GPCRs spanning the cell's plasma membrane connect extra- and intracellular environments, providing a direct mechanism for the transduction of extracellular messages into intracellular responses. In this way and together with their transmitters and effectors, GPCR systems function to modulate a broad spectrum of cellular phenomena dictated by the needs of the tissues and organs they serve. Common biological actions attributed to GPCRs include but are not limited to the following: modulation of neuronal firing, regulation of ion transport across the plasma membrane and within intracellular organelles, modulation of homeostasis, control of cell division/proliferation, and modification of cell morphology. When any of these fundamental processes go awry, the results can lead to acute or chronic human disease, a partial listing of which includes cardiovascular disease (β1-adrenergic receptor) (Drake et al., 2006), asthma (β2-adrenergic receptor) (Kawakami et al., 2004), and strokes and cerebral hypoperfusion (A2a-adenosine receptor) (Chen et al., 2007a; Duan et al., 2009). Other disease states directly linked to mutations in GPCRs include retinitis pigmentosa (rhodopsin), female infertility (follicle-stimulating hormone receptor), nephrogenic diabetes insipidus (vasopressin receptor), familial exudative vitreoretinopathy (frizzled receptors), and dominant and recessive obesity (melanocortin receptors) (for review, see Insel et al., 2007).
2. Molecular Properties.
Although the details of GPCR signaling in aggregate are complex, the basic tenets that describe the initial interaction of the receptor with its proximal partner, the G protein heterotrimeric complex, are straightforward. Upon adoption of an “active” conformation (most simply envisioned as the result of agonist binding), the intracellular domains of a GPCR interact with a membrane-associated GDP-charged G protein heterotrimeric complex (Gαβγ) (Oldham and Hamm, 2008). This heterotrimeric complex then undergoes GTP/GDP exchange with subsequent dissociation of Gα and Gβγ subunits that in turn interact with specific downstream intracellular effector systems (Fig. 1A). Activation of multiple heterocomplexes as well as Gα cycling through active and inactive configurations via a GTP hydrolysis cycle provides immediate amplification and temporal regulation of the initial receptor-ligand signaling event (Leskov et al., 2000; Heck and Hofmann, 2001; Minke and Cook, 2002; Bhandawat et al., 2005). In due course, through the process of desensitization, the active conformation of the receptor is blocked and signaling is attenuated by agonist dissociation and/or deactivation through interaction with β-arrestins in response to activation-specific phosphorylation by G protein-coupled receptor kinases and/or internalization (Hall, 2000; Bockaert et al., 2003). The immediate activities of these effector systems fall into four main categories: stimulation of cAMP production, inhibition of cAMP production, stimulation of phospholipase C with subsequent mobilization of intracellular Ca2+, and activation of plasma membrane proton flux. These phenomena are controlled by which class of Gα subunit is activated. There are at least 16 human Gα subunits, 5 Gβ subunits, and 11 Gγ subunits (Milligan and Kostenis, 2006) (Fig. 2). In addition to Gα-controlled events, the Gβγ subunits also can regulate their own effectors, including additional forms of adenylate cyclase as well as ion channels. The ramifications of signaling complexity implicit in the full range of combinatorial permutations within the heterotrimeric complex itself have yet to be fully examined (Jastrzebska et al., 2010).
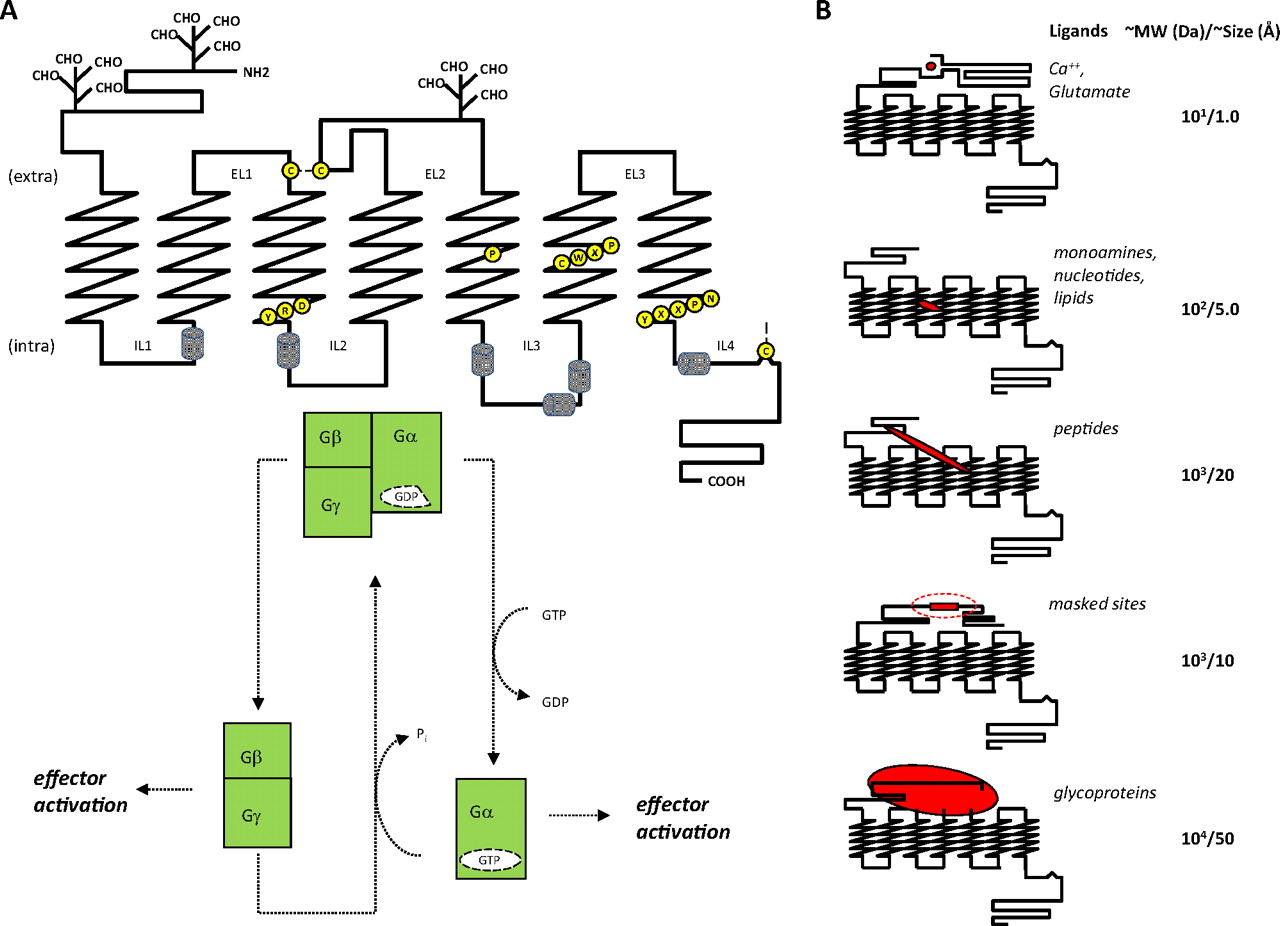
GPCR structure-function. A, principal hallmarks of GPCR structure include a serpentine transmembrane topology compsed of seven α-helical hydrophobic stretches interspersed by intra-and extracellular loops of indeterminate structure. This results in the presentation of an extracellular amino and cytoplasmic carboxyl terminus. The N terminus and extracellular loops can be glycosylated to varying degrees, and extracellular loops 1 and 2 are connected via a disulfide linkage, required for stability and function of the receptor. The C-terminal tail is anchored to the intracellular face of the lipid bilayer via palmitoylation to produce a short intracellular loop, which typically forms an α-helical structure. These receptors also display a series of conserved amino acid motifs thought to be involved in a rearrangement of receptor domains during ligand activated signal transduction. These include three conserved motifs that provide prominent micro-switches: 1) H-III's D(E)RY motif; Arg135 when unlocked from Glu247 interacts with H-V's Tyr223 to release constraints holding the receptor in the inactive state. 2) H-VI's CWxP motif; Trp265 undergoes rotamer isomerization needed for receptor activation. 3) H-VII's NPxxYx(5,6)F motif; Tyr306 undergoes rotamer isomerization during activation and Asn302 participates in an intrahelical H-bond network. Pro residues within H-V (Pro215), H-VI (Pro267), and H-VII (Pro303) are highly conserved and believed to form a staircase of transmembrane kinks required for the increase in rotational dynamics of these helices during activation. Rotation of these transmembrane helices brings disparate segments of intracellular loops in and out of proximity to the G protein complex causing its GTP-driven dissociation and subsequent regulation of various cellular effector cascades, the overall effect being an amplification of the original activating signal. B, the size and complexity of GPCR pharmacophores varies greatly, ranging upward from free atoms/ions to small molecules, small peptides, unmasked intrareceptor amino acid stretches, and even large glycoprotein hormones. Some correlation between receptor sequence and ligand class can be generated, which may reflect ligand-receptor recognition, but the range of sizes and diversity of ligand structures make it difficult to determine molecular mechanisms underlying ligand-receptor activation.

The GPCR signalosome: components and actions. The molecular components of GPCR signaling pathways interact to generate multiple effects and types of regulation. The full complement of endogenous ligands is currently unknown, and the structural nature of binding sites within the receptor is surprisingly varied, including conserved orthosteric sites and other sites such as nonconserved isolated ectopic and adjacent bitopic sites as well as remote allosteric sites that regulate the affinity of the activating sites. The size of the “druggable” GPCR genome currently stands at ∼750 but could increase as with the addition of taste and odorant receptors. Dimerization or oligomerization of GPCRs, with themselves or with other entities, provides additional specificity and complexity of pharmacological responses. The interaction of activated GPCRs with the G protein complex is influenced by both the ligand-defined stabilized receptor structure and the nucleotide-bound α-subunit of the G protein heterotrimer. Dependent upon the specific G protein in the complex, activation will result in one or more of four initial events, including cAMP production, cAMP reduction, intracellular Ca2+ mobilization, or regulation of proton flux. These cellular effects (both proximal and integrated) can occur partially or fully, can be positive or negative, and can be affected by positive or negative allosteric modulators. The large number of possible permutations provides some insight into the increasingly complex pharmacology exhibited by this superfamily.
Possibly because they share a relatively limited downstream effector ensemble, GPCRs themselves are similar with respect to their gross architectural topologies. Embedded within this structural homogeneity is the capability of receptor subtypes to distinguish chemical subtleties across and within structurally diverse transmitter and hormone families (Fig. 1B). This remarkable molecular recognition property combined with the breadth of specific pathophysiological conditions related to each receptor's tissue distribution make the GPCR superfamily a treasure trove of proven and potential drug discovery targets. In addition, plasma membrane-spanning GPCRs display their most important regulatory sites at or near the extracellular surface, making these sites readily available to circulating administered drugs. Such unfettered access mitigates many drug development concerns such as drug transport through the plasma membrane and intracellular drug metabolism.
3. G Protein-Coupled Receptor Drug Compendium.
As a biological target class, drugs of the GPCR superfamily are well represented in our current pharmacopeia (Kroeze et al., 2003; Bjenning et al., 2004; Doggrell, 2004; Dahl and Sylte, 2005), with an estimated 30 to 50% of marketed drugs acting directly on GPCRs or through GPCR-associated mechanisms (Flower, 1999; Robas et al., 2003). Although the most obvious GPCR modulators constitute some 26% of the top sellers and netted some $23.5 billion (9% of the global market share) of drug sales in the year 2000, these ∼150 marketed drugs surprisingly target only ∼20 of the ∼750 known GPCR subtypes (Vassilatis et al., 2003; Overington et al., 2006). Moreover, as of 2008, 5 of the top 15 generic drugs and 7 (Milligan, 2009) of the top 15 prescription drugs targeted GPCRs (McGrath et al., 2010). Of those, only the leukotriene drugs are “new” GPCR targets. This paradox does not imply that the bulk of the GPCR superfamily is therapeutically unimportant or “undruggable.” Instead, it reflects the comparatively long period needed to bring drugs to market relative to the short recent period during which postgenomic target access and high-throughput methods have been available. It follows that many drugs now in or emerging from the pharmaceutical pipeline have been enabled largely because of GPCR systems discovered or investigated in the past decade or so.
4. The Drug Discovery Process.
The research and development (R&D) process is lengthy and expensive, especially during the later clinical stages (Figs. 3 and 4). Two previous estimates of the costs of bringing a new drug to market range from $1.3 to $1.76 billion because of the adverse impact of program attrition, which continues well into the later stages of the development process (DiMasi et al., 2003; Paul et al., 2010). Such costs underscore the weight placed upon pursuing only well validated pathophysiological mechanisms and disease markets that are capable of delivering a return on the R&D investment. Of additional importance is the target-centric modus operandi of the R&D process. Together, these factors dictate that during the earliest stages of drug discovery, when therapeutic paradigms are being envisioned and refined, an over-riding research goal is the identification, characterization and validation of the association between specific molecular targets and specific disease states. Although the genomic catalog of GPCRs is complete, discussion as to the number and identity of “druggable” targets is still active. Basic approaches employed to explore the issue include 1) tissue expression profiling (including comparisons between healthy and disease states); 2) searching for regulation of transcripts and gene copies in disease models through microarray, quantitative polymerase chain reaction, and genomic deep sequencing analyses; 3) establishing gene-disease linkage through chromosome mapping and phenotypic analysis of transgenic animals; and 4) conducting in vivo/in vitro pharmacological studies with target-active prelead molecules when available. Absorption, distribution, metabolism, and excretion (ADME) and pharmacokinetic (PK) studies are increasingly being employed early on in the drug discovery process to assist in the interpretation of ongoing and future in vivo experimentation. If a GPCR drug target survives this initial validation gauntlet, it then proceeds to primary drug screening, where small molecules and/or biological compounds are tested as dictated by the therapeutic paradigm.

The drug discovery and development pipeline. To be therapeutically effective, a drug must be present in an adequate concentration at its site(s) of action within the body. In addition, the molecule must be safe—that is, eliminated unchanged or as a metabolite from the body without causing injury. The discovery and development “pipeline” is designed to obtain compounds conforming to these requirements. The earliest stages involving identification of linkage between a target and various disease states draws upon basic research conducted in the academic (gray) and pharmaceutical (blue) sectors. Hypotheses are validated as the concept enters the lead discovery phase of the pharmaceutical process. Upon validation, the preclinical work of finding molecules that specifically modulate a target and possess suitable pharmacokinetic, pharmacodynamic and toxicological profiles ensues. If such molecules are identified, formulation and manufacturing parameters are determined to prepare for clinical trials in human subjects. Once validation is achieved and a campaign is launched, the discovery process can take an additional 3 to 6 years before a compound enters the clinic or is terminated. The onset of clinical trials is heralded by submission to the FDA of an investigational new drug proposal (IND) that includes detailed protocols for the trials and criteria for success. Once accepted, phase I trials are initiated to establish safety in healthy human subjects. Upon successful completion of phase I trials, phase II ensues to establish the drug candidate's efficacy for treatment of its chosen disease and to assess its side effects, which together help establish an effective and safe dosage regimen. Phase III is an extension of phase II that involves a larger patient and control set to establish statistically significant safety and efficacy over time. If the results from phase III appear favorable, a new drug application (NDA) is filed with and reviewed by the FDA along with protocols and preparations for large-scale manufacturing, product launch, and long-term monitoring of the patient population (phase IV). Clinical trials are the most resource-intensive and typically take 6 to 7 years before a new drug reaches final review.

Attrition and costs. Idealized description of a typical HTS-driven small-molecule drug discovery campaign. Once validated, the target is enabled for HTS and subsequent profiling as part of the lead discovery process. The largest number of compounds is processed initially through single- or multitiered screens of libraries containing approximately a million separate chemical entities. Primary hits emerging from screening are then confirmed and characterized as they engage in hit-to-lead evolution, during which several thousand new analogs can be generated. The medicinal chemistry process continues on a refined number of advanced leads throughout the preclinical workup as modifications are made to achieve acceptable ADME, PD, and toxicological properties. Typically, projects seek to have several candidate compounds available (both primary and backup compounds) upon initiation of clinical trials, because the prospect of unforeseen liabilities remains throughout the remainder of the process. Costs for any one successful project can exceed one billion dollars. When one factors in the broader project failure rate encompassing all phases of the drug discovery pipeline, the costs and compound attrition are further amplified. Thus, there is considerable pressure to critically evaluate projects at all stages of discovery/development and, if warranted, terminate the process as early as possible.
Once the target is validated, the screening process begins in earnest and typically employs cell-based assays in a high-throughput screening (HTS) platform to interrogate corporate compound libraries consisting of hundreds of thousands to millions of separate chemical entities. These assays are typically engineered to be highly specific for a single disease-linked receptor. Primary hits arising from the screen are subsequently confirmed and evaluated for potency at their primary targets. These activity results then are considered together with the hit structure in making the decision to advance the primary hit into the lead development process. Structural properties of hits are also considered from the perspective of the “intellectual freedom to operate” needed to devise patentable chemical analogs of the prototype. Generic problems arising from reactive moieties that can render the compound toxic through irreversible covalent attack on either the target itself or other vital cellular proteins are usually apparent at this point and revealed through simple inspection of the compound's physicochemical properties. Clearly, narrowing the intrinsic specificity for a GPCR target must be an initial priority. Accordingly, characterization and management of drug cross-reactivity is an integral part of the hit-to-lead process that occurs immediately after hit discovery. Conceptually this can be organized according to site and gravity of action. Deliberations usually begin by considering cross-reactivity at sites associated with known health hazards. For example, cross-reactivity of a diverse group of drugs with the human ether-à-go-go-related gene potassium channel can result in sudden death caused by long QT syndrome (Sanguinetti and Tristani-Firouzi, 2006). The potential for such problems is typically discovered early for a given lead series by in vitro testing across a generalized panel of side-effect targets. Additional benefits can result from incorporating chemical properties that restrict drug distribution, as in the case of adverse CNS effects, for example, by keeping the compound from crossing the blood-brain barrier. Next in line are limitations imposed by a compound's pharmacokinetics involving its route(s) of administration, absorption from an administered site, distribution throughout the body as it gains access to the target tissue(s), metabolism and bioavailability via the liver's P450 redox and P-glycoprotein systems, and ultimate elimination of the parent drug with its metabolites. Poor ADME properties are the root cause of failure for ∼40% of drug candidates during clinical trials, so these parameters are extensively evaluated throughout lead development. Pharmacodynamic (PD) properties associated with the mechanism of action and involving interaction, number of lead compounds, and efficacy at the target in vivo are also of great concern and can help define the margins of risk versus safety as a therapeutic window.
In this fashion, chemical analogs of the initial screening hits are synthesized and tested to improve efficacy, selectivity, and potency and to diminish ADME/PK liabilities in a trial-and-error process based upon the projected therapeutic window of activity. Thus, during the journey from lead compound to drug candidate, each primary hit spawns hundreds to thousands of closely related derivatives; some are an improvement over the prototype, some are not, and all are deposited back into the corporate library for future use. Because the process is target-centric, the content of these libraries reflects the history of prior campaigns.
Iterative enhancements to technical tool boxes (e.g., assay methods or synthetic chemical processes) usually progress as well. Periodic assessments of and adjustments to the content and quality of libraries at large are also made, culling compounds with broad toxicity or degraded content. Such enhancements contribute to incremental improvement of drug discovery, most often favorably affecting operational efficiencies rather than fundamental biological or chemical characteristics of the platform. An underlying, but not always explicit, objective of this diligence is lowering the threshold needed to enable primary screens. Although a goal of the process is efficiency (i.e., handling more campaigns in a given time and resource frame), the process remains largely empirical, so the outcome is biased by the assay design and pre-existing library content.
The prospect of enabling structure-based drug design (SBDD) for GPCRs suggests that this empirical paradigm can be improved by enhancing or supplanting conventional HTS methods with new approaches that include defining and/or supplementing the content of the compound collection tested.
B. G Protein-Coupled Receptor Molecular Biology Challenges
1. Expansion of G Protein-Coupled Receptor Drug Targets into Class B, C, (D and E) Families.
Although GPCRs couple to G proteins, these receptors are also referred to as seven-transmembrane receptors, reflecting their seven-transmembrane-embedded helices and additional signaling mechanisms independent of G proteins (Pierce et al., 2002; Lefkowitz, 2004). The number of GPCRs in the human genome is estimated at ∼750 separate genes. These can be categorized into five major classes by comparisons of sequence and/or chemical structure of the ligand (Kobilka, 2007). At the primary structural level, homology among the superfamily is best observed at a limited number of conserved motifs that probably play similar functional roles (Mirzadegan et al., 2003). The three primary categories are as follows: class A (rhodopsin family), containing ∼700 members; class B (calcitonin family), containing ∼15 members; and class C (the metabotropic glutamate group), containing 15 members. Two ancillary categories consist of class D (adhesion family), containing 24 members, and class E (frizzled family), with 24 members. Class A includes those GPCRs activated by biogenic amines, catecholamines, glycoproteins, peptides, lipids, and nucleotides; class B contains GPCRs activated by calcitonin, calcitonin gene-related peptide, and secretin; and class C includes GPCRs activated by glutamate, GABA and Ca2+. Alternative categorizations have been proposed by the International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification based upon predicted structures, pharmacology and roles in physiology and pathology (Foord et al., 2005) (see also http://www.iuphar.org/nciuphar_arti.html; http://www.iuphar-db.org).
By virtue of their historical prevalence and relative ease of accessibility, class A receptors are best represented within the drug market and development pipeline. Class B and C GPCRs lag considerably behind because of challenges associated with their expression and pharmacological study, such as their larger overall and/or N-terminal sizes that tax expression systems and impede appropriate receptor-effector stoichiometry in screening assays. The physicochemical nature of class B and C GPCR ligands also makes assay development more problematic, because inclusion of these unstable, sticky, sometimes ubiquitous molecules is a typical prerequisite for proper control and interpretation of HTS results. Class D and E GPCR therapeutics are not yet represented in the market but, despite their few members, will likely find important uses, especially those involved in taste and adhesion.
2. Special Cases.
Scattered throughout branches of the GPCR superfamily are groups of receptors and derivatives that present special cases. These include putative olfactory receptors, orphan receptors, and receptor isoforms.
a. Olfactory Receptors.
Approximately half of the ∼750 receptors that detect exogenous transmitters (Fuchs et al., 2001; Glusman et al., 2001; Takeda et al., 2002; Venter et al., 2001; Zozulya et al., 2001), are dedicated to olfaction. These receptors have typically been considered therapeutically unimportant, but this assessment may prove to be untenable. Along with taste receptors, volatile odorant receptors form an important sensory collective that modulates a host of deeply rooted animal behaviors such as feeding, mating, and memory formation. Unfortunately, these receptors harbor their own molecular eccentricities in terms of expression requirements and poorly understood biochemistry, which at this time makes assay development and drug screening especially problematic.
b. Orphan Receptors.
The enabling of orphan receptors for drug discovery is a lingering problem compounded by difficulties inherent in creating screening assays without the availability of suitable pharmacological controls and the laborious task of biochemically seeking, purifying, and characterizing endogenous biologically active compounds (Howard et al., 2001). Although the latter is not a strict prerequisite for screening, it is a necessary component of the target validation process. Computational and/or molecular biological approaches to endogenous ligand discovery can also be employed but these depend upon the accuracy of their predictive methods along with a commitment to exhaustively screen large panels of orphan receptors against thousands of synthetic designs/compounds (Shemesh et al., 2008). Depending upon the assays employed, it may be impossible to ensure that a screen will report a valid positive hit. Also related to the orphan receptor problem is the need to understand the transmitter universe and its biochemical regulation more completely (i.e., to identify post-translational processing and/or metabolic transformation of previously known transmitter substances into different biologically active endogenous ligands).
c. Receptor Isoforms.
Receptor isoforms are not uncommon in the GPCR superfamily, but their functional importance and impact on GPCR-mediated signaling is poorly understood. These alternate forms arise through alternative splicing and variations in gene loci, including DNA insertion, deletions, and single nucleotide polymorphisms that can alter expression and/or function. Such variations can contribute subtly or dramatically to the onset or progression of disease and its responsiveness to therapeutics. At least 38 receptor subtypes spanning all five major GPCR families harbor these modifications. Although the structural and functional eccentricities of many isoforms have been characterized, their utility for drug discovery remains to be proven. Attempts to correlate specific isoform expression with individual pathophysiology may yet prove valuable by assisting in the design of clinical trials and perhaps unveiling novel interconnections between specific GPCR systems and physiological processes in the general population (Rohrer and Kobilka, 1998; Tang and Insel, 2005).
3. In Vitro Reconstitution of Monomeric Receptors.
A large body of work demonstrates that a monomeric receptor is sufficient to activate G protein (Meyer et al., 2006; Bayburt et al., 2007, 2011; Ernst et al., 2007; Whorton et al., 2008). However, functional activation by a monomeric receptor does not preclude the GPCR dimer from being the functional unit. The utilization of hetero- and homodimerization by the GPCR signaling process provides exquisite levels of sophistication needed to fine-tune the activated state. The combinatorial expansion available to the GPCR signaling repertoire through the formation of heterodimers and higher order oligomers presents yet another level of complexity that must be addressed in current drug design.
C. Increasingly Complex Pharmacology
Most drugs targeting GPCRs either directly activate (agonists) or inhibit the activation (antagonists) of these receptors by their endogenous ligands through steric competition at the receptors' highly conserved orthosteric ligand-binding sites. A legacy of our early concepts of receptor activation, this orthosteric perspective has resulted in the preponderance of competitive displacement assays that have until recently dominated drug discovery. The past decade, however, has revealed that GPCRs and their activation mechanisms are much more versatile and complex than previously imagined. Agonism and (silent) antagonism has given way to positive, neutral, and inverse agonism, varying degrees of efficacy, and positive/negative modulatory effects on orthosteric potency. Simple two-state (on/off) models of receptor activity have been superseded by more complex equilibria encompassing ectopic ligands, multiple receptor configurations and conformations, positive versus inverse agonism and the varying degrees of efficacy that each of these different ensembles produces (Fig. 5). The trend is clear. We must advance beyond purely orthosteric settings to take full advantage of the entire GPCR milieu and pharmacological repertoire.

Evolution of pharmacological complexity. Kinetic models of GPCR action have evolved from simple two-state models reflecting classic mass-driven chemical equilibrium principles (a) to more complex cubic ternary complexes (Weiss et al., 1996a,b,c; Hall, 2000) (b) that incorporate G protein interactions with both active and inactive receptor isomers and finally to a quaternary complex of GPCR allosterism (Christopoulos and Kenakin, 2002) (c), which includes an additional modulatory ligand that can cooperatively affect the binding of the orthosteric ligand and the subsequent functional interaction of the receptor with its preferred G protein partner. Principle kinetic constants (L, Kx) for each transition pair with various cooperativity factors (α, β, γ, δ, ε, ζ, η, θ, ι, κ, λ,…) describe particular steps. Discovery of additional biochemical phenomena such as receptor homo-/heterodimerization and the possibility that a given receptor can interact with multiple G protein partners suggests that even these extended allosteric models are insufficient to fully describe the behavior of GPCRs.
1. Allostery.
The blossoming complexity of GPCR pharmacology is perhaps nowhere better exemplified or formalized as in the area of allosteric action (Gao and Jacobson, 2006). Simple two-state models borrowed from classic mass-action chemical equilibria have since been extended to those that include G protein influences as described by complex cubic ternary complexes (Hall, 2000) and, more recently, to ternary and quaternary complexes that embody allosteric modulation of orthosteric ligand action (Fig. 5) (Bridges and Lindsley, 2008). It is said that allosteric phenomena provide “texture” to GPCR pharmacology by modifying the affinity or signal imparted by the receptor concomitant with binding of the orthosteric ligand (Leach et al., 2007). In revisiting old and validating new targets, the pharmaceutical industry has taken these new kinetic insights to heart while fine tuning our therapeutic paradigms to modulate the biological tone of endogenous ligands (Christopoulos and Kenakin, 2002; May et al., 2007; Keov et al., 2011). In addition to adjusting the natural tone and tempo of endogenous GPCR ligands, allosteric drugs can more readily achieve selectivity by acting outside a highly conserved orthosteric cavity (Kenakin, 2007; Raddatz et al., 2007).
Even as allostery is now well accepted and its value becomes increasingly more evident, there remain major challenges to its implementation in drug discovery (May et al., 2010). The ability to suitably engineer and tune assay platforms to detect and quantify allosteric effects is not yet routine. Furthermore, because of inherent difficulties in fitting complex mass-action models to experimental data, dissection and tracking of particular cooperativity factors relevant to a therapeutic paradigm during a structure-activity relationship (SAR) campaign can be impractical. To include allosteric effects in drug discovery, an operational approach is needed that seeks a middle ground wherein both mechanistic and empirical parameters are merged into a model that is a compromise between the thermodynamic ideal and the reality of the biological system in which it operates (Keov et al., 2011). The challenges of effectively implementing such platforms are significant.
Current descriptions employed in assay design do not explicitly accommodate more interactions than allosteric and orthosteric ligand binding on a single receptor-G protein complex. Moreover, the complexity escalates once other dimensions of GPCR pharmacology and regulation, such as ligand-directed signaling and receptor oligomerization, are included (Smith and Milligan, 2010). Allosteric modulators can produce complex effects that further complicate their use as therapeutics. Whereas in some cases these modulators may alter the target's binding affinity for endogenous or exogenous agonist ligands, this property may not necessarily result in greater therapeutic efficacy, because they may elicit a contradictory physiological effect at an off-target tissue or receptor or through changes in ligand affinity for individual receptor subtypes (May et al., 2010). Structural insights into the biochemical conformations that underlie these specific functional states could suggest entirely novel drug designs and guide lead development toward the most relevant pathophysiological pathways.
2. Receptor Oligomerization.
Homo- and heterooligomerization of GPCRs is now well accepted, and the functional impact of these types of interactions has been convincingly documented in a variety of systems (Dalrymple et al., 2008). Although the increasing number of instances in which this phenomenon can be demonstrated suggests it is the norm rather than an exception, more work is needed to define its occurrence in native settings and define the ligand-receptor-effector stoichiometry along with the possibility of half-site behavior and/or cross-receptor effects of antagonist, agonist, and allosteric ligands (Fig. 2). The possibility that homodimerization is indeed a native state does not contradict the long-standing empirical observation that most GPCRs reliably produce well behaved activity when heterologously expressed. However, it cannot be assumed without empirical evidence that a heterologous system equates to physiological state of the receptors. The prospect of heterodimerization as a prerequisite for function is, on the other hand, even more intriguing, because it raises the possibility of exploiting combinatorial degrees of specificity greater than can be afforded by either GPCR partner individually. Furthermore, the actuality that GPCRs function as dimers and that this intermolecular association likely occurs at the receptor's nonconserved helical periphery (Han et al., 2009) opens the possibility of designing novel drugs that act at these unique interfaces.
Although the functional unit of GPCRs has long been a matter of great debate (Bouvier, 2001; Chabre et al., 2003), evidence that the native state of GPCRs is a dimer or higher order oligomer continues to grow (Bouvier, 2001; Angers et al., 2002; Fotiadis et al., 2006; Milligan, 2009; Fuxe et al., 2010; Lohse, 2010). AFM images of native intact rod outer segment membranes revealed higher order oligomers of rhodopsin (Fotiadis et al., 2003, 2004; Liang et al., 2003). By comparison, a near-field screening optical microscopy study demonstrated that functional β2-adrenergic receptors are organized into small clusters of molecules, suggesting the importance of oligomerization for this receptor as well (Ianoul et al., 2005). Metabotropic glutamate receptors and a family of taste-specific class C T1/T2/T3 receptors have both been shown to function as obligate homo- and heterodimers, respectively (Rives et al., 2009; Prezeau et al., 2010). Analysis of the crystal packing present in the CXCR4, photoactivated rhodopsin, opsin, and squid rhodopsin structures reveal dimer interfaces that may recapitulate the physiological dimer or higher order oligomers present in the plasma membrane (dimer interfaces are detailed in Fig. 6) (Salom et al., 2006; Murakami and Kouyama, 2008; Park et al., 2008; Wu et al., 2010). Experiments that unequivocally demonstrate transactivation in both hetero- and homodimeric receptor pairs further support this notion (Salahpour et al., 2004; Terrillon and Bouvier, 2004; Waldhoer et al., 2005; Rivero-Müller et al., 2010). In heterodimers of signaling-deficient and ligand binding-deficient luteinizing hormone receptor, G protein activation is still observed, arguing for roles for each component monomer during the activation process (Rivero-Müller et al., 2010). The utilization of hetero- and homodimerization by the GPCR signaling process provides additional means of regulating the activated state. The combinatorial expansion available to the GPCR signaling repertoire through the formation of heterodimers and higher order oligomers in vivo presents yet another level of complexity that must be addressed in current drug design. The functional consequence of dimerization could be unique to each receptor or could be a common feature necessary (e.g., for intracellular trafficking or signaling for all GPCRs), but more work is needed to explain this phenomenon in physiological context.

Observed and calculated dimeric/oligomeric structures implicate various interfaces in GPCR structures. A, dimer observed in photoactivated rhodopsin structure (PDB IDs 2I37 and 2I36). This dimer interface relies primarily on contacts involving H-I and H-8. Opsin-derived structures also contain a very similar dimer interface as a crystal contact (PDB IDs 3CAP, 3DQB, 3PXO, 3PQR). A similar contact was also calculated to build up rows of parallel dimers observed in situ by AFM imaging. B, dimer observed in squid rhodopsin (PDB ID 2Z73). Contacts along H-IV and H-V are responsible for dimerization. C, crystallographic dimer observed in one of the β2-adrenergic receptor structures (PDB ID 3D4S). Contacts with cholesterol as well as with H-I form the basis of this dimeric contact. D and E, on the basis of biochemical results and AFM images of intact native murine rod outer segment membranes, a model (PDB ID 1N3M) was proposed that involves contacts along H-IV and H-V (D) as well as contacts along H-I (similar to A). An additional contact (E) using contacts between H-V and H-VI on one monomer contacts H-I and H-IV on the adjacent monomer. F, the dimer observed in several CXCR4 structures (PDB ID 3ODU) relies upon contacts along H-IV and H-V. The small diagram below each dimer shows the approximate positions of the ends of each helix on the intracellular face of each dimer. Helices are colored according as follows: H-I, red; H-II, orange; H-III, yellow; H-IV, lime green; H-V, dark green; H-VI, teal; H-VII, blue; and H-8, purple.
3. Ligand-Biased (Ligand-Selective) Signaling.
The historical notion that any given receptor subtype is preordained to act through one and only one receptor-defined Gα subtype-linked effector system has gradually yielded to the concept of agonist-receptor trafficking (i.e., the ability of a specific agonist to activate a select subset of the many possible signaling paths available to a given receptor-G protein system) (Kenakin, 2001; Hoffmann et al., 2008; Rajagopal et al., 2010; Vaidehi and Kenakin, 2010). Examples include the characterization of distinct signaling profiles for β1- and β2-adrenergic receptor ligands in the activation of adenylyl cyclase and mitogen-activated protein kinase pathways (Galandrin and Bouvier, 2006) and the opposing Gαi and Gαs effects of selective ligands for the α2-adrenergic system (Eason et al., 1992).
Such observations pervaded the assay engineering community for years and were long viewed as artifacts of heterologously overexpressed receptors in cell hosts with limited or imbalanced G protein complements. But this phenomenon is now understood to be an integral facet of GPCR behavior, termed “pluridimensional efficacy,” and an early kinetic formulation of the effect stands ready to be integrated into the expanding thermodynamic description of the GPCR system at large (Kenakin, 2010). From a practical point of view, the availability of naturally occurring pluridimensional effects suggests it should be possible to fine-tune the therapeutic action of a GPCR drug beyond the simple margins traditionally dictated by the target's tissue distribution, by also taking advantage of the selective activation allowed through the selective downstream activation (Bosier and Hermans, 2007; Conn et al., 2009). This ligand-selective signaling phenomenon underscores the complexity of GPCR signaling that must be addressed to effectively design GPCR therapeutics.
4. Constitutive Activity.
Originally viewed by many as an artifact of assay conditions, constitutive activity can now also be regarded as a physiologically important equilibrium extreme of the native spectrum of GPCR conformations (Lefkowitz et al., 1993; Costa and Cotecchia, 2005). Most easily considered in a mass-action context as an elevation in the amount of R* (activated state of the GPCR) available to interact with its effectors, the phenomenon can be experimentally generated by either changing the R-R* equilibrium through receptor mutagenesis or by increasing the total receptor population, R+R*, and hence elevating the amount of R* through overexpression. This can be observed in Leber congenital amaurosis, where defects in chromophore regeneration or delivery to opsin result in low-grade activation of the visual signaling pathway through the constitutive activity of the apoprotein opsin, which leads to the development of the disease state (Woodruff et al., 2003). When β2-adrenergic receptor is overexpressed in mouse heart, much higher basal rates of activation are observed, which eventually results in cardiomyopathy (Liggett et al., 2000).
The use of functional assays engineered to display elevated basal (constitutive) activity revealed that many drugs previously deemed to be simple (silent) antagonists did in fact possess intrinsic activity and led to their subsequent recategorization as inverse agonists. The clear in vivo efficacy that these inverse agonist drugs display suggests that the accompanying systemic tone resulting from this constitutive receptor activity can comprise a general feature of important GPCR-related biological feedback circuits. Although examples exist of a therapeutic preference of inverse agonists over neutral antagonists, as for example H3 inverse agonists for cognition (Schwartz et al., 2003; Arrang et al., 2007), the existence of constitutive receptor action as a prevalent in vivo phenomenon in either normal or pathophysiological states remains to be sufficiently cataloged (de Ligt et al., 2000; Milligan, 2003; Schwartz et al., 2003).
D. Assay Development
In working toward populating an assay tool box for any given target-focused drug discovery campaign, we face a practical dilemma: to know too little about many things or to know too much about a few. It is noteworthy that technology has helped to both mitigate and propagate this problem.
1. The Genomic Tool Box.
Closure of the genomic roster for the GPCR superfamily and others along with the establishment of a full catalog of corresponding clones has theoretically provided the building blocks for all assays needed to attack the therapeutic genome. In addition to the target proper, additional components such as assorted signaling proteins and cell lines that host engineered designs are now readily available. With the exception of certain GPCR subclasses (e.g., olfactory GPCRs and splicing variants), expression of GPCR proteins and their effectors for functional assessment is now relatively routine, although the details of such expression are important and can compromise discovery, for example by inadvertently limiting or biasing pharmacological results.
2. Screening Efficiency.
Over the past decade, the time and cost needed to execute large-scale GPCR screens has significantly diminished. Developments in HTS technologies have translated into time and cost savings and provided a variety of ultra-high-throughput assay options ranging from labeled and label-free queries of bimolecular interactions to image-based measures of multiparameter cell-based events. Integration of HTS detection platforms with robotics and informatics systems for materials handling and data capture/analysis together with visualization tools for data mining and report sharing has made execution of million compound screens and inspection of the results a timely, straightforward, and efficient process.
Resolution of the information content in such assays will vary depending upon how it is to be employed and the scope/precision required. Output typically ranges from 50 to 250,000 data points per week and may comprise assessment of hundreds of thousands of compounds against a single target by single point concentration (yes/no) activity measurements, as occurs during a primary screen, or assessment of hundreds of compounds in a concentration-dependent fashion against multiple targets, as occurs during ensuing SAR lead development. In these and other instances, archiving, management, and ready availability of such data constitute an important informatics estate for ongoing and future drug discovery programs.
3. Screening Mode.
The past decade has seen a shift away from traditional competitive displacement binding assays toward cell-based functional assays. This shift was encouraged by the need to probe more deeply into the functional activity of compounds at the outset, immediately distinguishing between agonist and antagonist hits. The switch was also encouraged by the problems inherent in introduction of labels into the orthosteric probes needed for competition binding assays and is, of late, vindicated by the increasing need to entertain nonorthosteric binding sites during the primary screening process.
To this end, first-generation functional assays employed measurements of second-messenger levels of any one of the canonical major GPCR pathways: cAMP induction for Gαs, inhibition of forskolin induced cAMP for Gαi, and phosphatidylinositol bisphosphate induction (or associated Ca2+ levels) by phospholipase C-induced Gαq pathways. The Gαq pathway is important to note, because when it was discovered that Gα15 and Gα16 subunits could promiscuously interact with many GPCRs to elicit Gαq-mediated second messengers, the idea of constructing generic platforms became obvious and widely employed (Simon et al., 1991; Offermanns and Simon, 1995; Stables et al., 1997; Zhu et al., 2008). This movement was further fueled by the availability of the industry's first truly high-throughput cell-based platform for Ca2+ fluorescence measurements, the fluorometric imaging plate reader. Additional tactics along these lines also took advantage of forced coupling through the use of Gαq chimeras fused to Gαs and Gαi adaptors. Reporter cell lines incorporating cassettes of response elements sensitive to Ca2+ or cAMP levels also became popular and were often used in conjunction with a promiscuous G protein cell host (Knight and Grigliatti, 2004). In all cases and in the interest of platform efficiency, the measured events were purposefully designed to be the same regardless of the GPCR studied and were validated based on preconceptions of signaling built into the assay. Thus, these assays reported what and only what the researchers were expecting to see; namely, they fail to address the possibility of ligand-biased signaling or the subtleties of efficacy arising from allosteric effects. The emerging complexity of GPCR pharmacology and its potential relevance for the specific regulation of pathophysiological conditions now suggests that such tactics were ill advised. More recent efforts to secure universal results rely more upon preserving the occurrence of the full spectrum of native downstream GPCR events [e.g., β-arrestins (Rajagopal et al., 2010)].
4. Integrative Assays.
Perhaps the earliest examples of integrative assays were ex vivo-based organ baths. It is thanks to their usage that some of the first GPCR transmitter substances were discovered (Rapport et al., 1948a,b). The concept of approaching pharmacological assays holistically, preserving more by modifying less, can be seen in modern “integrative” assays, which have been developed to mitigate the problems inherent in engineered artificiality and provide a panoramic window into the complex “signalosome.” Such cell-based methods include the microphysiometric measurement of cell metabolism (Salon and Owicki, 1995), the electrical measurement of changes in cell impedance (Verdonk et al., 2006; Peters and Scott, 2009) and the plasmon-based detection of cell mass redistribution (Fang and Ferrie, 2008; Fang et al., 2008; Lee et al., 2008). Such assays are a two-edged sword, providing a broad-based detection system but little to no information about the specific signaling pathways that are activated. Determination of the latter requires subsequent and sometimes nontrivial experimental dissection of the receptor-dependent phenomena (Kenakin, 2011).
5. New Generation Biochemical and Biophysical Assays.
Reappreciation of bimolecular interaction assays, whether composed of conventional radio- or fluoroligand displacement assays or newer label-free systems, promises to further simplify the identification/quantification of compound-receptor interactions through direct measurement of the immediate drug-target binding event. The nature of these assays makes them less affected by the practical problems of orchestrating and executing cell-based assays that, as stated above, suffer from cell viability issues, artifactual signaling introduced by cell engineering and the myriad downstream events that can confound their interpretation. Bimolecular interaction assays, on the other hand, provide the simplest and most direct route to the affinity number most sought during the early stages of drug development.
A particularly promising category of bimolecular assay that is prevalent in the drug discovery community are plasmon-based methods that have the additional advantage of being label-free. Conventional surface plasmon resonance (SPR) has been applied to detergent solubilized as well as purified GPCRs immobilized on a sensor chip to qualitatively detect ligand interactions and quantitatively assess the kinetics of their on and off rates (Navratilova et al., 2005). In addition to providing the kinetics of binding, SPR methods hold promise as rapid screening assays for GPCR constructs in X-ray crystallization trials and biophysical mapping of receptors as part of structure-function campaigns (Tollin et al., 2003; Hruby and Tollin, 2007). Alternatively, real-time plasmon waveguide resonance spectroscopy can be useful as both a kinetic tool and a means of predicting the pharmacological action of ligands. The PWR method offers increased sensitivity over traditional SPR by detecting plasmon shifts in two dimensions; it can report mass rearrangements associated with the conformational plasticity of the GPCR system that correlate with pharmacological activity (Devanathan et al., 2004; Alves et al., 2005; Hruby and Tollin, 2007; Georgieva et al., 2008).
E. Limitations of Current Compound Libraries
1. Chemical Space versus Biological Space.
The problem of comprehensively spanning all “druggable” chemical space for GPCR drug discovery, at least in an orthosteric setting, is confounded by the structural diversity of the endogenous transmitters. These activators range from small entities such as Ca2+, to chemically simple classic transmitters, to molecules with complex secondary structure such as peptide transmitters and glycohormones. From a drug design perspective, competing with the more complex ligands is especially challenging, because one must contend with combinatorially larger numbers of chemical interactions that involve both recognition and activity.
In addition, the distribution of content in corporate drug libraries across biological space is likely uneven. As for GPCRs, these collections have evolved through prior drug discovery campaigns directed largely toward the orthosteric modulation of class A targets. Because most corporations have been interested in the same targets, there is likely significant overlap across these corporate collections. Additional requirements that molecular content must possess in vivo drug-like qualities, as predicted for example by Lipinski rules (Lipinski et al., 2001) or as experimentally measured by PK and ADME, and the medicinal chemical “toolbox” employed for expansion of initial hits (Meanwell, 2011) also acts to confine content to isolated clusters of “well behaved druggable” space.
Somewhat different arguments about “druggable” space apply to the pursuit of large-molecule therapeutics (which, although not the same as replacement biologics, can be considered alongside them for drug development arguments). In these instances and with the exception of chemically centric protein designs, the problem of contending with the limited chemical space of a pre-existing library is even more profound; as the size of the recognition portion of a molecule increases, so too does the complexity of its interaction with the target. Compounds from pre-existing drug design campaigns would be perhaps even less likely to function as possible activators. To circumvent these limitations, techniques such as the generation of “avimers” comprising phage display or exon shuffling-based expression of multivalent protein interaction motifs have been proposed and are just now entering into the drug development pipeline (Silverman et al., 2005). Such complexity provides an opportunity for increasing selectivity through multivalent binding. but their large size may have negative implications for both PK and PD properties (Fig. 7).

Therapeutic modalities. Drug molecules can be broadly categorized as either small molecules or biologics, with development and FDA review after new chemical entity and biologic license application guidelines, respectively. Although GPCR drug discovery has historically been dominated by small molecule programs, this predominance is changing in part because of the increasing awareness that target receptors can have much larger endogenous ligands (e.g., glycohormones, viral entry proteins) and in part because of methods that merge antibody or other scaffolds with chemical and peptide moieties to provide robust delivery vehicles for active small molecule warheads. An application of this method is the newly developed “avimer” technologies, which are antibody mimetics that present multiple binding epitopes. SBDD methods and infrastructure benefit from both modalities by identifying novel sites accessible to small-molecule binding and providing purified GPCR proteins stabilized in specific conformations to capture large molecule drug candidates through more empiric screening methods.
2. Future Expansion of Chemical Screening Libraries.
The unevenness of library collections will ultimately smooth out as they become populated by discovery campaign data based upon new generation assays geared toward the search for nonorthosteric entities that possess new forms of pharmacological activity, such as ligands that target intermediary conformations associated with biased signaling and those that interfere with receptor dimers. Empirically, the current libraries at hand are what we must start with. It is unknown whether they can provide initial discovery points for the development of new chemical entities that operate in new pharmacological dimensions. Accordingly, arguments have been made for development of fragment-based libraries and their associated screening assays to maximize identification of building blocks for new drug molecules (Bartoli et al., 2007; Fattori et al., 2008). Clearly, greater insight into the atomic structure of specific GPCRs would help bypass these problems and open the prospect of a priori drug design against any region of these particular receptors. Identification of ligand-binding sites within specific conformations known to correlate with a desired pharmacological activity would further allow us to focus on ligands that stabilize specific protein “state” targets to achieve a desired effect. Recent advances in GCPR crystallography suggest this ambitious goal is fast becoming a real possibility.
III. History of G Protein-Coupled Receptor Structural Clarification
A. Functional Genomics Reveals the G Protein-Coupled Receptor Superfamily
Rhodopsin was the first GPCR purified to homogeneity in the 1970s for biochemical study, and it was in the visual system that functional coupling between GPCR and heterotrimeric guanylate nucleotide-binding protein was first observed in the early 1980s (Kühn, 1980; Fung et al., 1981). The determination of the amino acid sequence of rhodopsin by Hargrave et al. (1983) and Ovchinnikov et al. (1983) was a significant achievement and provided the starting point for models of its membrane topology. Shortly after this work, Nathans and Hogness (1983) used at-that-time novel molecular biological techniques to clone all human visual pigments that later permitted insights into the genetic basis of pathologic conditions in color vision (Nathans et al., 1986a,b). Cloning of rhodopsin and extrapolation of its putative molecular model to other GPCRs provided the prerequisites for understanding ligand binding and G protein interaction (Filipek et al., 2003b). This advance also provided early insights into G protein signaling and its ubiquitous presence in cells and tissues (Bitensky et al., 1984; Dixon et al., 1986). Analogous work performed by Dixon et al. (1986) and Fargin et al. (1988) employed the partial β2-adrenergic receptor protein sequence to design degenerate PCR primer sequences to clone and sequence the full-length β2-adrenergic receptor as well as the first orphan receptor (later identified as the 5-HT1A receptor). This and subsequent work suggested the existence of a new family of integral membrane proteins, all of which shared signaling pathways relying upon heterotrimeric G proteins and having in common a seven transmembrane α-helical architecture (Gilman, 1987; Baldwin, 1993, 1994; Baldwin et al., 1997; Filipek et al., 2003b). This homologous structure concept spawned a decade of homology and expression-cloning efforts, during which many of our initial molecular biological discoveries for the GPCR superfamily were made.
B. Electron Microscopy of Rhodopsin Provides a Conceptual Prototype of G Protein-Coupled Receptor Structure
The archaeal proton pump bacteriorhodopsin was initially hypothesized to be an analog of rhodopsin before the latter's three-dimensional structural determination by electron crystallography. Both proteins feature a covalently bound retinal chromophore linked through a Schiff base to a Lys residue, exhibit a light-dependent change in spectral absorption, and possess a seven-transmembrane helix architecture. Protein and DNA sequencing of rhodopsin (Hargrave et al., 1983; Nathans and Hogness, 1983, 1984; Ovchinnikov et al., 1983) and the β2-adrenergic receptor (Dixon et al., 1986) together with hydropathy plots facilitated construction of two-dimensional topology models (Argos et al., 1982; Hargrave et al., 1983; Ovchinnikov et al., 1983; Dixon et al., 1986) similar to those generated for bacteriorhodopsin. Electron crystallographic studies of two-dimensional crystals of bovine, amphibian, and invertebrate rhodopsins demonstrated that the arrangement of helices in rhodopsin differed substantially from those in bacteriorhodopsin (Schertler et al., 1993; Krebs et al., 1998; Davies et al., 2001). These results bolstered the idea that rhodopsin, and not bacteriorhodopsin, would serve as a model for all GPCRs, leading to its adoption for modeling helical arrangements within the transmembrane bundle of GPCRs (Baldwin, 1993, 1994; Baldwin et al., 1997; Unger et al., 1997). These structural results were further advanced by mutagenesis and biochemical studies allowing spacial assignment of post-translational modifications such as disulfide bonds (Karnik et al., 1988; Karnik and Khorana, 1990), palmitoylation (Ovchinnikov et al., 1988; Karnik et al., 1993), and phosphorylation (Palczewski et al., 1991; Ohguro et al., 1993, 1996, 1998). These biochemical studies together with specific mutations led to a clear demarcation of the border between loops and transmembrane regions (for review, see Menon et al., 2001; Sakmar et al., 2002; Filipek et al., 2003a; Hubbell et al., 2003; Palczewski, 2006). For more than 20 years, rhodopsin has served as the protypical GPCR for study owing to its relative ease of purification and the wealth of structural and biochemical information available.
C. Comparative Modeling Suggests Structure-Function Experiments
Elucidation of the first vertebrate and invertebrate GPCR structures (Palczewski et al., 2000; Li et al., 2004; Okada et al., 2004; Murakami and Kouyama, 2008; Shimamura et al., 2008; Stenkamp, 2008) opened the way to probe ligand-binding sites and assess structure-function relationships for these receptors (Lu et al., 2002; Hubbell et al., 2003; Park et al., 2004). The overall similarity in helical packing is expected to extend to other GPCRs whose structures have yet to be determined (Lodowski and Palczewski, 2009; Lodowski et al., 2009; Mustafi and Palczewski, 2009). Given the assumption that all GPCRs share grossly similar membrane structures, it is a reasonable expectation that this data can be used in conjunction with current three-dimensional templates to derive distinctive homology models.
Currently available structures of rhodopsin and other GPCRs thus present workable templates for producing homology models of the remaining GPCRs (Zhang et al., 2006; Michino et al., 2009). However, because of their low overall sequence homology, the imprecision of algorithms used to predict GPCR overall structure, and the location of small-molecule-binding sites within their transmembrane domains, this approach has not yet evolved sufficiently to produce models accurate enough to reliably predict pharmacophore sites. Even small differences in exact helical arrangements can greatly affect the predicted binding site for small-molecule ligands. Moreover, it should be considered that bovine rhodopsin remains the only truly “native” ligand-bound GPCR structure determined. The protein engineering in all current nonrhodopsin GPCR structures can disrupt and obscure the spatial relationships of important regulatory and functional motifs, such as changes in cytoplasmic loop three, which occur upon agonist activation or the breaking of the D(E)RY “ionic” lock (Dror et al., 2009). A related concern is the criteria used to assess the accuracy of existing models in the absence of high-resolution structures. For the many GPCRs, the low overall sequence identity between templates complicates accurate alignments and predictive demarcation of ligand-binding sites. Such approaches amount to pseudo-ab initio structural prediction (Schlyer and Horuk, 2006) and despite widespread usage, these methods have generally failed to provide models accurate enough for therapeutic ligand design (Deupi et al., 2007).
The recent agonist-bound GPCR structures provide little structural reason why particular compounds function as full agonists, partial agonists, or even antagonists, nor do they show consistent structural transformations that accompany the activation of a particular GPCR. The agonist and partial agonist bound β1-adrenergic receptor structures (Warne et al., 2011) as well as the tethered agonist-β2-adrenergic receptor structure (Rosenbaum et al., 2011) exhibit only small-scale changes compared with their antagonist-bound counterparts, consistent with conclusions on inactive state of receptors stated in section B above. Energy for such a transformation is typically between 8 and 12 kcal/mol, with only a small fraction of this energy provided by the ligand binding events per se. Because only small conformational changes are supported by the energetics of this process, structural differences between the activated state before G protein coupling and its inactivated state must be minimal, further emphasizing the high precision needed in a model if it is to have any utility. The recent β1-adrenergic agonist structure, the camelid antibody-agonist-β2-adrenergic receptor structure, the agonist bound A2a-adenosine receptor structure as well as the constitutively active rhodopsin and all-trans-retinal regenerated opsin structures provide additional snapshots as to the dynamic remodeling, which may become possible after the release of molecular restraints that accompany GPCR activation occurs (Choe et al., 2011; Rasmussen et al., 2011; Standfuss et al., 2011; Xu et al., 2011). Upon examination of the antibody-agonist-β2-adrenergic receptor structure, larger-scale displacements of helices H-V and H-VI are evident compared with the small-scale displacements in the β1-adrenergic receptor structures. It remains unclear as to how to dissect the relative influences on the observed structures of the antibody, crystal contacts/conditions, agonist binding, stabilizing mutations, and T4 lysozyme insertion in between H-V and H-VI. Although it is proposed that the antibody in the case of the antibody β2-adrenergic receptor structure and the Gt peptide in the opsin* and opsin derived Meta II-like structures acts as a G protein surrogate, the validity of this hypothesis remains to be seen (Choe et al., 2011; Rasmussen et al., 2011; Standfuss et al., 2011). Upon close examination, there is a lack of concordance in the degree of structural changes observed upon activation in the recently determined agonist-bound GPCR structures that probably reflects the structural plasticity needed for the varying levels of activity and regulation of the agonist induced response. Additional insight into the functional mechanics of these targets will likely be afforded by determining structures of important signaling states involving not only agonist-bound receptor but also G protein (Jastrzebska et al., 2010).
The problem becomes more difficult once the influence upon ligand binding from such issues as allosterism, ligand trafficking, and the conformational continuum that accompanies ligand binding are considered. As the diversity of available receptor-ligand structures increases and new functionally important commonalities (presumably) reveal themselves, it is hoped that the predictive performance of homology modeling will improve. More detailed knowledge of fold-space gained through structural genomics projects and recent attempts to computationally solidify more precise homology models will further extend the utility of homology modeling (Simons et al., 1999; Baker and Sali, 2001; Lee et al., 2001; Misura et al., 2006), ultimately allowing drug design in silico, circumventing the requirements for a starting crystal structure.
D. Structure-Function Campaigns Identify Hotspots: Common G Protein-Coupled Receptor Functional Moieties, Trigger Mechanisms, and Long-Awaited High-Resolution Three-Dimensional Structures
In addition to ground-state bovine rhodopsin, structures of heterologously and natively expressed GPCRs have been determined. Examples include light-activated rhodopsin (Salom et al., 2006), ligand-free bovine opsin (Park et al., 2008; Scheerer et al., 2008), heterologously expressed constitutively active mutant rhodopsin (Standfuss et al., 2011), proteolyzed squid rhodopsin (Murakami and Kouyama, 2008; Shimamura et al., 2008), human β2-adrenergic receptor-T4-lysozyme fusion protein with inverse agonist bound (Cherezov et al., 2007; Hanson et al., 2008), human β2-adrenergic receptor-T4-lysozyme fusion with camelid antibody and agonist bound (Rasmussen et al., 2011), mutant turkey β1-adrenergic receptor with full and partial agonists and antagonist bound (Warne et al., 2008, 2011), mutant human A2A-adenosine T4-lysozyme fusion protein with agonist and antagonist bound (Jaakola et al., 2008; Xu et al., 2011), C-X-C chemokine receptor type 4 T4-lysozyme fusion with multiple antagonists bound (Wu et al., 2010), and D3-dopamine-T4-lysozyme fusion with antagonist bound (Chien et al., 2010). Although overall sequence identity between these GPCRs is low (e.g., 15% identity between bovine rhodopsin and β2-adrenergic receptor), comparative alignments clearly reveal conserved amino acid residues and motifs known to be essential to GPCR function as well as an obvious conservation of the topology and seven-transmembrane (TM) architecture (Mirzadegan et al., 2003; Madabushi et al., 2004). This conservation of secondary structure provides the precise spatial positioning needed for the functional arrangement of these few sequentially discontinuous motifs. Thus, an examination of those receptors for which structural solutions exist reveals a remarkably conserved structural core expected to be applicable to the entire superfamily, with even higher degrees of similarity among subfamily A members (Mirzadegan et al., 2003; Lodowski and Palczewski, 2009; Lodowski et al., 2009; Mustafi and Palczewski, 2009) (Fig. 8).

Information flow during rhodopsin activation. Upon absorption of a photon of light, the 11-cis-retinylidene chromophore is isomerized to its all-trans-state, driving all subsequent activation steps. Deprotonation of the Schiff base linkage follows photoisomerization, and through small-scale changes within the transmembrane region, the activation signal is propagated to the D(E)RY (Glu134, Arg135, and Tyr136) region, resulting in disruption of the “ionic lock” and uptake of a proton from the cytoplasm (most likely onto Glu181, which protrudes toward the chromophore from the one of the β-strands of the plug domain), leading to fully activated meta II rhodopsin. Meta ll catalyzes nucleotide exchange upon the G protein α-subunit of transducin heterotrimers, propagating the activation signal inside the cell. Three regions important in activation and other GPCR functions are highlighted within the transmembrane region: the D(E)RY motif, the NPxxYx(5,6)F motif. and the chromophore-binding site. The three insets detail the interactions present within these conserved motifs. For ease of interpretation, helices are depicted in the following colors: H-I, red; H-II, orange; H-III, yellow; H-IV, lime green; H-V, dark green; H-VI, teal; H-VII, blue; and H-8, purple.
The tertiary structure of the receptor is demarcated by its ellipsoidal shell. Dimensions of the ellipsoid are ∼75 to 80 Å orthogonal to the membrane and ∼50 × 35 Å wide in the plane of the membrane. The surface area of those portions projecting from the membrane is ∼1200 Å2, the volume and surface area of the cytoplasmic projection typically exceeding the extracellular projection. For some GPCRs, the extracellular portion consists of a separate large ligand-binding domain (e.g., glutamate and Ca2+ receptors) and dominates the receptor's available hydrophilic surface area. The overall ellipsoidal cross-section of GPCRs results from the specific arrangement of the seven-transmembrane α-helices, each of which must be ∼20 residues long or more to completely traverse the lipid bilayer (Nyholm et al., 2007). Inspection of GPCR hydrophobicity plots and newly available three-dimensional coordinates show these helices to vary in length from 22 to 33 residues, including kinks in the helix as it bisects the plasma membrane.
These kinks and intrahelical packing cause the helices to bend and tilt away from the membrane normal vector (Fig. 8). The tilting and bending of helices, as well as other energetically unfavorable helical disruptions, are counteracted by internal hydrogen bonding between surrounding residues. Further bends or kinks are induced through Gly-Gly, Pro-Pro, or Gly-Pro segments, ensuring that the helical structure is disrupted. In general, the calculated tilt angle for each transmembrane helix (H) is 22 ± 12° (Nyholm et al., 2007). In rhodopsin, the strongest helical distortion is imposed by Pro267 in H-VI, one of the most highly conserved residues among all GPCRs. The presence of Pro291 and Pro303 in the region around the Lys296 retinal attachment site elongates H-VII. Pro303 is part of the highly conserved NPxxYx(5,6)F motif in subfamily A GPCRs.
The cytoplasmic face of the GPCR consists of three loops encompassing residues Gln64–Pro71, Glu134–His152 and Gln225–Arg252, the nontransmembrane H-8 and the C-terminal tail. Residues of the highly conserved D(E)RY motif found in almost all subfamily A GPCRs, is located in this region (Glu134-Arg135-Tyr136 in rhodopsin) (Mirzadegan et al., 2003). A second highly conserved motif is the NPxxYx(5,6)F motif, which is found near H-8.
1. The D(E)RY Motif within G Protein-Coupled Receptors.
The highly conserved D(E)RY motif forms an “ionic lock” at the cytoplasmic end of H-III thought to retain the GPCR in the inactive state through the salt bridge between the Arg residue in the motif and a conserved Glu or Asp residue in H-VI, thereby holding H-III and H-VI together (e.g., Arg135 forms a salt bridge with Glu247 in bovine rhodopsin) (Fanelli and De Benedetti, 2005). Mutagenesis data combined with FTIR analysis suggests that the ionic lock represents an energetic barrier that must be broken to achieve the activated state (Alewijnse et al., 2000; Fritze et al., 2003; Mahalingam et al., 2008; Schneider et al., 2010). Most but not all GPCRs contain this motif, suggesting that it plays an important but not wholly indispensible role in the activation process (Flanagan, 2005).
In rhodopsin, the carboxylate of Glu134 interacts with Arg135, thus positioning Arg135 to form a salt bridge with Glu247 and interact with Thr251 in H-VI. Recent work demonstrates the critical roles of Glu134 and Arg135 of the conserved D(E)RY motif in rhodopsin activation (Lüdeke et al., 2009) (Fig. 8). Disruption of the salt bridge between Arg135 and Glu247 is considered a hallmark of progression from the meta I to the meta II active signaling state. Protonation of the acidic Glu134 residue within this motif is thought to accompany activation (Scheer et al., 1997; Vogel et al., 2008; Lüdeke et al., 2009; Ye et al., 2009), because the protonation state of Glu134 is sensitive to its environment, so its protonation may accompany or even take part in the activation process (Mahalingam et al., 2008). Although changes in protonation states of these conserved acidic residues have been linked to the activation process, the mechanism linking the protonation state of the ionic lock to the deprotonation of the Schiff base linkage that accompanies attainment of the Meta II state is largely uncharacterized.
The bond between the Asp (or Glu) residue and the Arg within the D(E)RY region is absent in many of the recently determined structures of nonrhodopsin GPCRs. Each structure contains a bound agonist, an inverse agonist, or an antagonist and is stabilized by mutational modification of the receptor backbone. This observation led to the speculation that the actual lock might be unique to rhodopsin, so although the motif was present in most GPCRs, it cannot actually act as a functional “lock” (Cherezov et al., 2007; Rasmussen et al., 2007). This supposition prompted a series of molecular dynamics experiments for each of these receptors, all of which revealed that the ionic lock, even if absent (disrupted) in the crystal structure, quickly reforms once restraints imposed by the T4-lysozyme fusion and crystal lattice are removed (Dror et al., 2009; Lyman et al., 2009; Vanni et al., 2009; Jojart et al., 2010; Romo et al., 2010; Fanelli and Felline, 2011). This does not mean that the observed disruption of the ionic lock is an entirely artificially induced state; partial occupancy of a disrupted ionic lock state might explain the agonist-independent activation observed for these receptors (Fig. 8). Recent CXCR4 structures (Wu et al., 2010) add to the confusion as the Glu/Asp residue of the lock is a Lys residue in CXCR4. In the highest resolution structure (PDB ID 3ODU), Arg134 is bound to H-VI through two crystallographically observed waters, which could perform a role similar to that of the Glu residue in rhodopsin. Confounding matters further, the ionic lock is intact in the dopamine D3 receptor structure when bound to an antagonist, suggesting that the disruption of this motif may be an artifact in the other GPCR structures solved to date (Chien et al., 2010). In recent adrenergic receptor-agonist structures this motif was found to be disrupted, but because this motif was disrupted in the original antagonist-bound structures, it is difficult to gauge whether this disruption is a manifestation of the construct/crystallization conditions, a consequence of agonist binding or inherent agonist-independent activation, or some combination of these factors. Overall, the preponderance of biochemical evidence is that the ionic lock is a common feature of class A GPCRs and is likely important in transmitting conformational changes.
2. The NPxxYx(5,6)F Motif within G Protein-Coupled Receptors.
Another well conserved region among GPCRs is the NPxxYx(5,6)F sequence (NPVIY in rhodopsin) near the cytoplasmic end of H-VII, which is likely to be involved in G protein coupling. Side chains of the two polar residues in this region, Asn302 and Tyr306 in bovine rhodopsin, project toward the transmembrane core of the protein and Phe313 in H-8, respectively. The hydroxyl group of Tyr306 is close to Asn73 and is engaged in the interhelical hydrogen-bonding constraints between H-VII and H-II, which most likely emanate from water molecules. This hydrogen bonding interaction has been postulated to affect G protein coupling directly (Miettinen et al., 1997; Mills et al., 2000). Furthermore, hydrophobic interactions between Tyr and Phe residues link H-8 to the end of H-VII, allowing changes in the position of H-VII to induce movements of H-8 upon activation (Fritze et al., 2003). In higher resolution structures of rhodopsin, a cluster of three waters appears to link H-I, H-II, and H-VI (Okada et al., 2002; Faussner et al., 2005). Further in-depth examination of bound water interactions in this vicinity reveals that a similar network of waters interact with the Asn residue (Angel et al., 2009b). However, the interaction between Asn73 and Tyr306 observed in rhodopsin crystal structures has not been seen in any other determined GPCR structure, suggesting this may be a rhodopsin-specific interaction (Fig. 8), but the “local” structure within this region is similar in all GPCRs.
3. Conservation of Water and Water-Binding Sites within the Transmembrane Domain.
In rhodopsin, with the exception of Lys296, which is covalently modified with chromophore and residues Glu122 on H-III and His211 on H-V, which are involved in salt bridges, all charged residues in the transmembrane region interact with crystallographically observed waters. Conservation of a subset of these waters and associated charged residues has been observed in all high-resolution GPCR structures solved to date, suggesting that these waters form a network of allosteric effectors upon which the activation process relies (Fig. 9) (Angel et al., 2009a,b; Jardón-Valadez et al., 2009; Orban et al., 2010). Radiolytic footprinting studies indicate that these waters are not freely exchangeable with bulk solvent and that upon activation undergo significant rearrangements, further supporting their role as noncovalently bound prosthetic groups (Angel et al., 2009b; Orban et al., 2010). Further work probing the exact roles that these waters play in the activation process is required.

Homology of water-binding sites within the transmembrane region of GPCRs. Although overall sequence similarity, with the exception of globally conserved GPCR motifs, is low within the transmembrane region, comparison of the positions of crystallographically observed (ordered) water molecules reveals several clusters of solvent in similar positions. These similarities and the conservation of amino acid side chains that interact with these waters suggest a functional role for these water molecules. Water molecules from rhodopsin (PDB ID 1U19) are depicted in red, β2-adrenergic receptor (PDB ID 2RH1) in green, A2A-adenosine receptor (PDB ID 3EML) in yellow, β1-adrenergic receptor (PDB ID 2VT4) in blue, and CXCR4 (PDB ID 3ODU) in cyan.
4. Ligand-Binding Domains of G Protein-Coupled Receptors.
GPCRs are activated by chemically diverse classes of ligands that bind either to sites located within the receptor's transmembrane region, on its extracellular face, or within distinct extracellular ligand binding domains. To date, all structurally determined GPCRs are activated by binding within the transmembrane region. For rhodopsin, the 11-cis-retinal chromophore covalently linked to residue Lys296 in H-VII acts as an inverse agonist, locking the receptor in its inactive state. The retinal-binding cavity is closer to the extracellular surface than to the cytoplasmic surface; structural and mutational data reveal that the ligand-binding cavity is located in a grossly similar position for many GPCRs including those that bind amine ligands (e.g., the β2-adrenergic receptor). The β1- and β2-adrenergic receptors, which both bind epinephrine, constitute an intriguing case. The immediate residues that form the binding pocket for this hormone are identical, but the two receptors have distinctly different affinities for norepinephrine, suggesting that residues that do not directly interact with the ligand-binding site play a role in ligand selectivity (Fig. 10). The amino-terminal region and extracellular loops of rhodopsin form a plug covering the retinal-binding cavity and contacting the chromophore, shielding the retinal binding pocket from the extracellular milieu, a feature unique to rhodopsin. Because the large majority of GPCRs must bind ligand supplied through systemic circulation, this plug is absent and is a likely site for ligand entry.
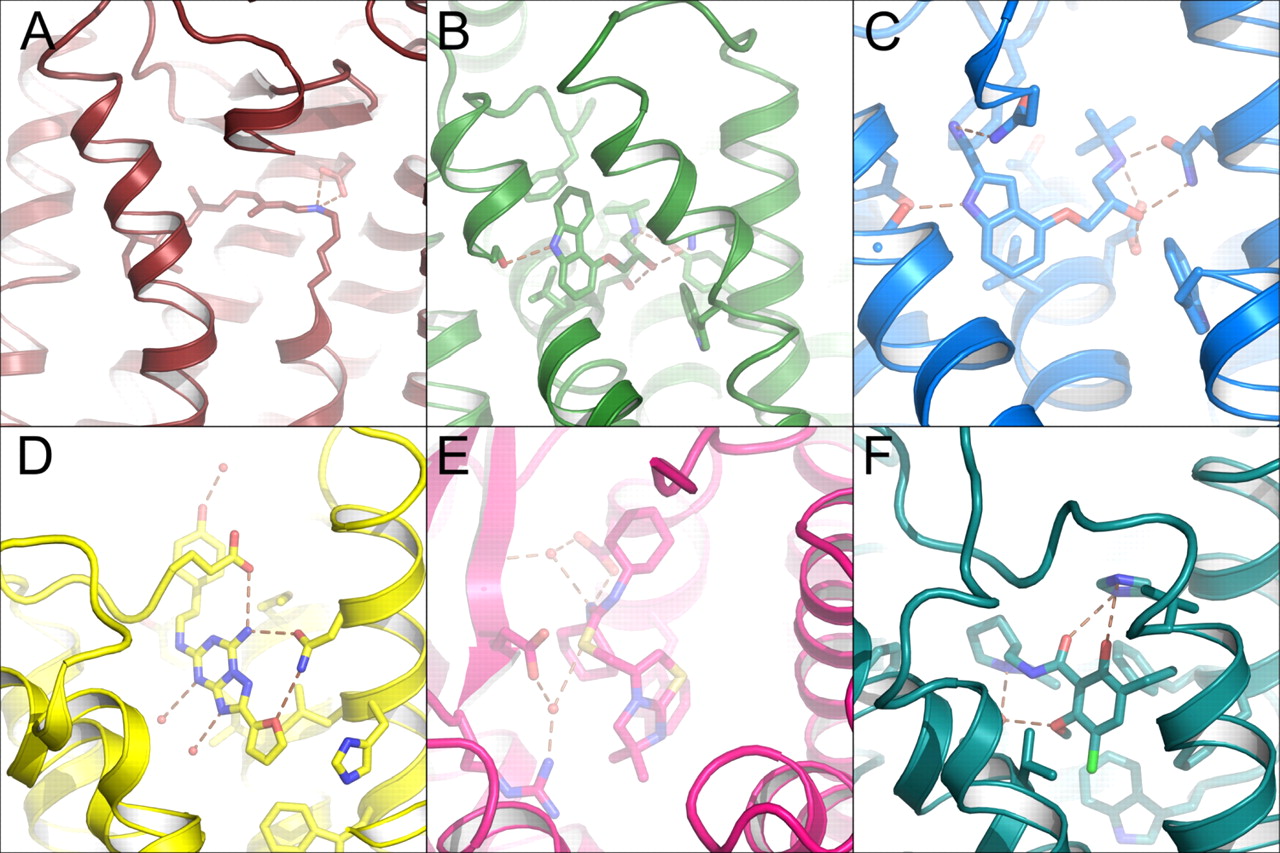
Ligand-binding sites within selected GPCR structures. For these sites, all polar contacts within 3.4 Å of each other are indicated by brown dashed lines and nonpolar side chains are also shown when they do not obscure the ligand. In some cases, portions of the transmembrane helices are removed for clarity. A, chromophore of rhodopsin (PDB ID 1U19) is a tethered ligand (through a Schiff-base linkage); the only polar contact in the inactive state is to the counter ion, Glu113. B, binding of the inverse agonist carazolol to the β2-adrenergic receptor (PDB ID 2RH1). C, antagonist cyanopindolol bound to the β1-adrenergic receptor (PDB ID 2VT4). D, binding of the A2a-adenosine receptor (PDB ID 3EML) to the antagonist ZM241385 implicates both solvent mediated interactions as well as direct interactions with amino acid side chains in ligand binding. E, structure of CXCR4 bound to the antagonist It1t (PDB ID 3ODU) uses both solvent and direct interactions to define the ligand-binding pocket. F, D3 dopamine receptor (PDB ID 3PBL) bound to the antagonist eticlopride.
For many GPCRs that recognize amino acid (e.g., metabotropic glutamate receptor), peptide (e.g., vasoactive intestinal peptide 1 receptor) or large protein hormones (e.g., thyroid stimulating hormone receptor), the ligand-binding site is located on a separate domain located in the extracellular space. Although several structures of these isolated domains have been determined in complex with their physiological ligands as well as with inhibitors, the mechanism by which such ligand binding is coupled to activation of the receptor remains unclear (Kunishima et al., 2000; Fan and Hendrickson, 2005; Parthier et al., 2007; Pioszak et al., 2008, 2010; ter Haar et al., 2010). Although the mechanism by which these GPCRs with large extracellular domains transfer their activation signal to the 7-TM portion of the GPCR has not been fully elucidated, some conclusions can be drawn. Experiments performed with metabotropic glutamate receptors mutated to have only one functional ligand binding domain indicate that agonist activates both protamers with equal efficiency (Brock et al., 2007). Because these disparate ligand-binding domains are linked to the 7-TM core via a single α-helix and these receptors are constitutively dimeric, it is likely that the activation signal is imparted through a mechanism that relies upon intersubunit rearrangement(s) to drive the changes in the 7-TM domain necessary to activate G protein.
Considering the transmembrane architecture that all GPCRs are expected to share, the conservation of motifs within the transmembrane domain, the variability of structural rearrangements that are observed in agonist bound GPCR structures, the conservation of ordered solvent binding sites within the transmembrane region, and likely mechanism of activation for class B and C GPCRs containing large extracellular ligand binding domains, it becomes possible to propose the following conclusions about the activation process: 1) Rather than a single distinct activated state that occurs upon binding of agonist, there exist multiple conformations of the protein backbone upon binding agonist, only a subset of which are capable of activating G protein. 2) Binding of agonist must act through a mechanism that begins not with large movements of helices but with the release of constraints that hold the GPCR in an inactive conformation. 3) A network of ordered solvent molecules plays an integral role in transmission of the activation signal through the transmembrane region. Despite the amazing level of progress in the determination of GPCR structures containing agonist compounds, more structural work is still needed. To fully understand the activation process, we still need the structures of intact class B and C GPCRs in addition to crystal structures that capture productive signaling states of the receptor in complex with G protein and agonist compounds (see Note Added in Proof).
IV. Advances in G Protein-Coupled Receptor Structural Determination
A. G Protein-Coupled Receptor Construct Design and Expression
The first hurdle in GPCR structural determination continues to be the production and purification of adequate amounts of homogenous, properly folded, and fully functional protein. Purification from native tissue, assuming the protein of interest is present in sufficient quantity, can provide fundamental advantages, such as presenting normal post-translational modifications that influence the folding of biologically relevant conformations. That said, only bovine rhodopsin has successfully been crystallized from its native source and with an intact sequence. All other GPCR structures thus far determined have required significant protein (re)engineering for stabilization. Cell-free expression, Escherichia coli, insect, yeast, and various mammalian cell line systems have been used with various success rates for GPCR expression. E. coli expression systems have been employed to produce heterogeneous preparations of neurotensin and olfactory receptors (Grisshammer, 2006, 2009; White and Grisshammer, 2007; Song et al., 2009). Yeast Saccharomyces cerevisiae and Schizosaccharomyces pombe expression systems have been used to express large quantities of A2a-adenosine receptor with ligand-binding characteristics near those of wild type (Niebauer et al., 2004; Niebauer and Robinson, 2006; O'Malley et al., 2009). Production through cell-free expression systems has proven useful for structural determinations of both soluble and membrane protein targets (Chen et al., 2007b), but large-scale use of these techniques for GPCR expression have yet to yield a GPCR structure (Klammt et al., 2007a,b; Junge et al., 2008, 2010). A variety of mammalian cell-based protocols using transfected and virally infected cells to express GPCRs for biochemical as well as structural determination studies have been published (Sen et al., 2003; Shukla et al., 2006a,b), but rhodopsin mutants expressed in transfected COS-1 cells constitute the only success for this expression methodology (Standfuss et al., 2007, 2011; Stenkamp, 2008). The most efficient and successful method to date, apart from native expression, has been the expression of truncated/stabilized/engineered GPCR constructs in baculovirus-infected insect cell hosts (Cherezov et al., 2007; Rosenbaum et al., 2007; Roth et al., 2008; Warne et al., 2008, 2009; Tate and Schertler, 2009). It is, however, entirely possible that adequate quantities of native GPCRs needed for structure determination may be produced through novel expression methods that may require further development (Zhang et al., 2005; Li et al., 2007; Salom et al., 2008).
It should be noted that the expense, equipment, and expertise needed for cloning and producing the quality and quantity of functional protein needed to begin crystallization trials, let alone to achieve a structural determination, often exceed the means of many academic laboratories. Large-scale protein production via outsourcing (e.g., Bio-Xtal consortium) has offered researchers a way to access large quantities of membrane protein without the need to invest in the associated infrastructure needed for their production. However, for reasons likely associated with the need to experimentally explore and control conformational flexibility, such contract services have yet to prove broadly useful for successful GPCR crystallography.
B. Solubilization and Purification of G Protein-Coupled Receptor Constructs
Once a suitable GPCR expression system has been optimized, the task of establishing a production pipeline for the purification of tens of milligrams of protein to the nearly homogeneous quality needed for structural determination must be accomplished. Because no standardized recipe/procedure is currently available for extracting a GPCR or any other integral membrane protein from its native environment and retaining its pharmacologically relevant conformation(s), a trial-and-error process must be undertaken. On the basis of the several successful crystallization campaigns reported to date, several general guidelines have emerged: 1) ligand or tag affinity purification of the target protein seems suitable for the isolation process. 2) Detergent selection and concentration together with their effects upon isolated receptor stability, homogeneity, and monodispersity must be considered in addition to detergent effects on ligand binding and receptor/G protein activation. Concomitant purification and screening of various detergents together with appropriate stability testing can provide a high-throughput mechanism to attack the empirical challenge of GPCR purification (Vergis et al., 2010). 3) Strategies that stabilize the GPCR are important. Purification in the presence of high-affinity antagonist compounds can assist in retaining activity and homogeneity through stabilization of the GPCR in an inactive state. A major complication in the crystallization of GPCRs is that the conformational flexibility integral to their function promotes inherently unstable purified receptor proteins, thus complicating the goal of generating uniform ordered crystal lattices. Indeed, heterologously expressed GPCR structures have all required at least point mutations or truncations, and most have required the presence of antagonist or inverse agonist compounds to restrain conformational flexibility and allow crystallization. Determination of agonist-bound GPCR structures also continues to rely upon mutational or other stabilization of the energetic minima associated with the activated agonist bound states or fortuitous stabilization via crystal contacts or binding/insertion of entire proteins or domains.
C. Predicting G Protein-Coupled Receptor Construct “Crystallizability”
Because of inherent difficulties in crystallization, a variety of predictors and screening methods have been proposed to determine the suitability of particular integral membrane protein constructs for crystallization. These include measurements of self-interaction and thermal denaturation to assess protein stability as a preliminary estimate of crystallizability. For all these predictors, it is assumed that by achieving more stable protein solutions and constructs, the likelihood of crystallization is increased. Although this hypothesis may generally hold, there are exceptions to the need for monodisperse, homogenous protein for crystallization (Mileni et al., 2010).
1. Assays.
Kawate and Gouaux (2006) used gel filtration chromatography of membrane protein-fluorescent protein fusions in various solubilizing detergents to screen for appropriate detergents based on the degree of protein aggregation and monodispersity. Stability of membrane proteins has also been assayed with the “thermo-fluor” stability assay, wherein thermal denaturation of protein in a solution of fluorescent dye is measured. Denaturation of the protein exposes hydrophobic core residues resulting in altered fluorescence (Ericsson et al., 2006; Vedadi et al., 2006). Lipidic cubic phase-melting temperature (LCP-tm) methodology allows thermo-fluor type measurements in solid lipid cubic phase crystallization matrices (Liu et al., 2010). Nonfluorescent methods for measuring thermal stability that rely upon residual ligand binding activity can also provide complementary data in the case of GPCRs, because incomplete denaturation can have profound effects upon ligand binding (Tate and Schertler, 2009; Warne et al., 2009), with the caveat that ligand binding is not necessarily a definitive measurement of receptor activity.
Light scattering data have also been proposed to provide predictive information for crystallization of soluble proteins. Constructs having narrow unimodal size distributions are much more likely to yield diffracting crystals; only a narrow range of values for the second osmotic virial coefficient or B value were found to be compatible with protein crystallization (D'Arcy, 1994; George and Wilson, 1994). Static light scattering studies of the E. coli integral membrane protein OmpF suggested that these values are predictive for membrane proteins as well (Hitscherich et al., 2000, 2001). The B value conflates a variety of factors such as temperature, ionic strength, and pH and estimates the extent of self-interaction of a protein in solution. B values positively correlate with increases in protein solubility. Self-interaction chromatography represents an easily usable method for determining B values. For example, iterative rounds of self-interaction chromatography and crystallization yielded significantly improved Allochromatium vinosum reaction center-1 crystals (Gabrielsen et al., 2010). Although the isolated cases in which virial coefficients have proven useful for improving crystal quality, the broad utility of the second virial coefficient as a predictor of GPCR or other membrane protein crystallizability remains to be determined.
2. Stabilization of G Protein-Coupled Receptors with Membrane Mimetics during Crystallization.
Although recent progress in structural determination of GPCRs has been remarkable, it is still difficult to make generalizations about optimal crystallization conditions. Details available for the currently limited number of successful campaigns, however, do provide clues as to how crystallization trials of GPCRs should to be conducted. Viscous lipidic cubic phases (LCP) composed of monoolein were first used in crystallizing the light responsive archaeal proton pump bacteriorhodopsin (Gouaux, 1998; Pebay-Peyroula et al., 2000; Nollert et al., 2001) and have been employed since to crystallize several other membrane proteins, including the β2-adrenergic, A2a-adenosine, CXCR4, and D3 dopamine receptors (Rosenbaum et al., 2007; Hanson et al., 2008; Jaakola et al., 2008; Warne et al., 2008; Chien et al., 2010; Wacker et al., 2010; Wu et al., 2010). Reconstitution of membrane proteins into this matrix from the detergent-solubilized state provides stabilization with a more “membrane-like” lipid environment. The dehydration of the cubic phase by precipitant drives formation of a lamellar phase from which protein crystals grow (Cherezov et al., 2002).
Other crystallization approaches for GPCRs also have met with some success. Rhodopsin/opsin and squid rhodopsin were grown from detergent-solubilized protein with ammonium sulfate as a precipitant, and the β1-adrenergic receptor was also grown from alkyl-glucoside/maltoside-solubilized protein with a low molecular weight polyethylene glycol (Okada et al., 2004; Salom et al., 2006; Murakami and Kouyama, 2008; Shimamura et al., 2008; Warne et al., 2008). Although it has not been extensively employed in the crystallization of GPCRs, reconstitution into lipid/detergent discs or bicelles has been used to generate high-resolution diffracting crystals of several integral membrane proteins and presumably results in a more membrane-like environment than LCPs (Faham and Bowie, 2002). Dimyristoylphosphatidylcholine bicelles were used successfully for the low-resolution structural determination of the antibody Fv-β2-adrenergic receptor complex solved by Rosenbaum et al. (2007) and Bokoch et al. (2010). Another formulation of the LCP/bicelle methodology, namely the lipidic sponge phase, has also been proposed as a crystallization matrix. This approach met with success in determining a 2.2-Å structure for the photosynthetic reaction center of Rhodobacter sphaeroides, offering improvements in resolution and order over a previous structure determined from LCP grown crystals. However, this approach has not yet been employed successfully in GPCR structure determination (Wadsten et al., 2006; Johansson et al., 2009; Wöhri et al., 2009).
3. Expanding Soluble Domains of G Protein-Coupled Receptors Using Nanobodies and Antibody Fv Fragments.
The rationale for this method is that, because integral membrane regions of membrane proteins are inherently hydrophobic, fewer specific hydrophilic interactions exist around which a crystal lattice can be built. By adding antibody fragments, it is possible to increase the probability of these interactions and thus drive crystallization. Successful examples involving these techniques include voltage-gated potassium channels and the SecYEβ membrane antiporter (Zhou et al., 2001; Jiang et al., 2003; Tsukazaki et al., 2008). With GPCRs, this was first exemplified by the β2-adrenergic receptor complexed with Fv fragments of an activating antibody, although structures of limited resolution were obtained (Day et al., 2007; Rasmussen et al., 2007; Bokoch et al., 2010). The utilization of the single chain “nanobodies” derived from Camelid spp. (Tereshko et al., 2008) for stabilization/crystallization was successfully implemented by Rasmussen et al. (2011) to crystallize an agonist-bound T4-lysozyme-β2-adrenergic receptor construct in complex with a nanobody.
D. Recent G Protein-Coupled Receptor Successes
Determining X-ray crystallographic structures of integral membrane proteins historically has been much more difficult than those of soluble proteins. Less than 1% of the unique protein structures currently deposited in the PDB represent integral membrane proteins, and this number decreases to less than 0.1% when only mammalian membrane protein structures are considered. Early attempts to crystallize GPCRs were successful only with natively expressed protein (rhodopsin). More recent studies have relied upon heterologously expressed GPCRs artificially stabilized through protein fusions and/or point mutations and often involving complexation with antagonist/inverse agonist compounds. It is also possible to dispense with the transmembrane regions altogether and focus on the soluble extramembrane domains, which will provide structural guidance for SBDD of certain ligands.
1. Rhodopsin and Related Structures.
Rhodopsin was the first GPCR to have its atomic structure determined (Palczewski et al., 2000) (PDB ID 1F88). Incremental increases in resolution and model completeness (PDB IDs 1L9H, 1GZM/3C9L) eventually yielded the 2.2-Å structure of bovine rhodopsin (Teller et al., 2001; Okada et al., 2002, 2004), which remains the highest resolution structure of any GPCR structure determined to date (PDB ID 1U19). Additional structural work on rhodopsin produced largely superposable structures of the inactive photocycle end-product, opsin, as well as opsin complexed with a peptide derived from the Gα subunit of transducin (Park et al., 2008; Scheerer et al., 2008) (PDB IDs 3CAP and 3DQB). Further work with crystals grown under these conditions revealed that treatment with all-trans-retinal results in a meta II-like state (PDB IDs 3PXO and 3PQR) (Choe et al., 2011). These structures superpose well with the original opsin structures with a RMSD of only ∼0.5 Å, and all exhibit the same outward movement of the ends of H-V and H-VI compared with ground state structures. An additional, constitutively active, heterologously expressed rhodopsin structure solved by Standfuss et al. (2011) also superposes well with the opsin structures (RMSD ∼0.6 Å) and exhibits a similar displacement of H-V and H-VI. The structure of invertebrate (squid) rhodopsin (PDB IDs 2Z73 and 2ZIY) was determined after proteolysis of the receptor to remove a large cytoplasmic domain (Murakami and Kouyama, 2008; Shimamura et al., 2008). By carefully controlling light exposure and crystallization conditions, several photoactivated structures of bovine rhodopsin were determined, including the early photointermediates batho- and lumirhodopsin (Nakamichi and Okada, 2006a, b) (PDB IDs 2HPY and 2G87) as well as a photoactivated rhodopsin that exhibited all the spectral and biochemical characteristics of meta II rhodopsin, the physiologically activated state (Salom et al., 2006) (PDB ID 2I37). Additional work with synthetic retinoids resulted in the structure of 9-cis-rhodopsin (Nakamichi and Okada, 2007) (PDB ID 2PED) (Fig. 11).
Structural coverage along the continuum of states that comprise the activation of rhodopsin. The structural plasticity necessary for transmission of the activation signal from receptor to G protein is evident even in the ground state structures of rhodopsin; although all three groups of structures are grossly similar, each structure captures distinctly different conformers of the opsin backbone although each is spectroscopically identical and each exhibits little to no activity toward G protein. Although slight differences are scattered throughout the structure, the major differences observed are in the third cytoplasmic loop (C-III), the loop connecting H-V and H-VI, the ends of which are proposed to undergo structural rearrangement upon activation. The early photointermediate structures (denoted in pink), which are spectroscopically distinct from both ground state and later photointermediates superpose well with only the PDB ID 1U19 (P41) structures, again with major differences observed only in the C-III loop compared with PDB IDs 2I35/2I36 (P3112) or PDB IDs 3C9L/1GZM (P64) structures. The structure of PDB ID 2I37 (which was the first crystal structure to exhibit the characteristic absorbance at 360 nm indicative of deprotonation of the Schiff base linking the chromophore to Lys296, a hallmark of attaining the activated state) demonstrated only small- to medium-scale shifts in structure that were confined to the C-II and C-III loops, rather than the large-scale rigid body movements proposed by earlier studies. The observed structural changes could best be explained as being due to a loss of constraint within the transmembrane region, resulting in altered protein backbone dynamics. Because ground state crystals in the same unit cell and space group (PDB ID 2I36) were available, the direct comparison with photoactivated rhodopsin (PDB ID 2I37) revealed structural changes that accompanied photoactivation apart from structural changes due to differences in crystallization conditions/unit cell contacts. These crystals were capable of returning to the ground state upon storage in the dark but were incapable of surviving treatment with hydroxylamine to remove chromophore. From the other end of the spectrum, the Ernst group has to great success used the phototransduction cascade end product, opsin, as a structural target and starting point for probing the activation of rhodopsin. The initial crystal structure of opsin (PDB ID 3CAP) and a following structure opsin with a peptide derived from the C terminus of the α subunit of transducin (Gt) (PDB ID 3DQB), although mostly colorless, contain density within the chromophore active site that may correspond to precipitant, buffer, detergent, or hydrolyzed chromophore when composite omit maps are calculated from the deposited data, which was fortuitous as the instability of opsin state in the detergent solubilized state would further complicate crystallization. The PDB ID 3CAP and PDB ID 3DQB structures both exhibited larger movements of H-V and H-VI (and the connecting C-III loop) than observed in the PDB ID 2I37 structure. The structure of opsin in the Gt peptide bound structure was postulated by the authors to be the conformation of opsin in its G protein-interacting state. By treating these very same crystals with all-trans-retinal (PDB IDs 3PXO and 3PQR), the authors were able to obtain crystals that were spectrally indistinguishable from the PDB ID 2I37 crystals. Standfuss et al. (2011) were also able obtain an additional crystal form of photoactivated rhodopsin from heterologously expressed, constitutively active opsin “regenerated” with all-trans-retinal (PDB ID 2X72). All of the opsin derived structures are identical (with the exception of the observed chromophore) within the precision of structures determined at this resolution (RMSD ≈ 0.4 Å). These crystals were incapable of being pushed to the ground state by reconstitution with 11-cis-retinal. Recent solid-state NMR studies suggest the existence of multiple conformational states after photoactivation with only a subset competent to transduce the signal to G protein (Struts et al., 2011). These multiple activated states (highlighted with a blue-gray box in the figure) underlie a fundamental structural disconnect between the two groups of activated state structures. It will only be through direct structural observation of the complex or complexes between rhodopsin and Gt that the precise nature of the interactions between activated rhodopsin and G protein will be observed.
2. Reconciling Activated Structures of (Rhod)opsin with Biochemistry and Biophysics.
Even before the advent of the first activated structures of rhodopsin, it became apparent that the traditional phototransduction cascade, which relies on spectral absorption changes in the retinal chromophore, was not sufficient to completely characterize the conformation of the protein portion of rhodopsin. RMSD between ground state structures is ∼1.0 Å, and structural differences are observed in cytoplasmic loop 3 in all ground state rhodopsin structures determined to date, indicating its conformational flexibility. The phototransduction cascade composed of temperature and chemically trapped photointermediates has been of great utility in the understanding of the activation process of rhodopsin and by extension all GPCRs in general (Matthews et al., 1963; Yoshizawa and Wald, 1964; Thorgeirsson et al., 1993). However, the structural plasticity of even spectrally identical ground state structures suggests that absorption is insufficient to characterize the conformational states of rhodopsin. Trying to “shoehorn” all of the photoactivated rhodopsin structures into these artificially isolated, photointermediate states is perhaps an academic exercise that has little bearing upon our understanding of the activation process. Each determined structure simply provides a structural snapshot along the continuum of conformational states that rhodopsin occupies after photoactivation. There is no requirement that the structure has progressed through any particular intermediate state.
EPR spin-label experiments using heterologously expressed, spin-labeled rhodopsin were interpreted to suggest that major conformational alterations on the order of 15 to 20 Å occur within the transmembrane domain upon transition of rhodopsin from an inactive to an active conformation (Farrens et al., 1996; Hubbell et al., 2003). Such large-scale alterations within the helical bundle have been called into question after isolation of various photointermediate states of rhodopsin that display characteristic absorbance maxima (Matthews et al., 1963; Yoshizawa and Wald, 1964; Thorgeirsson et al., 1993) More recent EPR studies have revised these initial estimates for conformational alterations to ∼6- to 10-Å displacements (Altenbach et al., 2008; Hornak et al., 2010). Furthermore, the chemical differences between the Meta I and Meta II states simply involve the changes in protonation state, and each of these states is in equilibrium. Were large scale movements of entire helices before G protein binding involved, it is thermodynamically unlikely that the equilibrium between the states could be potentiated by a simple protonation/deprotonation event(s). This line of reasoning can be extended, taking into the changes observed in retinal dynamics that occur upon photoactivation and the multiple activated states that this implies (only a subset of which are capable of activating G protein) (Struts et al., 2011).
The activated rhodopsin structures determined to date can be grouped into two broad categories: those derived from photoactivated rhodopsin (PDB ID 2I37) and those determined from the end product of the phototransduction cascade, opsin (PDB IDs 3PXO, 3PQR, 3CAP, 3DQB, and 2X72) (Park et al., 2008; Scheerer et al., 2008; Choe et al., 2011; Standfuss et al., 2011). Although the PDB ID 2I37 structure exhibits small-scale changes in the conformation of cytoplasmic loops and the ends of H-V and H-VI (Salom et al., 2006), the changes in these regions of opsin-derived structures are of a larger magnitude. It is important to consider that all opsin-derived protein structures begin with material at the end product of the phototransduction cascade and neither addition of the Gt peptide nor addition of chromophore has an appreciable change upon the structure (RMSD for the transmembrane region of all opsin structures is ∼ 0.4 Å). When these observations and structures are coupled with the retinal dynamics observed upon photoactivation by solid state NMR (Struts et al., 2011), it becomes clear that spectrophotometric analysis and these structures are not sufficient to fully describe the activation process. Measurements of the displacement of cytoplasmic loop three, H-V and H-VI observed in opsin-derived structures upon activation agree quite well with the later EPR-based estimates, with the caveat that these measurements were performed with heterologously expressed opsin mutants, which were then regenerated. Although the PDB 2I37 crystals could reisomerize their chromophore when removed from light, reforming the ground state, the opsin-derived crystals are incapable of being regenerated with 11-cis-retinal to form the ground state. Furthermore, treatment of the photoactivated (PDB ID 2I37) crystals with hydroxylamine to form opsin resulted in a loss of diffraction similar to the loss of diffraction seen in the opsin crystals treated with 11-cis-retinal chromophore (O. Ernst, personal communication). This hints at a fundamental disconnect between the PDB ID 2I37 photoactivated structure and the opsin-derived structures, because it is not possible to take either class of crystals to the opposite endpoint of the phototransduction cascade. Thus, neither class of crystal structures is sufficient to describe the dynamic and conformational state(s) of the receptor that pass the activation signal from the chromophore pocket to the cytoplasmic face where G protein binding and activation occurs.
Comparisons of agonist-bound nonrhodopsin GPCRs all exhibit some movement of the H-VI and to a lesser extent H-V, but the movement is smaller in magnitude than that observed between ground-state rhodopsin and opsin-derived structures. Were the activation process to rely upon release of internal restraints that lead to a profound change in receptor dynamics, these sorts of variations can be explained. The influences that crystallization conditions, crystal contacts, and thermostabilizing mutations could simply remove energetic barriers present in the wild-type receptor, thus exhibiting a larger degree of structural plasticity needed to fully bind and activate G protein (summarized in Fig. 11).
Comparative analysis of all high-resolution structures of rhodopsin and other GPCRs reveals a subset of crystallographically observed waters found in similar positions within the transmembrane bundle (Angel et al., 2009a). These “homologous” waters and their interactions with highly conserved and functionally important residues such as the D(E)RY and NPxxYx(5,6)F motifs thought to play crucial roles in receptor activation (Mirzadegan et al., 2003) suggests that these waters are functionally important or even essential to the mechanism of activation (Angel et al., 2009a,b; Jardón-Valadez et al., 2009) (Fig. 9). Furthermore, these internal waters do not freely exchange with bulk solvent in ground-state rhodopsin, meta II, or opsin states, further supporting the notion that these waters are noncovalent cofactors integral to the activation process. This however does not imply that the transmembrane region is completely impervious to bulk solvent. Indeed, the Schiff base can be deuterated in unactivated rhodopsin placed in D2O (Deng et al., 1994), and the water used for chromophore hydrolysis is derived from bulk solvent (Jastrzebska et al., 2011), suggesting that only a subset of these solvent molecules are tightly bound. Further work is needed to elucidate the exact role(s) of these ordered waters in the GPCR activation process.
3. Adrenergic Receptor Structures.
In 2007, the report of the X-ray structure for the human β2-adrenengic receptor marked a breakthrough in the structural study of liganded GPCRs. The structure was solved by two different techniques (Rasmussen et al., 2007; Rosenbaum et al., 2007). In one approach, an antibody Fv fragment–β2-adrenergic receptor complex was generated that yielded low-order diffracting crystals with a well-defined Fv entity but a considerably less ordered β2-adrenergic receptor portion (PDB IDs 2R4R and 2R4S). In a second approach, since used to determine the majority of nonrhodopsin GPCR structures to date, a T4 lysozyme fusion was introduced within the third cytoplasmic loop of heterologously expressed β2-adrenergic receptor to aid crystallization in a modified lipid cubic phase matrix and resulted in high-order diffracting crystals (Cherezov et al., 2008; Hanson et al., 2008; Roth et al., 2008). In addition to the high-resolution 2.4-Å carazolol-bound structure (PDB ID 2RH1), a series of adrenergic receptor structures have been solved that contained a variety of antagonist and inverse agonist compounds (Hanson et al., 2008; Roth et al., 2008; Wacker et al., 2010) (PDB IDs 3D4S, 3NYA, 3NY9, and 3NY8). These structures revealed a common binding mode and contact residues for such compounds, suggesting further avenues for structure-based drug design using these structures as restraints.
The crystallization of the turkey β1-adrenergic receptor involved a series of mutations/truncations designed once again to constrain the flexibility of the purified receptor and facilitate the formation of the crystalline lattice. The effects of individual modifications were preliminarily assessed by thermal denaturation analysis and when combined as an ensemble into to a new construct produced diffracting crystals (Tate and Schertler, 2009; Warne et al., 2009). Cocrystallization, as has been true with all nonopsin GPCR templates determined to date, with an antagonist (cyanopindolol, in this case) was needed to obtain crystals, which diffracted to 2.7 Å (Warne et al., 2008). In this case, standard crystallization methodology (i.e., vapor diffusion rather than LCP methods) was employed. Comparison of structures revealed similar binding modes of antagonist to both β1- and β2-adrenergic receptors (Lodowski et al., 2009).
Crystallographic structure determination of both β1 and β2 adrenergic receptors has continued with the solution of several full and partial agonist structures using variations upon the above crystallization schema. Warne et al. (2011) recently published a series of five full and partial agonist-bound structures of thermostabilized turkey β1-adrenergic receptor that exhibit only small-scale structural changes compared with the structure of turkey β1-adrenergic receptor with antagonist bound. These changes are consistent with the scale of structural changes observed in the low-resolution photoactivated rhodopsin structure (Warne et al., 2011). Rasmussen et al. (2011) and Rosenbaum et al. (2011) have also recently solved two agonist bound structures of their T4-lysozyme inserted human β2 adrenergic receptor by two different crystallization schemes but with conflicting results. Although both structures use T4 lysozyme-inserted mutant protein, one structure uses a covalently attached ligand and shows very little change upon agonist binding compared with their antagonist bound counterparts (Rosenbaum et al., 2011). The other uses a complex with a camelid nanobody and exhibits structural changes on the scale of those seen in opsin compared with ground-state rhodopsin (Rasmussen et al., 2011).
4. A2a-Adenosine Receptor Structure.
Again, a T4-lysozyme chimera coupled with the antagonist 4-(2-(7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl)phenol (ZM241385) and use of a cholesterol-saturated LCP crystallization method yielded crystals of the A2a-adenosine receptor refracting to 2.6 Å (Jaakola et al., 2008) (PDB ID 3EML). As expected and in agreement with all other GPCRs determined to date, the binding site for this orthosterically competitive ligand was located within the transmembrane region, grossly overlapping binding sites determined for adrenergic receptor ligands and the rhodopsin chromophore. More recently, an agonist bound structure of a T4-lysozyme A2a-adenosine receptor structure was determined (Xu et al., 2011). Compared with the antagonist bound structure, this agonist-bound structure shows smaller changes in the conformation of H-V and H-VI than seen in opsin-derived structures.
5. C-X-C Chemokine Receptor Type 4 Structures.
The Stevens group determined the structure of the chemokine GPCR, CXCR4, also as a T4 lysozyme chimera in complex with the small molecular antagonist IT1t (6,6-dimethyl-5,6-dihydroimidazo[2,1-b][1,3]thiazol-3-yl)methyl N,N′-dicyclohexylimidothiocarbamate as well as with a cyclic peptide antagonist (CVX15), (Wu et al., 2010) (PDB IDs 3ODU, 3OE9, 3OE6, 3OE8, and 3OEO). This receptor is of great importance to human health, because it is involved in both HIV viral entry and cancer metastasis. It is noteworthy that these structures completely lack the amphipathic helix H-8 present in all other GPCR structures determined to date that functions in receptor activation. All CXCR4 crystal structures determined to date exhibit a crystallographic dimer that is quite similar to that seen in squid rhodopsin (PDB ID 2Z73) as well as in models of rhodopsin oligomerization derived from atomic force microscopy data (PDB ID 1N3M) (Fig. 6).
6. Dopamine D3 Receptor Structure.
The T4-lysozyme fusion technique was also used to obtain a crystal structure of the D3 dopamine receptor in complex with the D2/D3 selective antagonist eticlopride (R-22) (Chien et al., 2010). Because dopamine signaling is involved in cognition and emotion, structures of the dopamine receptor can outline possible structure-based drug design targets for modulation of these activities. Although in many other of the T4 lysozyme fusion constructs, the ionic lock was found to be disrupted, here it was found to be intact, suggesting that this crystal structure more fully recapitulates the inactive state of the receptor.
7. Structures of G Protein-Coupled Receptor Extracellular Domains.
In contrast to GPCRs that have their ligand-binding domains situated within the transmembrane region, many GPCRs contain large extracellular ligand-binding domains. These extracellular domains can bind the entire size range of ligands seen in all classes of GPCRs, from single ions in the case of the Ca2+ receptors to entire proteins in the case of the follicle-stimulating hormone receptor (Figs. 1 and 7). Crystallization of these domains ex situ can provide information regarding ligand recognition. Several subtypes of metabotropic glutamate receptor extracellular ligand-binding domains have been studied (Kunishima et al., 2000; Muto et al., 2007) (PDB IDs 3HSY, 3MQ4, 3LMK, 3FUZ, 2E4U, 1LSR, 1EWK, and at least 15 others) and have defined ligand binding modes and localized structural changes that occur upon both agonist and antagonist binding. All of these glutamate GPCRs are obligate dimers. The distance observed between each monomer fits well with the spacing observed for rhodopsin oligomers within rod outer segment membranes (Fotiadis et al., 2003, 2004) and with dimers observed in rhodopsin, opsin, and CXCR4 structures (Salom et al., 2006; Lodowski et al., 2007; Park et al., 2008; Scheerer et al., 2008; Wu et al., 2010), providing further evidence for the physiological existence of GPCRs as dimers or higher order oligomers (Fig. 6).
Ligand-binding domains from GPCRs that recognize larger soluble protein/peptide hormones have also been determined. These include follicle-stimulating hormone complexed with the follicle-stimulating hormone receptor (Fan and Hendrickson, 2005) (PDB ID 1XWD), thyroid-stimulating hormone receptor in complex with an activating antibody (Sanders et al., 2007) (PDB ID 3GO4), parathyroid hormone receptor complexed with parathyroid hormone (Pioszak et al., 2010) (PDB ID 3L2J), calcitonin gene-related peptide receptor (ter Haar et al., 2010) (PDB IDs 3N7P, 3N7R, and 3N7S), corticotropin-releasing factor receptor 1 (Pioszak et al., 2008) (PDB IDs 3EHS and 3EHT), and frizzled 8 receptor (Dann et al., 2001) (PDB ID 1IJY). What is immediately obvious upon examination of these isolated ligand-binding domains is that although they share little to no structural homology there must be some shared mechanism by which these disparate domains can be linked to the transmembrane portion of the receptor. This raises the concern that because the structures for these domains have been solved without the contextual constraints of the GPCR functional core, usage of such ligand-binding domain structures, although promising from a SBDD standpoint, should be approached with the additional caveat that the modes of ligand binding observed may not fully recapitulate that seen in intact receptor.
E. The Need for More G Protein-Coupled Receptor Structural Work
With many distinct GPCR structures now available to us, there is a good body of information for delineating common features present within the transmembrane region of class A GPCRs that give some insight into their ligand binding and possibly their activation. Further structures will strengthen confidence in such common features but could reveal a subclass of GPCRs with different modes of ligand binding within the TM region. However, certain approaches to structure determination of GPCRs will be particularly helpful for increasing our understanding of how ligands bind GPCRs, how this binding event is coupled to receptor activation, and how these activated receptors consequently catalyze G protein activation. First, a greater diversity of GPCR templates, especially those of class B and C receptors, is needed. Second, continued characterization of agonist-bound structures will help define determinants of endogenous ligand binding and the structural changes that accompany GPCR activation. This information would be particularly valuable in the rationale design of therapeutic ligands and allosteric modulative compounds. Third, and most obviously, structures of agonist-bound GPCRs in complex with their intact appropriate heterotrimeric G proteins provide the best chance to capture the structural changes that enable G protein activation. (Please see Note Added in Proof for recent structural work on GPCR-G protein complexes.)
V. Ramifications for G Protein-Coupled Receptor Drug Discovery
A. Brief History of De Novo Approaches
1. Examples of Success.
Most drugs in our collective medicine chests have been and still are discovered by serendipitous methods (Kubinyi, 1999). Contemporary medicines have emerged from broad screens of large and complex collections of chemical or biological substances that, though individually diverse in origin and content, share en mass an important trait—a low percentage hit rate. Cursory inspection of compound and program attrition common to the pharmaceutical sector underscores the long odds faced during drug discovery. Still, the content and effectiveness of our pharmacopeia is a testament to the use of empirically based screening. No doubt it is foolish to depend solely on serendipity, but we cannot discount it either. Accordingly, our use of random screening will not likely disappear, nor should it, because it continues to be a proven approach for the discovery of novel therapeutics.
Yet for those with a more calculating disposition, the pursuit of new drugs through de novo design presents a compelling logic. Benefiting from advances in biophysics and computational methods, this discipline has steadily advanced over the past 30 years and produced scores of groundbreaking therapeutics. These approaches can be broadly categorized into ligand- and structure-based methods depending upon the origin of the blueprint that guides the actual synthesis of molecules. Ligand-based approaches are based on quantitative structure-activity relationship (QSAR) studies of pre-existing molecules known to specifically interact with a target system of interest. These data are distilled to generate an idealized pharmacophore model that is then used to design new candidates for synthesis. Structure-based methods, on the other hand, rely upon a high-resolution map of a target's putative ligand-binding sites, to provide a mirror-image blueprint for the a priori design (or modification) of entirely novel molecules. Obviously, these two methods are complementary. Both have had their share of success, and both are used throughout the pharmaceutical industry along with computer-aided docking, visualization strategies, and empirically driven assay platforms, often deriving QSAR information from data generated by high-throughput screens.
An early success for the ligand-based approach was the design of highly selective small molecule antagonists against integrin receptors for the treatment of thrombosis, cancer and osteoporosis. This work used conformationally constrained cyclic peptides bearing a similar small binding motif to inform the design of small molecule leads that had remarkably high selectivity for closely related integrin receptor subtypes (Engleman et al., 1996; Samanen, 1996; Samanen et al., 1996; Keenan et al., 1997).
Perhaps the most often cited early example of a successful “rationally designed” drug is the small molecule captopril, an angiotensin-converting enzyme inhibitor for the treatment of hypertension. Its design benefited from a prior decade's QSAR study of the inhibitory action of bradykinin potentiating factor on the conversion of angiotensin as well as from insights gained from the three-dimensional structure of the closely related enzyme carboxypeptidase A (Harrold, 2008). Other early examples of purely structure-based approaches are the carbonic anhydrase inhibitor dorzolamide for the treatment of glaucoma (Greer et al., 1994), the HIV protease inhibitors saquinavir, indinavir, ritonavrir, and nelfinavir (Lu et al., 1995; Kaldor et al., 1997; Vacca and Condra, 1997) and the neuraminidase-inhibiting influenza medications zanamivir and oseltamivir (von Itzstein et al., 1993; Kim et al., 1999). To date, more than 40 drugs have been derived from structural insight into their proper targets (Kuhn et al., 2002).
2. Strengths and Weaknesses.
Despite their successes, de novo approaches, especially those involving structure-based methods, have historically lacked the high throughput and reliability of more traditional screening methods. Because these design-based methods rely on the time-consuming elucidation of new target structures (typically through either X-ray or NMR methods), successes were (and continue to be) hard won. Progress is subject to the difficulties inherent in each step of the structural process, beginning with securing adequate amounts of suitable target protein and proceeding through isolating concentrated samples of pharmacologically relevant target conformations, crystallizing said conformers, achieving high resolution X-ray diffraction or NMR data, and finally solving the structure. For integral membrane proteins, each step has been additionally aggravated by the target's requirement for its supportive membrane environment and the complications that inclusion of such hydrophobic material confers on the isolation and crystallization processes. The pursuit of compounds targeting integral membrane proteins has shown encouraging benefits from significant technical advances in both the front- and back-end stages of high-throughput structural biology platforms.
Virtually comprehensive cDNA target collections, standardized heterologous expression systems for large-scale protein production and high-throughput parallel purification procedures have all facilitated the generation of multiple target constructs and production of their respective proteins as an entree to crystallization trials (Waldo et al., 1999; Stevens, 2000; Lesley, 2001; Gilbert and Albala, 2002). At the other end of the process, multiple-wavelength anomalous dispersion, microfocused synchrotron beamlines, beamline automation, and automated structure solution methods enable data collection and structure determination in a fraction of the time than was required just a few years ago (Guss et al., 1988; Hendrickson et al., 1990; Garman, 1999; Perrakis et al., 1999; Abola et al., 2000; Adams and Grosse-Kunstleve, 2000; Muchmore et al., 2000). Although incorporation of these methodological improvements into the drug discovery process has helped to streamline the efficiency of drug discovery for soluble protein targets, crystallization still remains one of the greatest challenges for integral membrane protein targets.
B. Future Directions for Integral Membrane Proteins in Drug Discovery
1. The Current Industry Climate for G Protein-Coupled Receptor Drug Discovery.
Despite the pharmaceutical sector's increasing investment in technical infrastructure and a steady flow of information about basic biological processes from academia, the rate of drug discovery for all target classes is in steady decline (Fig. 12) (Kola and Landis, 2004). As would be expected, this fact is cause for concern for both potential patients and for those in the drug discovery business, where it is the focus of much analysis and debate. From a larger perspective, it may well be that current and future targets, especially systemic and age-related maladies, are more complex than diseases we have successfully tackled before. The usefulness of intervention paradigms based upon ascribing a given pathology to the action of a single target protein is waning. For example, common therapy for hypertension can involve inhibition of a kidney transporter, a GPCR, and a soluble blood enzyme. Going forward, the involvement and therapeutic control of multiple molecular systems spatially and temporally intertwined in pathophysiological conditions will probably become the norm. Accordingly, the mapping of these complex systems-level relationships will increasingly preoccupy drug discovery scientists as they hypothesize new therapeutic paradigms (Butcher et al., 2004).

Return on investment. Output of new chemical entities (NCE) has declined over a 10-year period despite an increase in overall R&D spending and a steady average development time for those drugs that eventually prove successful. Factors contributing to this problem involve increased governmental regulatory requirements such as the (now) larger patient pools needed to satisfy FDA standards. More fundamental issues are also likely involved, including the increasing complexity of diseases targeted and the associated slowing of target validation as well as the departure from conventional mechanisms of action and the incomplete coverage of chemical space in our current screening libraries.
Even within individual target classes and certainly for GPCRs, the problem of target/activity complexity pervades our considerations. Drug discovery for this superfamily has focused on the generation of compounds that directly orthosterically compete with endogenous substances to either activate or block the action of the receptor. However, this approach is becoming less and less productive as we pursue receptor-ligand systems involving more elaborate recognition and activation relationships, such as peptidergic and glycohormone subtypes involving protein-protein interactions generally considered intractable for small molecular intervention. There is also the problem of building selectivity into compounds that operate within the highly conserved agonist sites of closely related receptor subtypes. There is thus an increasing need to consider nonorthosteric modes of action as the basis for new types of GPCR therapeutics. An a priori understanding of the three-dimensional structure of such sites could both suggest nonobvious modifications for orthosteric leads and reveal allosteric opportunities for the design of entirely novel molecules and mechanisms of action.
2. Requirements for the Optimal Use of Structural Studies.
To provide timely guidance, structure-based drug design must lead the medicinal synthesis curve. That is, it must prospectively suggest new designs rather than limiting itself to retrospectively explaining how old designs work. Understanding how molecules already “in-hand” bind and regulate their targets is valuable, of course, but to achieve its maximum potential, structural information must act as a springboard to suggest innovative compound designs unlikely to be included in a medicinal chemist's usual to-do list. Given the speed and rigor with which medicinal chemistry and screening now operates, this is indeed an ambitious goal.
A key requisite for progress will be the ability to rapidly solve apo-protein crystals and ligand-protein cocrystals, ideally before commitment and expenditure of substantial medicinal chemistry resources. Given the internal competition of projects for medicinal chemistry support, such insights would be welcome and could be exploited to make the synthesis aspects of drug development more efficient. Initially, tactical arguments for targeting specific GPCRs for structural determination are likely to be reminiscent of debates about the value of mounting HTS campaigns for targets with speculative therapeutic validity. Such deliberations eventually eased as efficiencies in enabling and executing HTS improved, the ratio of information return over a smaller experimental investment increased, and the prospect of securing chemical tools to assist in the validation and prioritization of target programs became better accepted. Likewise, one can expect some push back in a proposed mainstream pursuit of GPCR structures until they can be more routinely and easily solved. At that point the expectation that comparative structures should be part of a program, even at an early project stage, will likely become the norm and further encourage a priori generation and selection of drug designs targeting unconventional binding sites and mechanisms of action.
VI. Unresolved Issues and Concluding Remarks
The study of GPCR pharmacology has been the subject of structure-function conjecture since its inception and has long awaited the type of intuitive insights that only high-resolution atomic coordinates can provide. The accelerating success in generating such structures observed over the past few years suggests that we can now begin to discern the molecular basis of GPCR function. Although these breakthroughs have been impressive and the promise of their utility is great, a variety of hurdles remain that must be overcome as we reduce the art of GPCR structure determination to the practice of drug discovery.
A. Caveats about Static Crystal Structures
The increasing availability of GPCRs structures will undoubtedly provide insights into the structural basis of their molecular function. It is important however, to appreciate caveats associated with individual structures, the most obvious of which is that they represent only a “snapshot” of one of many conformations available to the receptor. This issue is especially relevant to 3D coordinates derived from X-ray crystal structures. In such cases, these structures “freeze” at a thermodynamic minimum, which may not fully recapitulate the receptor's native membrane environment but rather reflect restraints imposed by its crystallization matrix. This concern is amplified by the use of modified receptor constructs and/or ligands to stabilize receptors for crystallization. Use of crystal structures derived from specifically stabilized ligand-receptor complexes can predispose the resulting drug designs to those ligand-defined conformations, thus limiting the de novo approach. Furthermore, in the case of ligand-induced stabilization methods and dependent upon the method employed, invoking the use of an a priori ligand molecule suggests that this area of chemical space has been previously probed, further diminishing the novelty of subsequent drug designs. Modified receptor protein further removes the solution from in vivo reality and can increase the risk of guiding design efforts down unfruitful paths. A partial answer to such conundrums can be to structurally solve and compare a variety of discrete conformational solutions to provide a kinescopic view of the protein's dynamics within its full range of function. Such merging and comparison of dynamic states would benefit from other forms of structural information such as provided by solution and solid state NMR and dynamic computational modeling. The problem, of course, is to crystallize such discrete conformations.
B. Partnering with Computational Methods
The accuracy of homology models for GPCRs built from templates that are in turn based upon actual three-dimensional coordinates of a previously solved but different GPCR structure becomes increasingly less predictive as the receptors diverge. This observation is especially applicable to drug design when sub-Ångstrom tolerances and “second shell” contacts provide the basis for selectivity and action. Thus, efforts to improve homology modeling algorithms and merge them with de novo structure assignment methods by, for example, taking into consideration template information, knowledge of fold space derived from structural genomics initiatives, and improved ab initio structure prediction techniques, should ultimately provide us with computer-aided design tools that are both predictive and intuitively effective. Such methods development will be facilitated by additional GPCR structure determinations to provide more points of reference and to validate model based predictions across GPCR subtypes, types, and classes. Applications of static models to in silico screening using compound and fragment-based libraries has become a popular exercise but is limited by current docking algorithms that focus on preconceived-binding domains of the receptor, effectively eliminating allosteric opportunities. Such constraints are largely driven by processor limitations that require a practical shrinkage of the possible binding box. Our need to understand and exploit important functional phenomena such as allosteric modulation and collateral efficacy inherent in the purposeful conformational adaptability of GPCRs seems more suited to the application of molecular dynamic (MD) simulations inclusive of all of the system's protein, lipid bilayer and water atoms. Such MD simulations are also limited by processor resources available and the accuracies of current atomic force fields and MD algorithms. As these factors improve, these tools will naturally merge with extant GPCR structural information to improve drug design that is conformation specific.
C. Rate of Structure Determination
Structure determination of specific GPCR drug targets runs the risk of being retrospective. To be unequivocally guiding, the process of moving from target selection to structure availability must proceed rapidly enough to provide nonobvious drug designs that would have been unlikely to be identified through random screening or QSAR and SAR methods. The rate of generating new designs from structures compared with the pace of discovering new molecules via comprehensive synthetic analog campaigns is thus a common point-counterpoint for all de novo versus randomization debates. Typically, the rationale for obtaining a structure is more compelling when either chemical starting points do not exist, such as when no hits emerge from a HTS or where SAR guided synthesis fails to improve selectivity across related targets, or other off-target phenomena cannot be avoided. Since the initial structures for rhodopsin and β2-adrenergic receptor were reported, the pace of new GPCR structural determinations has rapidly accelerated. If target structures can routinely be solved within 12 months of project inception, this would compare favorably with the lead time it typically takes to enable and execute a HTS study for a given target. This suggests that as the pace of structural determination increases, structurally informed drug design can be merged with screening-based hit identification and QSAR campaigns not only to bootstrap new projects but also to provide guidance for ongoing efforts that have encountered insurmountable chemistry challenges.
D. Structure versus Function
Functional (i.e., screening) approaches to GPCR drug discovery that seek to exploit a textured (i.e., allosteric) approach to pharmacological action will become increasingly powerful provided cell-based assays that are more refined than the current norm are employed. Such progress will also require that ligands (both controls and screening compounds) that modulate these new areas of pharmacological space currently exist in our screening collections. The novelty of such space is encompassed by both the specifics of the target and the specifics of the conformational state that is to be modulated. It seems reasonable to expect that in some if not most cases, such space is poorly populated in our current collections. De novo approaches on the other hand are not beholden to a priori molecular content but rather require insight into whether or not the structure (i.e., conformation) under study is relevant to the disease-defined mechanism of action (i.e., target-effector pathway). Both approaches can be synergistically combined, providing a powerful tool for future GPCR-targeted drug discovery.
E. Cocrystallization with Ancillary Protein(s)
As crucial components of GPCR signaling system and equal partners in the kinetics of their action, ancillary G protein subunit structures, especially the Gα subunit, in complex with their receptor partners will help shed light on structural determinants of GPCR activity. More specifically, the use of G proteins to capture pathway-specific conformations could reveal binding opportunities associated with ligand trafficking in either the receptor's orthosteric domain or elsewhere on more accessible and less conserved regions of the GPCR. Furthermore, structures of protein complexes such as GPCR-G protein, GPCR with receptor activity modifying proteins, GPCR-GPCR dimers can suggest a means of allosteric regulation of GPCR function by interfering with heretofore-untargeted protein-protein interactions. The structures of these GPCR complexes will reveal novel “druggable” sites present for targeting via SBDD.
F. Impact upon Compound Libraries
Clearly, a high resolution view of the internal and surface landscape of GPCRs should assist in the identification and design of novel pharmacophores and as such will constitute an invaluable resource for SBDD. This is especially true for a potentially large number of allosteric designs whose corresponding chemical space is most probably unoccupied in existing libraries. Without such guidance, these sectors of pharmacologically active compound space will only slowly become populated through the creeping expansion of pre-existing, distant, and orthosterically oriented structural neighborhoods. The current trend in generation and utilization of target-focused libraries for drug discovery is typically rationalized by invoking the savings realized in screening a smaller number of compounds with a higher en mass hit rate. This scenario can be true when the focused library is tasked with providing structures that act through mechanisms (and sites) similar to those affected by the populated prototypes. But it cannot account for allosteric/nonorthosteric sites. Realizing that GPCRs constitute the core component of a still poorly understood allosteric system, the design of screening libraries would benefit from as few assumptions as possible when selecting chemical structures for its population. In the case of novel mechanisms of action and allosteric targets, diversity cannot be achieved by reference to prior knowledge. Given the almost infinite number of possible compounds that encompass all of chemical space, the efficient pursuit of diversity would benefit from the reciprocal mapping of novel target binding sites, sites uniquely provided through structural data. Such spatial leads could then be embraced on a manageable scale by focused library approaches to rationally but more efficiently increase performance and diversity.
G. Synergy with Biologics
As regards the generation of very-large-molecule drugs for GPCRs (e.g., therapeutic antibodies), use of GPCR structures becomes more complicated because of the manner in which these therapeutic molecules bind to targets and the in vivo methods employed for antibody generation. Multiple recognition sites spread over a receptor's surface make structural information enlightening but difficult to employ on a purely synthetic basis. Still, one could envision instances where knowledge of disparate epitopes constituting a conformational lock might be employed to construct an artificial mimetic for use as an in vivo immunogen or capture reagent. The fact that such a protein would be stabilized in a particular conformation relevant to the targeted molecular mechanism would be of particular benefit and a significant advance over current methods.
H. Other Synergies
Availability of purified receptor proteins generated as part of the drug discovery process will undoubtedly constitute a windfall for the enabling of other drug discovery tools. It is expected that this protein will also be used for additional assays including NMR and fluorescence based measurements of receptor-ligand dynamics, new generations binding assays via measurement of plasmon resonance and molecular exclusion and various other proteomic/biophysical approaches to characterize and map structure-function aspects of the receptor.
I. Selection of Therapeutically Validated Targets
However promising and inherently powerful a structure-based drug design approach may be, its ultimate value can only be realized if it can operate within the drug discovery pipeline at large. As such, the breadth of application of a structural approach will inevitably face constraints beyond its scope, perhaps the most important of which is the identification and validation of GPCR targets. To these ends, concerted efforts to exhaustively catalog and manage information about normal versus pathological tissue distribution, knock-in/knock-out phenotypes, chromosomal mapping of disease linkages, and generation and correlation of microarray and gene copy information with disease models have only grudgingly given up the secrets embedded in their data, perhaps because of the immensity of the task and the difficulty of formulating therapeutic paradigms from such diverse information. Application of systems analyses to these information layers, although still in its infancy, promises to elucidate complex causal relationships and reveal key regulatory points for therapeutic intervention. Given the emergence of pluridimensional GPCR potency and the prospect that structure-based drug design could be instrumental in designing molecules to selectively regulate one of the many interactions and pathways of a given GPCR, it seems reasonable to expect that a retrospective analysis of past GPCR programs will ensue. Salvage of failed targets and programs might then be possible through consideration of the broader range of molecular phenomena now known to occur within the GPCR signaling paradigm.
In conclusion, the likelihood that the challenges and opportunities discussed in this review will be met and bested seems reasonable, and the convergence of GPCR structural methods with therapeutic exploitation of newly emerging modes of GPCR pharmacology will significantly and favorably affect the field of GPCR drug discovery.
Acknowledgments
This work was supported by National Institutes of Health National Eye Institute [Grant EY019718 (to D.T.L.), EY008061, EY009339 (to K.P.)]; the National Institutes of Health National Institute of General Medical Sciences [Grant GM079191] (to K.P.). K.P. is John H. Hord Professor of Pharmacology. We thank Drs. Leslie T. Webster, Jr., and Michael E. Magurie for extremely helpful comments on the manuscript.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Salon, Lodowski, and Palczewski.
Note Added in Proof
Since the submission of the manuscript for this article, several articles of note regarding structure have been published that shed light on agonist and antagonist binding as well as the activation of GPCRs and mechanisms of G protein binding. A structure of a T4L-histamine H1 receptor fusion bound to the subtype-specific antagonist doxepin [(3Z/E)-3-(dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine] has been determined, exhibiting the subtleties of antagonist specificity (Shimamura et al., 2011). Additional work with thermostabilized A2a-adenosine receptor in complex with the antagonist compounds caffeine and xanthine have further characterized the diversity of the binding site (Doré et al., 2011; Xu and Stevens, 2011). Further work with these thermostabilized A2a-adenosine receptor constructs has produced structures bound to the agonists adenosine and 5′-N-ethylcarboxamido adenosine, revealing the variability of structural changes within the transmembrane region compatible with agonist binding, as well as proposing mechanisms by which the binding of agonist may effect attainment of the activated state or states (Lebon et al., 2011). A 20-Å molecular envelope calculated from single particle analysis of negatively stained electron microscopic images of the complex between native purified rhodopsin and transducin has been isolated, revealing the pentameric structure of the receptor-G protein complex and providing a snapshot of the activation complex (Jastrzebska et al., 2011). A structure of an agonist-bound T4L-β2 adrenergic receptor in complex with a Gs-containing heterotrimer stabilized by a camelid antibody has also been determined that may show some of the structural interactions between GPCR and G protein necessary for recognition and nucleotide exchange (Rassmussen et al., 2011).
Footnotes
All authors contributed equally to this work.
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.003350.
↵1 Abbreviations:
- ADME
- Absorption, distribution, metabolism, and excretion
- AFM
- atomic force microscopy
- CXCR
- C-X-C chemokine receptor
- FDA
- US Food and Drug Administration
- GPCR
- G protein-coupled receptor
- Gt
- photoreceptor G protein (transducin)
- H
- transmembrane helix
- HTS
- high-throughput screening
- IT1t
- 6, 6-dimethyl-5,6-dihydroimidazo[2,1-b][1,3]thiazol-3-yl)methyl N,N′-dicyclohexylimidothiocarbamate
- LCP
- lipidic cubic phase
- MD
- molecular dynamics
- PD
- pharmacodynamics
- PDB
- Protein Data Bank
- PK
- pharmacokinetics
- QSAR
- quantitative structure-activity relationship
- R&D
- research and development
- RMSD
- root-mean-square deviation
- SAR
- structure-activity relationship
- SBDD
- structure-based drug design
- SPR
- surface plasmon resonance
- TM
- transmembrane.
- © 2011 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
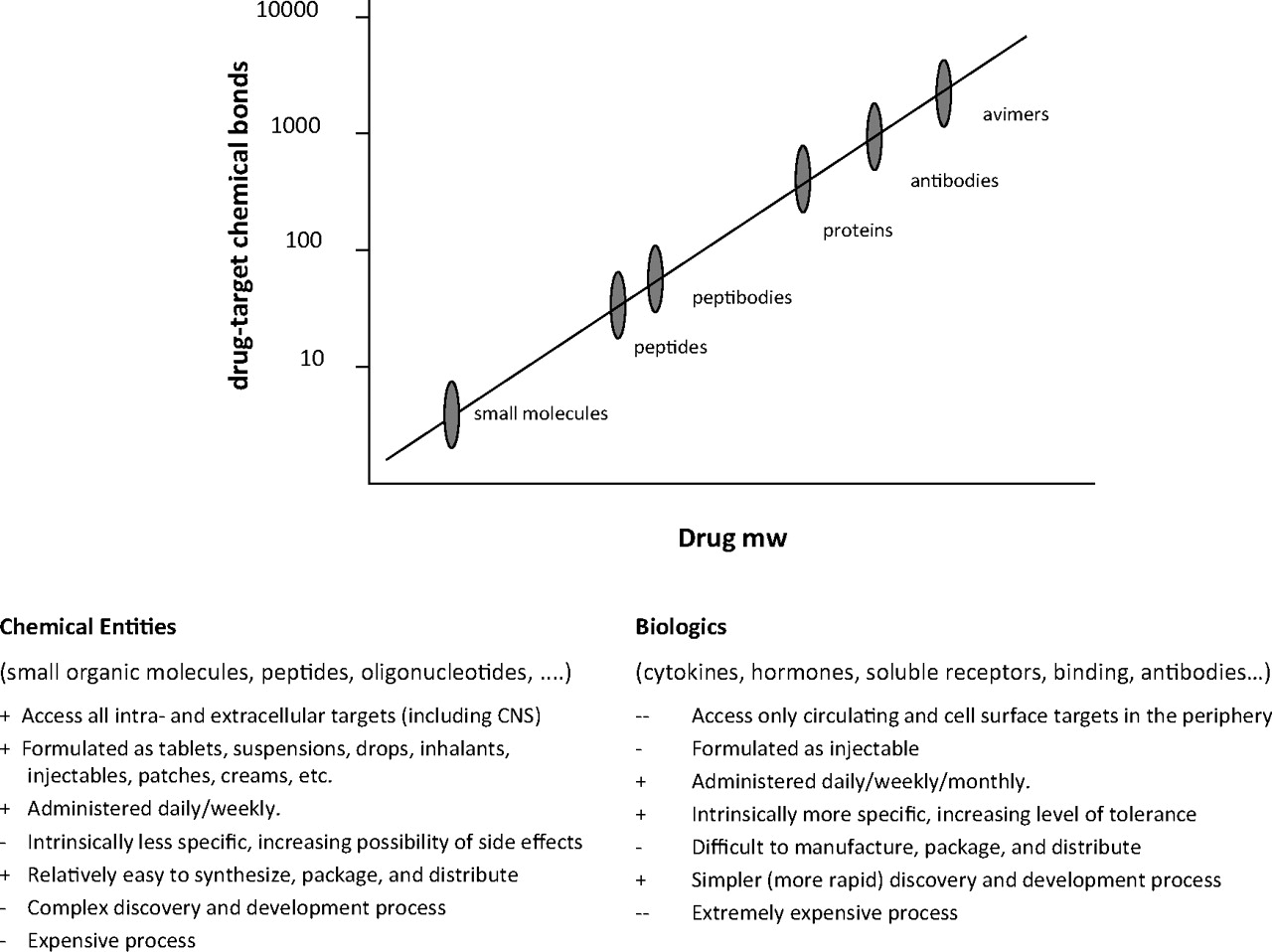{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Current Challenges and Opportunities in G Protein-Coupled Receptor Drug Discovery
- III. History of G Protein-Coupled Receptor Structural Clarification
- IV. Advances in G Protein-Coupled Receptor Structural Determination
- V. Ramifications for G Protein-Coupled Receptor Drug Discovery
- VI. Unresolved Issues and Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Note Added in Proof
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters