


Abstract
Regulation of gene expression represents a major component in antidepressant drug action. The effect of antidepressant treatments on the function of cAMP-responsive element binding protein (CREB), a transcription factor that regulates expression of several genes involved in neuroplasticity, cell survival, and cognition, has been extensively studied. Although there is general agreement that chronic antidepressants stimulate CREB function, conflicting results suggest that different effects may depend on drug type, drug dosage, and different experimental paradigms. CREB function is activated by a vast array of physiological stimuli, conveyed through a number of signaling pathways acting in concert, but thus far the effects of antidepressants on CREB have been analyzed mostly with regard to the cAMP-protein kinase A pathway. A growing body of data shows that other major pathways, such as the calcium/calmodulin-dependent kinase and the mitogen-activated kinase cascades, are involved in activity-dependent regulation of gene expression and may also be implicated in the mechanism of action of antidepressants. In this article the available evidence is reviewed with an attempt to identify the reasons for experimental discrepancies and possible directions for future research. Particularemphasis is given to the regulation of brain-derived neurotrophic factor (BDNF), a CREB-regulated gene, which has been implicated in both the pathophysiology and pharmacology of mood disorders. The array of different results obtained by various groups is analyzed with an eye on recent advancements in the regulation of BDNF transcription, in an attempt to understand better the mechanisms of drug action and dissect molecular requirements for faster and more efficient antidepressant treatment.
I. Introduction: Regulation of Gene Expression and Neuroplasticity
Activity-dependent regulation of gene expression represents a general mechanism whereby neurons and neuronal networks adapt their short- and long-term responses to environmental stimuli. Several families of proteins, called transcription factors, carry out this function by binding to specific domains in the promoter region of various genes and by stimulating mRNA transcription (Kandel, 2001; Lonze and Ginty, 2002; West et al., 2002). A limited number of genes, estimated to be in the range of 15 to 300, show activity-dependent up-regulation in the nervous system, whereas the number of genes down-regulated is much lower, suggesting that gene induction is the favored process for long-term neuronal adaptations (West et al., 2002). A large number of transcription factors that regulate a wide variety of physiological processes have been identified; among them cAMP-response element binding protein (CREB1), a member of a family of transcription factors including CREB, cAMP-responsive element modulator, and activating transcription factor 1, stands out as the most widely expressed and most thoroughly investigated so far. Initially it was found that stimulation of adenylyl cyclase by forskolin leads to phosphorylation of CREB on a specific residue, Ser133 (Seamon et al., 1981); soon it was discovered that the kinase responsible for CREB phosphorylation was protein kinase A (PKA) and that this event represented a key signal for induction of somatostatin transcription (Gonzalez and Montminy, 1989). Subsequently it was found that CREB-dependent transcription is activated when CREB is phosphorylated on Ser133 by a number of protein kinases. Depolarization of neurons or elevation of cAMP strongly stimulates the phosphorylation of CREB at Ser133, whereas the Ser133Ala mutation blocks CREB-dependent transcription (Gonzalez and Montminy, 1989; Sheng et al., 1991).
Activation of CREB was shown to be involved in a large variety of processes in both the developing and mature nervous system, including proliferation of neuronal precursors, outgrowth of neuronal processes, learning and memory in invertebrates and vertebrates, induction of neurotrophic and neuroprotectant cellular programs, and regulation of circadian rhythms (Fig. 1) (Kandel, 2001; Mabuchi et al., 2001; Reppert and Weaver, 2001; Kida et al., 2002; Pittinger et al., 2002). Activation of CREB is induced by a vast array of physiological stimuli, including activation by neurotransmitters of G protein-coupled receptors or ionotropic receptors, growth factors, extrasynaptic glutamate, light, and a variety of stressors (Tan et al., 1996; De Cesare et al., 1999; Mayr and Montminy, 2001). It is intriguing that the same mechanism of transcriptional activation can be induced by so many different physiological stimuli and be used to induce a wide host of cellular and functional responses. The number of genes with CRE sequences in their promoter region is now in excess of 100 and includes different products such as the neurotrophin brain-derived neurotrophic factor (BDNF), the rate-limiting enzyme in catecholamine biosynthesis tyrosine hydroxylase, the antiapoptotic protein Bcl-2, the presynaptic protein synapsin I, the Alzheimer's disease-related protein presenilin, and the circadian clock gene Per1 (Shieh et al., 1998; Tao et al., 1998; Riccio et al., 1999; Mitsuda et al., 2001; Travnickova-Bendova et al., 2002; Conkright et al., 2003; Impey et al., 2004; Zhang et al., 2005). The most likely explanation for the vast array of downstream processes induced by CREB function is a selective activation of different genes in response to co-ordinated action of cell- and stimulus-specific transcription factors and coactivators (see the discussion on BDNF induction in section V.), although the mechanisms whereby specific gene transcription is achieved are not yet clear. Different environmental stimuli may induce different CREB-mediated responses through the activation of different signaling mechanisms converging on CREB. Indeed, it was shown recently that even the same stimulus (calcium elevation) can induce a different response, depending on the route of calcium entry into the cell (Dolmetsch et al., 2001).

Several physiological stimuli, including activation by neurotransmitters, growth factors, extrasynaptic glutamate, light, and a variety of stressors can induce CREB activation. On the other hand, activation of CREB is involved in a large variety of processes, including proliferation of neuronal precursors, outgrowth of neuronal processes, learning and memory in invertebrates and vertebrates, induction of neurotrophic and neuroprotectant cellular programs, and regulation of circadian rhythms.
Because of the growing appreciation for the importance of CREB and CREB-regulated gene expression for cell survival, neuroplasticity, and cognition, this area has become a major focus for the investigation of long-term action of psychotropic drugs. A number of researchers in recent years analyzed the effect of various kinds of antidepressant treatment on expression level and Ser133 phosphorylation of CREB in the brain, on the signaling mechanisms that regulate phosphorylation and transcriptional activity of CREB, and on the activation of expression of CREB-regulated genes, particularly BDNF. These topics and their relevance for both the understanding of antidepressant drug action and the search for novel drug targets will be analyzed in this article. Other recent reviews summarized the work in this field (Coyle and Duman, 2003; Conti and Blendy, 2004).
II. The Action of Antidepressants on cAMP-Responsive Element Binding Protein Expression and Function: Overview of Present Evidence and Hypotheses
In a number of seminal studies, Duman and colleagues investigated the effect of antidepressant treatments on CREB expression and function, which contributed to drawing the attention of other authors to this particular topic. First, these authors (Nibuya et al., 1996) found that chronic but not acute administration of several drugs, representative of different classes of antidepressants, as well as electroconvulsive treatment (ECT), up-regulated the expression of CREB mRNA in hippocampus. They also showed that following repeated administration of fluoxetine (FLX), a selective serotonin reuptake inhibitor (SSRI), or ECT, the amount of CREB bound to CRE regulatory element, as detected by gel-shift analysis, was increased. These results were confirmed by a different group (Frechilla et al., 1998). All of these findings are consistent with the hypothesis that the time-dependent therapeutic effect of these drugs is linked to a modification of gene expression. However, it has been shown that the transcriptional activity of CREB is mainly induced by its phosphorylation (Mayr and Montminy, 2001; Lonze and Ginty, 2002; West et al., 2002). Therefore, in subsequent work Duman and colleagues (Thome et al., 2000) investigated both the CRE-mediated gene transcription and the phosphorylation of CREB. They used transgenic mice carrying a CRE-LacZ reporter gene construct to assay changes in CRE-mediated gene expression, finding region- and drug-selective effects. All drugs increased CRE-mediated gene expression in amygdala; FLX and tranylcypromine (TCP) (an inhibitor of monoamine oxidase) increased it in cerebral cortex and hypothalamus and TCP only in dentate gyrus (DG) and CA3 hippocampal areas. CREB phosphorylation was studied by immunohistochemical analysis following FLX or desipramine (DMI) treatment; only FLX increased phospho-CREB levels in all areas except CA3. DMI was effective only in DG, although at a smaller extent than FLX. These results suggested that in several brain areas, particularly cerebral cortex, FLX was more effective than DMI in inducing CREB phosphorylation and gene transcription. However, in some regions induction of CREB phosphorylation was not accompanied by transcription of reporter gene, a result suggesting that additional cofactors may be necessary in different areas for antidepressant-induced CRE-mediated gene transcription.
Additional evidence for an up-regulation of CREB in antidepressant action came from further work, which showed that viral-mediated overexpression of CREB in hippocampal DG produces an antidepressant-like effect in behavioral tests (Chen et al., 2001a). It was speculated that the antidepressant action of CREB could be due to the induction of BDNF, one of the genes regulated by CREB. Indeed, bilateral infusion of BDNF into the DG also produced an antidepressant effect in the same experimental paradigms (Shirayama et al., 2002). CREB expression was investigated following chronic treatment with FLX or DMI of mice with impaired glucocorticoid receptor function. Whereas in wild-type (wt) mice both drugs increased expression in hippocampus but not in cerebral cortex, in transgenic (tr) mice FLX increased expression in hippocampus, and both drugs did so in cerebral cortex (Blom et al., 2002). These results on the regulation of CREB by antidepressants were complemented by data from postmortem brain analysis, showing that the level of CREB protein was higher in the temporal cortex of depressed patients treated with antidepressants at the time of death than in temporal cortex of untreated patients (Dowlatshahi et al., 1998) and that mRNA and protein CREB levels as well as CRE-binding activity were significantly reduced in brains from suicide subjects (Dwivedi et al., 2003).
On the other hand, additional studies showed an opposite effect, namely a decrease in CREB expression and/or function following antidepressant treatment. Schwaninger et al. (1995) assayed CRE-directed gene transcription following incubation of HIT cells with several representative antidepressants for 1 h. All drugs dose dependently inhibited CRE-directed gene transcription (induced by depolarization) with IC50 values between 70 nM and 1.73 μM; DMI markedly decreased phospho-CREB. These effects were attributed to inhibition of the depolarization-induced increase of intracellular free calcium, a phenomenon described also by other authors (Shimizu et al., 1994), which could reduce the activation of calcium/calmodulin (CaM)-dependent protein kinases (CaM kinases). Indeed, we found that short-term (1 h) incubation of primary neuronal cultures with either FLX or RBX dose dependently blocks the activation of CaM kinase II (Tiraboschi et al., 2004a). This suggests that the observed inhibitory effect of drugs on CREB phosphorylation and function is a short-term effect that in the long run may in turn induce adaptive changes in the opposite direction of CREB- and CRE-mediated gene transcription, as shown above.
Based on the well-known desensitization of central β-adrenergic receptor by chronic tricyclic antidepressants (Vetulani and Sulser, 1975), Manier et al. (2002) proposed that this phenomenon is accompanied by significant deamplification of β-adrenoceptor-coupled cAMP-PKA signaling and asked what the consequences on downstream CREB phosphorylation are. They measured phospho-CREB in the frontal cortex of rats treated with DMI or reboxetine (RBX) and found a slight, but not significant, increase in nuclear phospho-CREB after 10 days of treatment and a significant decrease after 21 days. Furthermore, they assayed CREB phosphorylation induced by isoproterenol in human fibroblasts treated for 48 h (to mimic long-term treatment in animals) with either DMI, RBX, or venlafaxine (a serotonin and noradrenaline reuptake inhibitor) (50 μM). Both DMI and RBX, but not venlafaxine, significantly reduced phospho-CREB level. These results, not in line with the studies above, suggested that pronoradrenergic antidepressants decrease, rather than increase, cAMP-PKA-mediated signaling. Although in this work a selective serotonergic drug was not tested and total CREB levels were not reported (Manier et al., 2002), these data reinforced the message from other studies (see above), drawing attention to possible selectivity of action on CREB and CREB-related pathways of different classes of antidepressants.
Our own work suggested that the diffused up-regulation of CREB mRNA induced by antidepressants does not necessarily transduce into an up-regulation of protein level and that proserotonergic drugs induce CREB phosphorylation more efficiently than pronoradrenergic drugs (Tiraboschi et al., 2004b). In a study on the action of different drugs on the main signaling pathways regulating CREB transcriptional activity (see below) we treated rats chronically with either FLX, DMI, or RBX and measured mRNA/protein expression for CREB and its Ser133 phosphorylation, respectively, by quantitative real time polymerase chain reaction and Western blot, in hippocampus and prefrontal/frontal cortex. Whereas CREB mRNA was increased by all drugs in both brain regions, CREB protein level in cell nucleus was increased slightly only by DMI in hippocampus and more markedly by DMI and RBX in prefrontal/frontal cortex. On the other hand, CREB phosphorylation in both regions was markedly and selectively increased by FLX, a result particularly evident when net phosphorylation was calculated by normalizing the phospho-CREB signal for total CREB protein (Tiraboschi et al., 2004b).
Furthermore, a study using CREB-deficient mice showed that although induction of BDNF was abolished in CREB mutant mice after chronic administration of DMI, the behavioral and endocrine responses to both DMI and FLX were similar in wild type and mutant mice (Conti et al., 2002).
Overall, if one looks at the different results obtained by various studies, the following may be argued:
-
There are conflicting results for CREB expression and function. They could be produced by different experimental paradigms (modality of treatment, drug dosage, different animal strains, etc.).
-
In many cases, chronic antidepressant treatments exert a seemingly stimulatory action on CRE-dependent gene transcription and on CREB expression and/or phosphorylation.
-
CRE-dependent gene transcription, CREB expression, and, even more, CREB phosphorylation show area- and drug-dependent effects. These effects were found mainly in hippocampal cell fields and in cortical areas (particularly frontal cortex). Data from both the Duman group (Nibuya et al., 1996; Thome et al., 2000) and our own (Tiraboschi et al., 2004b) suggest a greater efficacy of SSRIs (FLX) in inducing CREB phosphorylation compared with pronoradrenergic drugs. The data from Manier et al. (2002) might be in line with this axiom. Additional data from treatments with different SSRIs would be required to substantiate this evidence.
However, the main questions are: Why are these effects on CREB drug-selective? And why are there so many different and conflicting results? We propose here that the main reason for this is the complexity of pathways and mechanisms regulating CREB. Thus far the up-regulation of CREB expression and function observed in response to chronic antidepressants was mainly ascribed to an up-regulation of the cAMP-PKA signaling cascade (Duman et al., 1999; Manji et al., 2003). However, as we report below, recent in vitro and ex vivo evidence showed that, different from peripheral tissues, other signaling pathways are primarily involved in the regulation of CREB phosphorylation in brain. Henceforth we will briefly review the present evidence on the up-regulation of the cAMP-PKA cascade by antidepressants. Then, in the following section, we will summarize the present knowledge on main signaling pathways responsible for CREB regulation in the nervous system. In this article we do not examine the action of antidepressants in brain areas different from hippocampus and cerebral cortex, such as nucleus accumbens, in which activation of CREB may have depressive effects (Newton et al., 2002).
A. The cAMP-Protein Kinase A Cascade in the Action of Antidepressants: Available Evidence
As reported in the Introduction, several independent lines of investigation showed that activation of G protein coupled-receptor-stimulated adenylyl cyclase increases levels of cellular cAMP, promotes dissociation of regulatory subunits from PKA holoenzyme, and favors diffusion into the nucleus of catalytic subunits, which phosphorylate and activate CREB (Mayr and Montminy, 2001). Accordingly, when the evidence that antidepressants up-regulate CREB function came along (Nibuya et al., 1996), it was proposed that this was due to an increased activation of the cAMP-PKA cascade, consequent to increased functional output of β-adrenergic receptors and serotonin receptors positively coupled to adenylyl cyclase, even though some of these receptors were actually down-regulated by antidepressants. In the remaining part of this section we will critically review the available evidence on the modulation of cAMP-PKA cascade by antidepressants; it can be anticipated that we found no substantial evidence thus far in favor of a robust activation of nuclear PKA, at least not to an extent implying a primary role for this cascade in antidepressant-induced CREB transcriptional up-regulation. On the other hand, a number of studies in recent years suggested the presence of abnormalities in both CREB and the cAMP-PKA cascade in affective disorders as well as in animal models (Dowlatshahi et al., 1998; Perez et al., 2000; Dwivedi et al., 2003; Kohen et al., 2003). However, even though this evidence made it more likely that the therapeutic effects of antidepressants are achieved by modifying this pathway, independent evidence must be obtained by evaluating the outcome of drug treatments. The main lines of evidence for an up-regulation of the cAMP-PKA cascade by antidepressant treatments are the following:
1. Nuclear Protein Kinase A.
An early study reported that several different antidepressants as well as ECT altered the cellular distribution of PKA in rat frontal cortex, decreasing soluble and increasing particulate activity (Nestler et al., 1989). Fractionation of cellular compartments from brain of rats treated with imipramine (IMI) (a tricyclic antidepressant) revealed that PKA activity, assayed as the level of histone phosphorylation in the presence of cAMP, was increased in the nuclei-enriched fraction (P1) and decreased in the cellular cytosol, suggesting that IMI induces translocation of the catalytic PKA subunit from cytoplasm to the nucleus. This simple experiment was not replicated for several years. Recently we measured basal and cAMP-stimulated PKA activity with an assay using kemptide (a selective peptide substrate) in total extracts and nuclei-enriched fractions (P1), following 2-week treatments with FLX, DMI, or RBX (Tiraboschi et al., 2004b). As reported above, in these conditions FLX up-regulated phospho-CREB more efficiently than pronoradrenergic drugs, particularly in prefrontal/frontal cortex. In total extracts, basal PKA activity was increased by RBX in both hippocampus and prefrontal/frontal cortex and to a lesser extent by FLX and DMI in prefrontal/frontal cortex, whereas cAMP-stimulated activity was unchanged in both areas. In nuclear fractions, basal PKA activity was increased by RBX in hippocampus and much less in prefrontal/frontal cortex; stimulated activity was increased by RBX and DMI in hippocampus but was changed little or not at all in prefrontal/frontal cortex. Overall, nuclear PKA activity seemed to be somewhat increased only in hippocampus and only by pronoradrenergic drugs. Vice versa, in prefrontal/frontal cortex, the up-regulation of the kinase activity observed in the total extract was not found in the nuclei, ruling out a possible translocation of PKA. In summary we did not find evidence for drug-induced PKA translocation to the nucleus (Tiraboschi et al., 2004b). However, it is possible that PKA translocated at earlier times, and at the end of treatment the kinase activity was back in the cytosol. This question should be investigated with time course experiments (for a discussion on these aspects, see Popoli et al., 2001).
2. Protein Kinase A in Microtubules.
One of the more convincing lines of evidence for an up-regulation of the cAMP-PKA cascade by antidepressants came from the work of Perez et al. (2000). In a series of different studies they demonstrated that chronic antidepressant treatments activate PKA in the microtubule compartment, enhancing the binding of cAMP to the RII regulatory subunit of PKA and increasing the phosphorylation of microtubule-associated protein MAP2, a major substrate of PKA (Perez et al., 1989, 1991). Increased disassembly of microtubules could be a functional consequence of these changes (Miyamoto et al., 1997). However, this change in PKA activation is restricted to the microtubule compartment and does not implicate changes of PKA in the neuronal nucleus.
3. G Protein-Adenylyl Cyclase Coupling.
Another convincing line of evidence came from the work of Rasenick and colleagues (Ozawa and Rasenick, 1989; Donati and Rasenick, 2003). In both in vivo and in vitro experiments, they clearly demonstrated that chronic antidepressant treatments facilitate the activation of adenylyl cyclase by increasing the coupling of Gsα with this enzyme. They were able to show that this effect of antidepressants occurs even in the presence of down-regulation of β-receptors (Chen and Rasenick, 1995). It was proposed that the increased coupling with adenylyl cyclase is due to a membrane redistribution of Gs and to an altered interaction with tubulin-based cytoskeleton (Donati and Rasenick, 2003).
4. Protein Kinase A and Dopamine- and cAMP-Regulated Phosphoprotein of Mr 32,000.
Recently Greengard and colleagues (Svenningsson et al., 2002) investigated the effect of FLX on expression and phosphorylation of dopamine- and cAMP-regulated phosphoprotein of Mr 32,000 (DARPP-32), a cytosolic PKA substrate, in mice. They found that acute FLX treatment increased the phosphorylation of DARPP-32 at the PKA consensus site, whereas chronic treatment with the drug increased DARPP-32 mRNA and protein expression but not net phosphorylation of DARPP-32 in hippocampus and frontal cortex.
In summary, there is consistent and robust evidence for an up-regulation of this signaling cascade by antidepressant treatments at the membrane level, with increased coupling of Gsα and adenylyl cyclase and at the level of microtubules, with increased binding of cAMP and activation of PKA. However, there is no consistent evidence for a robust activation of nuclear PKA that may mainly or exclusively account for the up-regulation of CRE-dependent gene expression observed by independent studies. Therefore, if the cAMP-PKA cascade is not the main pathway activating CREB transcriptional activity in response to antidepressants, what are the pathways involved? Furthermore, why is CREB function selectively activated by different drugs?
The results with the cAMP-PKA cascade and the different efficacy of proserotonergic and pronoradrenergic antidepressants in the stimulation of CREB phosphorylation (Thome et al., 2000; Tiraboschi et al., 2004b) suggest a higher complexity for this phenomenon, with variable effects of different drugs, drug dosages, and experimental paradigms on different signaling cascades impinging on CREB (Mayr and Montminy, 2001; Lonze and Ginty, 2002; West et al., 2002). There is now a wide range of characterized signaling cascades that, in response to various kinds of stimuli, regulate CREB phosphorylation and function (Fig. 2). In the following section we will briefly review the field to try to understand the relevance of available evidence for antidepressant drug action.

Schematic representation of signaling pathways modulating CREB activation. CREB activates transcription of target genes in response to a vast array of stimuli, including neurotransmitters, hormones, growth factors, synaptic activity, stressors, and inflammatory cytokines. These stimuli activate a variety of intracellular signaling pathways, leading to activation of a number of protein kinases such as PKA, Ca2+/CaMKIV, and MAPK. Modified from De Cesare et al. (1999), with permission from Elsevier.
III. Signaling Pathways and Protein Kinases Regulating cAMP-Responsive Element Binding Protein Phosphorylation and Function
The phosphorylation of Ser133 is a central event in the activation of CREB and of CRE-dependent gene transcription (Seamon et al., 1981; Gonzalez and Montminy 1989; Sheng et al., 1991; Kandel, 2001; Mayr and Montminy, 2001; Lonze and Ginty, 2002; West et al., 2002). However, it is becoming increasingly clear that recruitment of different kinases, depending on the type of stimulus, as well as of different phosphorylation events on CREB and other transcriptional coactivators may lead to different patterns of gene expression (Deisseroth and Tsien, 2002). Several lines of evidence support the notion that CREB can be phosphorylated by a vast array of protein kinases, which in turn are activated by a variety of signals, as summarized in Fig. 2.
A. Neurotransmitter and Hormone Signaling to cAMP-Responsive Element Binding Protein
Neurotransmitters and hormones regulate CREB phosphorylation mainly by activating G protein-coupled receptors. Binding of ligand to cognate receptor leads to activation of a coupled heterotrimeric G protein, whose activated subunits stimulate adenylyl cyclase and increase the cellular concentration of cAMP. Some isoforms of adenylyl cyclase are calcium-sensitive; therefore, levels of cAMP may also be stimulated by increases in intracellular calcium. Most of the cellular effects of cAMP are conveyed through the stimulation of PKA; binding of cAMP to PKA regulatory subunits allows detachment of catalytic subunits from holoenzyme and phosphorylation of substrate proteins. In the case of CREB, the kinase must translocate to the nucleus and phosphorylate Ser133 on CREB to stimulate transcriptional activity. In PC12 cells, PKA was translocated to the nucleus 15 to 30 min following forskolin stimulation, and similar kinetics was observed for phosphorylation of Ser133 and somatostatin gene transcription (Hagiwara et al., 1992, 1993). Several cAMP-regulated genes are transiently induced; transcription of somatostatin peaks at 30 min following forskolin stimulation and is back at the baseline level after 4 h (Hagiwara et al., 1992). Dephosphorylation of Ser133 follows similar kinetics and was shown to be promoted by at least two different phosphatases, protein phosphatase (PP) 1 and PP2A, respectively, in PC12 and HepG2 cells (Hagiwara et al., 1992; Wadzinski et al., 1993). The efficiency of gene transcription is correlated with duration of Ser133 phosphorylation. Recently the role of other signaling pathways to CREB began to be understood; in particular it was shown that CaM kinases cascades and Ras-mitogen-activated protein kinase (MAPK) cascades may have a primary role in the regulation of neuronal CREB (see below).
B. Synaptic Activity and Calcium Signaling to cAMP-Responsive Element Binding Protein
Synaptic activity within the brain encodes incoming information from the environment. To give rise to durable traces, changes in synaptic activity are transformed in long-lasting structural and functional changes of neurons. These latter changes are mostly due to the synthesis of new gene products. Intracellular calcium signaling has a central role in the transduction of synaptic activity into gene expression changes. Calcium fluxes may be altered by various mechanisms, including activation of voltage-gated calcium channels (VGCCs), activation of ligand-gated ion channels, such as the N-methyl d-aspartate (NMDA) glutamate receptor, activation by neurotrasmitters of G protein-coupled receptors that activate phospholipase C, in turn inducing a rise in intracellular calcium by stimulation of release from internal stores. Early studies demonstrated that depolarization and various neurotransmitters activate the expression of immediate early genes in a calcium-dependent manner (Morgan and Curran, 1986; Sheng et al., 1990; Bading and Greenberg, 1991). A promoter element responsive to calcium (CaRE) was identified in the c-fos gene (Fisch et al., 1987; Sheng et al., 1988). Subsequently, CREB was identified as the CaRE-binding factor (Sassone-Corsi et al., 1988; Sheng et al., 1990), and it was found that, in addition to functioning as a cAMP-inducible transcription factor, CREB also works as a calcium-inducible factor (Dash et al., 1991; Sheng et al., 1991). Several protein kinases can phosphorylate CREB, upon direct or indirect activation by calcium (see Fig. 2). The best characterized CREB kinases activated by calcium are the calcium/CaM-dependent kinases (CaMKs) I, II and IV. Both inhibitors of CaM and of CaM kinase enzymatic activity were shown to block depolarization-induced transcription of c-fos (Morgan and Curran, 1986; Bading et al., 1993).
However, a number of independent studies showed that CaMKIV is the major calcium/CaM-dependent kinase regulating CREB function, even though CaM kinase II is more efficient in phosphorylating CREB. Indeed, it was shown that CaMKIV has a more pronounced nuclear localization compared with other CaM kinases, that its kinetics of activation correlates with that of CREB phosphorylation, that cotransfection of constitutively active CaMKIV drives CRE/CREB-dependent gene expression, and that inhibition of CaMKIV function inhibits depolarization-induced CREB phosphorylation on Ser133 (Enslen et al., 1994; Ghosh et al., 1994; Nakamura et al., 1995; Bito et al., 1996; Ho et al., 2000; Kasahara et al., 2001; Cullen and Lockyer, 2002).
More recently, independent studies suggested a primary role for CaMKIV in activity-dependent neuronal gene expression. In an elegant study Bito et al. (1996) used selective inhibitors for CaMKIV and several other protein kinases to identify the kinase responsible for CREB phosphorylation activated by electrical stimulation of hippocampal neurons. In this experimental paradigm phospho-CREB formation was unaffected by inhibitors of PKA, protein kinase C, cGMP-dependent kinase, MAPK, and tyrosine kinase, but blocked by KN-93 (a selective CaMK inhibitor). Interestingly, they found that both PP1 and PP2B (calcineurin, a calcium/CaM-dependent phosphatase) are involved in the duration of CREB phosphorylation, implying that the same stimulus, carried by calcium/CaM, is responsible for both phosphorylation and dephosphorylation of CREB.
Transcription of BDNF is induced by neuronal activation and was shown to be required for activity-dependent survival of cortical neurons (Ghosh et al., 1994). Shieh et al. (1998) identified a signaling pathway whereby calcium regulates the expression of BDNF. They found that constitutively active mutants of CaMKIV can induce BDNF expression in the absence of extracellular stimuli. Interestingly, they identified two different calcium-responsive elements in the BDNF promoter (named CRS-I and BIII-CRE) and found that the second regulatory element, which depends on the phosphorylation of CREB by CaMKIV, is indispensable for induction of BDNF expression in postnatal but not embryonic neurons. This result suggested that the CaMKIV pathway is necessary for activity-dependent survival of adult neurons.
Kasahara et al. (2001) investigated CREB phosphorylation following induction of LTP, a widely used paradigm of synaptic plasticity, in the CA1 region of hippocampus. They found early activation of CaMKIV and delayed activation of MAPK during induction of LTP, accompanied by a gradual increase in CREB phosphorylation. Both CREB phosphorylation and potentiation of synaptic strength were blocked by inhibitors of calmodulin (calmidazolium) or CaM kinases (KN93). An inhibitor of the MAPK pathway blocked CREB phosphorylation, although it did not block LTP. On the other hand, PKA inhibition did not block either CREB phosphorylation or LTP. It was concluded that the CREB phosphorylation required for hippocampal LTP in CA1 is mainly dependent on the activation of CaMKIV and, to a lesser extent, of MAPK, but independent on the activation of PKA. The results of these studies were substantially confirmed recently by Lee et al. (2005), who made the interesting finding that whereas sustained CREB phosphorylation induced by synaptic activity or a low NMDA concentration (1–5 μM) is due to transient activation of CaMKIV and sustained activation of Erk-MAPK, transient CREB phosphorylation induced by an excitotoxic NMDA concentration (50 μM) is due neither to these kinase cascades nor to any other individual cascade known. They also found that sustained phosphorylation of CREB does not require persistent kinase pathway activity.
Overall, two aspects should not be overlooked. 1) Changes in calcium fluxes may induce CREB phosphorylation by various pathways. First, they may activate the CaM kinase cascade, by stimulating CaM kinase kinase, which activates nuclear CaMKIV, which in turn phosphorylates CREB. Second, they may activate the cAMP-PKA pathway, by stimulating the calcium/CaM-dependent isoforms 1 and 8 of adenylyl cyclase. Third, they may activate the Ras-MAPK pathway by several mechanisms (Cullen and Lockyer, 2002) (Fig. 2). 2) A primary role is emerging for the CaM kinase cascade and CaMKIV in neuronal activity-dependent gene expression as well as in regulation of learning and cognitive functions, as exemplified by the studies above and by the finding that targeted gene disruption of CaMKIV in mouse brings about defective CREB phosphorylation and activation, impaired synaptic plasticity, and impaired fear memory (Ho et al., 2000; Wei et al., 2002). These aspects should be kept in mind when the effect of antidepressant treatments on CREB phosphorylation and activation is considered. Thus far there is only one study investigating the action of antidepressants on CaMKIV (Tiraboschi et al., 2004b).
C. Growth Factor- and Stress-Induced Signaling to cAMP-Responsive Element Binding Protein
Growth factors and neurotrophic factors were shown to regulate gene transcription by inducing phosphorylation of CREB Ser133 (Ginty et al., 1994; Reusch et al., 1994). As shown above for calcium/synaptic activity, phosphorylation of CREB in response to growth factors may also be induced by using different pathways (Fig. 2). The best characterized of these is the Ras/Erk pathway; binding of nerve growth factor to the TrkA receptor, via the Grb2/Shc adaptor complex activates the guanine-exchange factor Sos, leading to subsequent activation of the small GTPase Ras (Patapoutian and Reichardt, 2001). In turn, Ras activates the downstream kinases mitogen-activated protein kinase kinase and Erk1/2; activated Erk stimulates the final effectors that directly phosphorylate CREB in the nucleus. So far two families of kinases that carry out this task have been identified; one of these is Rsk1/2/3. Although Rsk2 was identified as the main kinase responsible for CREB phosphorylation in response to nerve growth factor stimulation, all three members of the family were shown to activate CREB (Xing et al., 1996; De Cesare et al., 1998). Two other structurally related kinases, Msk1 and Msk2, can also be activated by Erk and phosphorylate CREB (Deak et al., 1998; Arthur and Cohen, 2000). In addition to the Ras-Erk pathway, tyrosine kinase-coupled receptors may activate a second pathway, the phosphoinositide 3-kinase/Akt pathway (Fig. 2). This pathway was recently implicated in the control of CREB phosphorylation in neurons and in synaptic plasticity (Lin et al., 2001).
CREB phosphorylation can also be induced in response to stressful stimuli, such as UV irradiation, heat shock, and hypoxia, or to inflammatory cytokines such as tumor necrosis factor. The signaling routes may involve alternative pathways such as p38 MAPK, which activates the downstream target MAPKAP-K2, but may also involve molecular effectors in common with growth factor signaling, i.e., Msk1/2 (Fig. 2). Few or no data are available on the action of antidepressants on these signaling cascades, whereas the effects of mood stabilizers have been more extensively studied (see below).
IV. The Complexity of cAMP-Responsive Element Binding Protein-Regulating Signaling and the Action of Antidepressants
As reported in the previous section, CREB function may be regulated by many convergent and cross-talking pathways, and each of them may potentially be involved in the action of antidepressants (Fig. 2). Independent lines of evidence have shown that in neurons activity-dependent phosphorylation of CREB at Ser133 is induced by sequential activation of the CaM kinase cascade and the Ras-MAPK cascade (Bito et al., 1996; Kasahara et al., 2001). In the first few minutes following stimulation, CaMKIV activation is crucial for rapid phosphorylation of CREB; however, subsequent activation of the Ras-MAPK cascade in the following several minutes is necessary for induction of sustained CREB phosphorylation (West et al., 2002). This chain of events was found to be induced by synaptic activity and consequent changes in calcium fluxes carried out through VGCCs and NMDA receptors. It would be interesting to investigate the involvement of these kinases in the action of antidepressants on CREB function, even though care must be exercised in the evaluation of long-term drug effects on these mechanisms, which could be quite different from those of acute neuronal stimulation. A study performed with cultured astrocytes found a rapid activation of Erk1/2 and of Erk-dependent gene expression by fluoxetine (Mercier et al., 2004). By contrast, the effects of mood stabilizer substances (lithium and valproate) on this kinase cascade are better documented. Chronic treatment with either lithium or valproate increased activation of Erk1/2 and phosphorylation of CREB in rat hippocampus and frontal cortex (Einat et al., 2003). Inhibiting the Erk pathway with a blood-brain barrier-penetrating inhibitor decreased immobility time and increased swimming time of rats in the forced-swim test (a behavioral test for antidepressant activity). The behavioral changes were prevented with chronic lithium pretreatment. Similarly, valproate increased Erk1/2 activation in cultured cortical neurons (Hao et al., 2004). Lithium was also shown to antagonize the glutamate-induced decrease of phospho-CREB in granular neurons by increasing the activity of mitogen-activated protein kinase kinase (the Erk phosphorylating kinase) and reducing protein phosphatase 1 activity (Kopnisky et al., 2003).
In the studies reported above (Tiraboschi et al., 2004b), we found that all antidepressant drugs used for treatment (FLX, DMI, and RBX), independently of their primary mechanism of action, increased the enzymatic activity of nuclear CaMKIV (Fig. 3), due to increased phosphorylation on Thr196 by CaM kinase kinase (a main mechanism of activation for CaMKIV). This was found in the same area (prefrontal/frontal cortex) in which FLX, but not pronoradrenergic drugs, markedly increased CREB phosphorylation in the same animals. Still in the same area, we found that FLX, but not pronoradrenergic drugs, increased the protein expression level of Erk1/2. At the same time a reduction of Erk1 phosphorylation (activation) was observed (in both prefrontal/frontal cortex and hippocampus), suggesting a complex and multifaceted regulation of Erk by antidepressants. It can be speculated that the differential action of these drugs on CREB depends on a balance between the activation of CaMK and MAPK cascades.
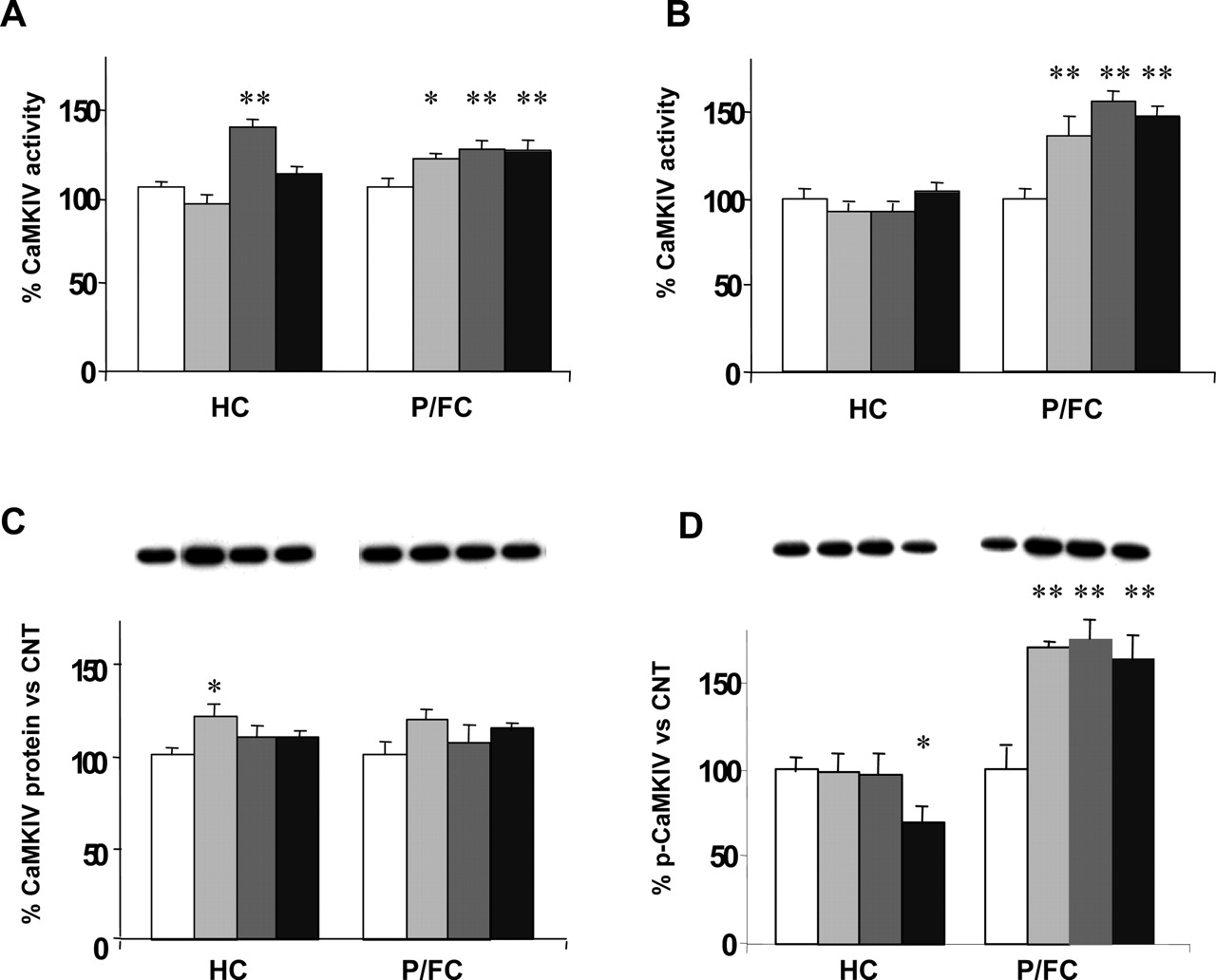
Chronic antidepressants increase nuclear CaMKIV activity in P/FC. A, CaMKIV enzymatic activity in total homogenates. Enzymatic activity was measured by assaying phosphate incorporation in peptide substrate following immunoprecipitation of CaMKIV. Only DMI increased the kinase activity in hippocampus (HC), whereas a slight but significant increase was observed with all drugs in prefrontal/frontal cortex (P/FC). B, CaMKIV enzymatic activity in nuclear fractions. No changes were detected in HC, but all drugs tested induced a marked increase of CaMKIV activity in P/FC. C, total CaMKIV protein levels in nuclear fractions. A slight increase, statistically significant only in HC, was observed after chronic treatment with FLX. In P/FC, although there was a trend toward an increase of CaMKIV protein levels, no significant changes were detected. Representative immunoreactive bands are shown. D, Thr196 phosphorylation of CaMKIV in nuclear fractions. A marked increase after treatment will all drugs was observed in P/FC; p-CaMKIV significantly decreased with RBX in HC. Data are shown as percent mean ± S.E.M. of control. *, p < 0.05; **, p < 0.01. Representative immunoreactive bands are shown. Modified from Tiraboschi et al. (2004b), with permission from Nature Publishing Group.
Even though these results represented the first evidence for the involvement of these two kinase cascades in the chronic action of antidepressants, they do not explain: 1) why FLX seems to be more efficacious in inducing CREB phosphorylation; 2) how CaMKIV, which is normally just transiently activated, could be persistently activated following 2 weeks of treatment; and 3) what the downstream effectors of Erk responsible for CREB phosphorylation are. It has been speculated that sustained phosphorylation of neuronal CREB is due to cooperative action of different pathways occurring in concert (West et al., 2001), and this may well be the case for the chronic action of antidepressants. Furthermore, it was proposed that different stimulatory inputs, conveyed by different signaling cascades, may activate different subsets of CREB target genes (Lonze and Ginty, 2002); differences in the routes to CREB phosphorylation and in their combination may account for different readouts. As an example, Hardingham et al. (1999) found distinct kinetics of CREB phosphorylation, depending on calcium influx through either L-type VGCCs or NMDA receptors. We speculate that these mechanisms could explain the conflicting results obtained for treatment with the same drugs (different dosages, routes of administration, or experimental conditions) and for drug-selective effects on CREB (e.g., proserotonergic versus pronoradrenergic drugs). Future studies to unravel the complexity of signaling pathways activated by antidepressant treatments will undoubtedly contribute to our understanding of the multifaceted action of these drugs on CRE-dependent gene expression.
One last element of complexity that should be considered is the phosphorylation of additional residues on CREB. The prevailing view is that, after cellular stimulation, the activated pathways transmit the signal to the nucleus phosphorylating CREB on Ser133. This event leads to recruitment of the transcriptional coactivator CREB-binding protein to CREB, thus promoting the transcriptional activation. However, it has been reported that phosphorylation of CREB at Ser133 is not always sufficient to activate CREB-dependent transcription. CREB can be phosphorylated also on two other residues, Ser142 and Ser143, and recent studies suggest that phosphorylation of Ser142 is required for the activation of CREB in response to membrane depolarization and for in vivo regulation of circadian clock (West et al., 2001; Gau et al., 2002). In light of the evidence showing that phosphorylation of Ser142 or Ser143 blocks the interaction of CREB-binding protein with phosphorylated Ser133 (Parker et al., 1998), it has been proposed that CBP and CREB could interact in other regions. Another possibility is that phosphorylation of Ser142/143 may lead to recruitment of alternative coactivators, thus inducing the formation of different protein complexes with CREB (West et al., 2001). It has been shown that phosphorylation of Ser142 or Ser143 is induced only by stimuli that lead to calcium influx, such as membrane depolarization or glutamate stimulations (Kornhauser et al., 2002). Therefore, it is possible to speculate that phosphorylation of CREB on Ser142 or Ser143 induces the formation of an alternative CREB complex that can transcribe in response to different stimuli, thus finely modulating gene expression.
V. cAMP-Responsive Element Binding Protein Target Genes: The Example of Brain-Derived Neurotrophic Factor
A. Evidence of Antidepressant Action on Brain-Derived Neurotrophic Factor
Among the genes regulated by CREB, the one whose expression has been investigated more following antidepressant treatments is by far BDNF, the most abundant neurotrophin in the brain. In the last several years increasing preclinical and clinical evidence suggested involvement of BDNF in both the pathophysiology of affective disorders and the mechanism of action of antidepressant drugs (Duman et al., 1999; Chen et al., 2001b; Manji et al., 2001, 2003; Neves-Pereira et al., 2002; Sklar et al., 2002; Egan et al., 2003).
One of the first lines of evidence suggesting involvement of BDNF in the treatment of depression was the finding that local infusion of BDNF into the midbrain led to an augmentation of serotonergic activity within the brain (Siuciak et al., 1994). Subsequently, it was reported that local infusion of BDNF in the midbrain has antidepressant effects in two behavioral models of depression, the forced-swim and learned-helplessness paradigms, thus suggesting that increased expression of endogenous BDNF may have antidepressant effects (Siuciak et al., 1997).
These data, together with the first evidence for involvement of BDNF in the pathophysiology of stress-related mood disorders (Smith et al., 1995), prompted other authors to test the hypothesis that antidepressant treatments regulate the expression of BDNF and its receptor TrkB. Nibuya et al. (1995) demonstrated for the first time that chronic ECT as well as antidepressant drugs, representative of different classes (tricyclic antidepressants, SSRIs, monoamine oxidase inhibitors, etc.) enhance the induction and prolong the expression of BDNF and TrkB mRNA in treated animals. In particular, both acute and chronic ECT (although at different extents in the various brain regions) increased the induction and prolonged the duration of BDNF and TrkB mRNA. No significant change was observed after acute antidepressant administration, and only chronic (21 days) antidepressant treatments modified BDNF and TrkB. In frontal cortex only TCP, but not the other drugs, significantly increased BDNF mRNA. Conversely, in the hippocampus all of the tested antidepressants (TCP, sertraline, DMI, and mianserin), although at a different extent, significantly increased BDNF mRNA, and all but mianserin (an atypical antidepressant) increased TrkB mRNA (Nibuya et al., 1995). Furthermore, chronic, but not acute, administration of ECT or antidepressant drugs completely blocked the down-regulation of BDNF mRNA in the hippocampus in response to restraint stress. A confirmation of these data came from a subsequent study, showing that chronic treatment with FLX or TCP increased BDNF and TrkB mRNA expression in hippocampus, but not in the piriform cortex (Nibuya et al., 1996).
In contrast, it was later reported that acute, short-term, or chronic administration of either a norepinephrine reuptake inhibitor (DMI or Org 4428) or rolipram, a phosphodiesterase inhibitor, alone did not influence BDNF mRNA levels in hippocampus. A significant increase in BDNF mRNA levels was detected only after coadministration of a phosphodiesterase inhibitor and DMI or Org 4428 (Fujimaki et al., 2000). As summarized in Table 1, on the basis of these findings, several groups investigated the effects of different antidepressant treatments on BDNF expression, leading to somewhat conflicting results and thus suggesting a more complex picture than expected from the early studies.
Effects of antidepressant treatments on BDNF expression
In line with the results obtained by Nibuya et al. (1995, 1996), it was reported that prolonged treatment with TCP increased BDNF mRNA in hippocampus, whereas treatment with IMI did not modify BDNF mRNA levels (Russo-Neustadt et al., 1999). Interestingly, it was also shown that physical exercise significantly increased BDNF expression level and that its combination with antidepressants led to a potentiation of BDNF mRNA expression in the hippocampus, also in conditions in which antidepressant alone (IMI) appeared to have no significant effects (Russo-Neustadt et al., 1999). The same group subsequently assessed the level of specific BDNF mRNA transcripts (containing exon I and II) and their regulation, at several time points, by physical exercise and TCP, both alone and combined (Russo-Neustadt et al., 2000). The transcript-containing exon I was increased after both exercise and antidepressant administration and was enhanced to a greater degree by long-term interventions. On the other hand, the BDNF transcript-containing exon II appeared to be up-regulated after a short time, particularly by exercise. The combination of the two interventions led to a progressive potentiation and acceleration of BDNF mRNA levels, thus suggesting an apparent cooperative regulation of BDNF transcription by the two interventions and implying convergent molecular mechanisms.
Results conflicting with those mentioned above have recently been reported. Miró et al. (2002) found a significant reduction of BDNF transcripts in several brain structures after acute treatment with FLX, along with an increase in two cortical areas (frontoparietal and anterior cingulate cortex). After a 14-day treatment, BDNF mRNA levels were higher in the pontine nuclei but decreased in all other brain structures analyzed. Xu et al. (2003) investigated the effects of two different dosages (5 and 10 mg/kg) of amitriptyline (a tricyclic antidepressant) or venlafaxine on the protein expression level of BDNF. Consistent with earlier reports on mRNA levels, an increase of BDNF protein levels following chronic administration of both drugs at the lower dose was found in the hippocampal pyramidal cell layer but, in contrast with previous data, not in the dentate granule cell layer. Intriguingly, no increase in BDNF was detected after the higher dose of amitriptyline and a reduction after venlafaxine, suggesting that the direction of BDNF change may be related to the dose of drug administered (Xu et al., 2003).
A recent study was designed to clarify the influence of postdrug interval on the effect of acute and repeated treatment with antidepressants on BDNF gene expression (Coppell et al., 2003). The main finding was that antidepressants inhibiting either serotonin reuptake (FLX, paroxetine, or sertraline) or monoamine oxidase (TCP) but not those acting on norepinephrine reuptake (DMI and maprotiline) or mianserin affect BDNF gene expression in the rat hippocampus in a biphasic manner following repeated drug injections. In particular, BDNF mRNA was down-regulated 4 h after the last injection but up-regulated at 24 h. A decrease of BDNF mRNA was also seen 4 h after acute administration (Coppell et al., 2003). To get more insight into the onset of BDNF mRNA response to antidepressants and the concomitant effects on corresponding BDNF protein, the same group investigated the effects of FLX by measuring BDNF mRNA and protein at 24 h after acute administration or after 4, 7, 14, and 21 days of treatment (De Foubert et al., 2004). A single FLX administration had no effect on BDNF mRNA in any of the brain regions analyzed. Four days of once-daily oral FLX administration decreased BDNF mRNA in hippocampus, medial habenular, and paraventricular thalamic nuclei. Whereas 7 days of treatment nonsignificantly increased BDNF mRNA, a marked and region-specific increment was detected following 14 days of FLX. Moreover, regional and total BDNF protein levels were measured in hippocampus by immunohistochemistry and ELISA, respectively. Although increased, BDNF protein levels in hippocampal areas were not significantly higher until 21 days of treatment, when the numbers of BDNF-immunoreactive cells in the CA2 and CA3 regions of hippocampus were significantly increased. However, the ELISA method, which perhaps is not sensitive enough, failed to show any significant change in the total hippocampal BDNF protein level. Overall, these results suggested that a significant increase of BDNF mRNA at 14 days is followed by a significant increase of BDNF protein 1 week later in selected hippocampal areas. Although the authors did not address this seemingly long lag time, this result could be explained by two considerations. First, after 14 days of FLX treatment there was a trend for an increase in BDNF-immunoreactive cells, with values nearly identical to those found 1 week later. Second, it is possible that BDNF protein increased gradually during the 3rd week of treatment, but the measurement of BDNF at the 14- and 21-day time points did not allow detection of in-between changes in protein expression. The BDNF gene seems to be expressed differently, depending on the length of FLX treatment, thus contributing to the slow onset of antidepressant action observed in depressed patients (De Foubert et al., 2004). In another recent study, Dias et al. (2003) investigated whether different antidepressant treatments (ECT, TCP, DMI, or FLX) recruit single or multiple BDNF promoters to regulate mRNAs. The results showed that both acute and chronic ECT acts mainly on exon I- and II-containing BDNF mRNAs in hippocampal and cortical subfields. Acute treatment with antidepressants led to region-specific decreases in distinct exon-specific BDNF transcripts. In contrast, chronic administration of TCP and DMI enhanced exon II and exon III mRNA, respectively, in discrete hippocampal and cortical subfields. Chronic FLX treatment did not have significant effects on the exon-specific BDNF transcripts. These results show that distinct antidepressants differentially regulate BDNF mRNAs through a region-specific recruitment of the four BDNF promoters, thus suggesting that different signaling mechanisms may be responsible for the regulation of BDNF transcription (Dias et al., 2003).
Other researchers investigated the effect of antidepressant treatments on BDNF mRNA and protein. In one study BDNF protein levels were measured by ELISA in several brain areas after acute and chronic treatment (4 days and 2–3 weeks) with ECT and antidepressant drugs (TCP, FLX, and DMI) (Altar et al., 2003). ECT produced rapid, large, and widespread increases in BDNF protein in all cerebral regions studied. Much smaller and more delayed increases in BDNF were produced by the monoamine oxidase inhibitor TCP in frontal cortex and neostriatum but not in hippocampus. None of the other drugs tested significantly modified BDNF protein levels. BDNF mRNA levels were also measured in a transgenic animal model of depression characterized by impaired type II glucocorticoid receptor function after prolonged treatment with FLX and DMI (Vinet et al., 2004). In wt mice neither FLX nor DMI modified BDNF expression. A significant increase in BDNF mRNA was observed in treated tg animals; indeed, both FLX and DMI up-regulated BDNF expression in DG, and FLX also significantly increased BDNF mRNA in the CA3 region (Vinet et al., 2004). Another recent study by Jacobsen and Mork (2004) investigated the effect of escitalopram (ESC), DMI, and ECT on BDNF mRNA and protein expression in rat brain. No significant changes were observed in frontal cortex and CA3 after the different treatments. Only ECT and DMI significantly increased BDNF mRNA in DG, whereas ESC did not modify BDNF expression. Regarding BDNF protein levels, ECT increased BDNF protein levels, ESC significantly reduced BDNF in both frontal cortex and hippocampus, and DMI was devoid of effect. Overall, this work did not support the notion that increased BDNF expression is a common feature of antidepressant treatments and suggested that mRNA expression was not indicative of BDNF protein levels (Jacobsen and Mork, 2004).
The effects of FLX on BDNF and TrkB were recently investigated in cultured astrocytes (Mercier et al., 2004). The authors, examining the time course of BDNF mRNA after addition of 40 μM FLX to cultured cells, reported an increase in BDNF and TrkB mRNA detectable within 2 h of FLX treatment and a maximum after 6 to 8 h. In a recent study by Itoh et al. (2004), the effects of IMI in combination with rolipram on BDNF protein level were assessed in learned helplessness (LH) rats. LH rats have significantly lower levels of BDNF than control rats in both frontal cortex and hippocampus; when animals were treated with one of the two drugs alone a trend toward an increase was observed, but a significant increase in the BDNF level was detected only after chronic treatment with the IMI-rolipram combination. The co-administration was also more effective in reducing the depressive behavior of LH rats than either drug alone.
In the same study showing selective stimulation of CREB phosphorylation by FLX (Tiraboschi et al., 2004b), we found a marked reduction of BDNF mRNA level (unpublished data) in hippocampus and prefrontal/frontal cortex of rats chronically treated with RBX or DMI, with no change after FLX administration (all drugs, 10 mg/kg per day, administered 14 days with osmotic minipumps). Activation of BDNF signaling in mouse brain by antidepressants was also investigated by measuring TrkB phosphorylation (Saarelainen et al., 2003). Acute as well as chronic antidepressant treatments (IMI and FLX) induced autophosphorylation and activation of TrkB in cerebral cortex and hippocampus, without modifying its protein level. In addition, recently Monteggia et al. (2004) investigated the effects of subchronic DMI treatment in mice with a conditional BDNF knockout (KO). The study showed that whereas saline-treated control and BDNF KO mice exhibited a similar behavior in the forced-swim test, DMI treatment significantly reduced immobility time in control mice but not in BDNF KO mice, thus suggesting that BDNF may be essential for mediating some aspects of antidepressant efficacy.
What conclusion can be drawn with so many different and sometimes conflicting results? The only treatment that consistently increased BDNF mRNA and protein levels was ECT. As an example, let us summarize the results obtained with two representative drugs most used in these studies (and for clinical therapy), FLX and DMI (Table 1). Three FLX studies showed increased expression of BDNF mRNA in hippocampus or selected hippocampal areas after chronic treatment (Nibuya et al., 1996; Coppell et al., 2003; De Foubert et al., 2004). One showed elevated BDNF mRNA in tg but not in wt mice (Vinet et al., 2004). Two studies showed no changes in BDNF exon-specific transcripts or protein, respectively (Altar et al., 2003; Dias et al., 2003). One study showed decreased BDNF mRNA in hippocampus (Miró et al., 2002). With respect to DMI, two studies showed increased BDNF mRNA expression (Nibuya et al., 1995; Dias et al., 2003), one only in tg but not in wt mice (Vinet et al., 2004). Three studies showed no changes in either mRNA (Fujimaki et al., 2000; Coppell et al., 2003) or in protein (Altar et al., 2003). Jacobsen and Mork (2004) found increased mRNA only in the DG, and no change in protein in any area examined. Although in a large number of studies these drugs up-regulated BDNF expression, there is a roughly equivalent number of studies in which this result was not replicated. The situation with other drugs is not dissimilar (Table 1). Furthermore, the few studies in which BDNF protein was also measured showed that mRNA expression is not indicative of BDNF protein levels. This finding suggests that a number of experimental factors, which may vary among different studies, account for these discrepancies. Some of these factors could be the following:
-
Type of stimulus (drug versus physical exercise; drug versus ECT) (e.g., Russo-Neustadt et al., 1999, 2000; Altar et al., 2003)
-
Type of drug and drug dosage (e.g., Russo-Neustadt et al., 1999; Fujimaki et al., 2000; Coppell et al., 2003; Dias et al., 2003; Xu et al., 2003; De Foubert et al., 2004; Jacobsen and Mork, 2004)
-
Route of administration (e.g., Miró et al., 2002; Tiraboschi et al., 2004b)
-
Time window of BDNF measurement with regard to treatment duration (e.g., Coppell et al., 2003; De Foubert et al., 2004).
However, part of the reason for this could be inherent to the mechanism itself whereby the production of BDNF is regulated (see section V.B.). The few studies carried out thus far, investigating the action of different drugs and environmental stimuli on the transcription of specific BDNF exons, showed that different drugs or drug/physical exercise combination may selectively influence transcription of distinct exons (Russo-Neustadt et al., 2000, 2004; Dias et al., 2003). In the following section we discuss briefly some features of the regulation of BDNF promoter III (best known so far among the different promoters) to exemplify how the understanding of specific regulatory elements may help to detect the relevant signaling pathways involved and their activation by different drugs or environmental stimuli. A thorough dissection of these mechanisms will be crucial for understanding how antidepressant therapeutic action is conveyed through BDNF signaling, and what optimal features a treatment should present to be able to exert neurotrophic action. It should also be added that, although some studies indirectly suggest that antidepressants actually increase the release of BDNF (Aloyz et al., 1999; Saarelainen et al., 2003), an independent demonstration of this effect is still missing.
B. Regulation of Brain-Derived Neurotrophic Factor Expression: An Additional Layer of Complexity
In this last section we will briefly analyze some regulatory features in the transcription of BDNF gene, in an attempt to understand whether these features may in some way account for the different experimental results and help to identify tentative landmarks for optimization of treatment. BDNF is a small secreted protein which, upon binding to TrkB cognate receptor, promotes the activation of signaling pathways that both rapidly modify local synaptic targets and also have long-term effects on gene transcription (West et al., 2001). Consistent with a role of BDNF as a mediator of neuronal adaptive changes, the expression of BDNF is extremely responsive to electrical activity in the brain (Tao et al., 1998). For a review of the role of BDNF in synaptic plasticity in adult brain see Poo (2001).
The BDNF gene has a complex organization with four initial exons that can be spliced to a single 3′ exon containing the coding domain for the BDNF protein (Timmusk et al., 1993). Each of the four splice variants can use one of two alternative polyadenylation sites within the 3′ untranslated region, generating a total of eight distinct transcripts, all of which encode the same protein. This gene structure is referred to the rat gene, as described by Timmusk et al. (1993); however, more recently eight exons have been characterized in the human and mouse gene for BDNF (Liu et al., 2005). The different function of these transcripts is not known, but it is possible that they are differentially targeted and translated within the cell. Transcripts containing exons I, II, and III are expressed throughout the brain, whereas exon IV transcripts are expressed primarily outside the brain. Exon III- and IV-containing transcripts display immediate early gene properties, whereas transcription of exons I and II requires protein synthesis and is likely to be regulated by long-term interventions, such as antidepressant treatment (Lauterborn et al., 1996). Thus far particular attention has been paid to exon III because it has been demonstrated that exon III-containing transcripts are the most highly induced following membrane depolarization (West et al., 2001).
Differential activation of BDNF promoters I and III (PI and PIII) was found to be induced in cultured neurons by different pathways of calcium signaling (Tabuchi et al., 2000). PIII was activated by calcium influx through either L-type VGCCs or NMDA receptor, whereas PI was activated only by calcium influx through L-type VGCCs. Furthermore, it was found that the efficiency of transcription was strictly dependent on concentration of KCl used for depolarization. As reported above, duration of CREB phosphorylation was found to be longer when calcium influx occurs through L-type VGCCs rather than through NMDA receptor (Hardingham et al., 1999). These findings combined give an idea of how distinct stimuli may differentially affect the signaling machinery and evoke different gene expression readouts. With such versatile and flexible mechanisms it is not strange that even small differences in the modality of drug treatment may produce a wide panel of different experimental results. But how could we use the present knowledge to improve our experimental set-ups?
The functional organization of BDNF promoter III was recently described in detail (West et al., 2001). Initially, a CREB binding site was identified in BDNF-PIII at -35 bp to the transcription initiation site (Shieh et al., 1998; Tao et al., 1998), which may be phosphorylated in response to both cAMP and calcium stimulation. However, activity-dependent transcription of BDNF exon III in neurons was found to be calcium-dependent (Tao et al., 2002); mutation of a region upstream of the CRE/CaRE sequence blocked transcription, suggesting the presence of additional regulatory elements. Further deletion analysis and detailed point mutagenesis revealed the presence of two other regulatory elements required for calcium-induced transcription of exon III, that were named CaRE1 and CaRE2 (calcium-response element) (West et al., 2001, 2002). Transcription factors that bind to these regulatory elements were recently identified. The calcium-response factor CaRF is a stimulus-selective transcription factor that binds CaRE1 (Tao et al., 2002). Interestingly, CaRF-dependent transcription of BDNF is selective for neurons, because neither CaRF activity nor BDNF expression was detected in PC12 cells after membrane depolarization (Tao et al., 2002), suggesting that a specific activation of CaRF might also contribute to the neuron-selective expression of BDNF. It was shown that CaRE1, isolated from the BDNF promoter, selectively enhances transcription in response to membrane depolarization but not to a cAMP increase (West et al., 2002). Two more factors, upstream stimulatory factors 1/2, that bind CaRE2 element were identified (Chen et al., 2003). Mutation of any one of the three CaREs nearly eliminates calcium induction of the BDNF promoter III, suggesting that a coordinated activation of the four transcription factors on the promoter is necessary for an efficient transcriptional initiation. This cooperativity requirement may subserve the integration of a number of signaling events into a correct sequence, leading to transcription of BDNF. In a framework like this, elevation of cellular cAMP is not sufficient to induce BDNF expression. Efficient transcription occurs only if the four transcription factors (CREB, CaRF, and upstream stimulatory factor 1/2) sit at the same time on the BDNF promoter III, perhaps by recruiting a cofactor that binds all four factors (West et al., 2001). These and other less investigated transcription factors could also be relevant targets for novel antidepressants.
It may be envisaged that increasing knowledge of BDNF gene regulation will in the near future enable better understanding of the regulation of this crucial gene by psychotropic drugs. It will be necessary to define better what combination of signaling pathway stimulation must be induced by treatments. One clue may come from the finding that different types of stimulation (e.g., drug treatment, physical exercise, and their combination) lead to distinct patterns of regulation of exon I and II (Russo-Neustadt et al., 2000). This kind of analysis could be carried out also for different drugs, drug dosages, and time windows following treatment; a main task would be to define minimal requirements for efficient BDNF transcription, as well as the behavioral outcome of increasing expression of different BDNF exons. Finally, different combinations of stimuli should be tried to achieve fast and efficient induction of a right combination of BDNF isoforms that may be associated with antidepressant effect.
VI. Conclusions
CREB-regulated gene expression is a major target of antidepressant drug action. The way whereby different signaling pathways are involved in this function and antidepressants induce long-term changes in gene expression is beginning to be unraveled. To achieve a better knowledge of these pharmacological mechanisms it is crucial that the action of antidepressants on the various signaling cascades, and the outcome as to actual gene expression readout, is carefully dissected. We are confident that the near future will bring substantial advancements in the understanding of the mechanisms of antidepressants, and that this knowledge will help to improve their efficacy and achieve faster onset of therapeutic action as well as, perhaps, identifying new targets for better therapies.
Footnotes
-
↵1 Abbreviations: CREB, cAMP-responsive element binding protein; PKA, protein kinase A; CRE, cAMP-responsive element; BDNF, brain-derived neurotrophic factor; ECT, electroconvulsive treatment; FLX, fluoxetine; SSRI, selective serotonin reuptake inhibitor; TCP, tranylcypromine; DG, dentate gyrus; DMI, desipramine; wt, wild-type; tg, transgenic; CaM, calmodulin; RBX, reboxetine; IMI, imipramine; DARPP-32, dopamine and cAMP-regulated phosphoprotein of Mr 32,000; PP, protein phosphatase; MAPK, mitogen-activated protein kinase; VGCC, voltage-gated calcium channel; NMDA, N-methyl d-aspartate; CaRE, calcium-responsive element; CaMK, calcium/calmodulin-dependent protein kinase; Erk, extracellular signal-regulated kinase; Rsk, ribosomal S6 kinase; Msk, mitogen- and stress-activated protein kinase; ELISA, enzyme-linked immunosorbent assay; ESC, escitalopram; LH, learned helplessness; KO, knockout. CaRF, calcium response factor; Org 4428, 1,3,4,13b-tetrahydro-2,10-dimethyl-dibenz[2,3:6,7]oxepino[4,5c]pyridin-4a(2H)-ol.
-
This work was supported by a grant from the National Alliance for Research on Schizophrenia and Depression (U.S.) to M.P., by a European Union (6th Framework Program) grant for project GENDEP (Contract LSHB-CT-2003-503428) to M.P., and by grants from the Ministry of University and Scientific Research (PRIN Grants 2001054224 and 2003053993) and the Ministry of Health (Italy) to M.P., G.R., and J.P.
-
Article, publication date, and citation information can be found at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.58.1.7.
- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction: Regulation of Gene Expression and Neuroplasticity
- II. The Action of Antidepressants on cAMP-Responsive Element Binding Protein Expression and Function: Overview of Present Evidence and Hypotheses
- III. Signaling Pathways and Protein Kinases Regulating cAMP-Responsive Element Binding Protein Phosphorylation and Function
- IV. The Complexity of cAMP-Responsive Element Binding Protein-Regulating Signaling and the Action of Antidepressants
- V. cAMP-Responsive Element Binding Protein Target Genes: The Example of Brain-Derived Neurotrophic Factor
- VI. Conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters