


Abstract
The heme-heme oxygenase system has recently been recognized to possess important regulatory properties. It is tightly involved in both physiological as well as pathophysiological processes, such as cytoprotection, apoptosis, and inflammation. Heme functions as a double-edged sword. In moderate quantities and bound to protein, it forms an essential element for various biological processes, but when unleashed in large amounts, it can become toxic by mediating oxidative stress and inflammation. The effect of this free heme on the vascular system is determined by extracellular factors, such as hemoglobin/heme-binding proteins, haptoglobin, albumin, and hemopexin, and intracellular factors, including heme oxygenases and ferritin. Heme oxygenase (HO) enzyme activity results in the degradation of heme and the production of iron, carbon monoxide, and biliverdin. All these heme-degradation products are potentially toxic, but may also provide strong cytoprotection, depending on the generated amounts and the microenvironment. Pre-induction of HO activity has been demonstrated to ameliorate inflammation and mediate potent resistance to oxidative injury. A better understanding of the complex heme-heme
I. Introduction
The ubiquitous heme1 molecule serves as the functional part of a wide variety of crucial proteins and is involved in various cellular processes such as gene transcription/translation, cell differentiation, and proliferation (Abraham et al., 1983, 1988; Beri and Chandra, 1993; Sassa and Nagai, 1996; Ponka, 1999). It is therefore of fundamental importance for life and has fascinated many investigators for decades.
Heme oxygenase (HO2), the heme-degrading enzyme, however, obtained only scarce attention after its discovery in 1968 (Tenhunen et al., 1968), but during the last 10 years, this enzyme has rapidly gained interest from a fast growing group of scientists. The potential properties of the heme-heme oxygenase system and the multiple and diverse functions of its downstream effector molecules are mesmerizing (Lane, 1998; Maines, 2000). Recently, an overwhelming body of evidence indicates that the heme-heme oxygenase system is tightly involved in the regulation of many physiological as well as pathophysiological processes, such as cytoprotection, apoptosis, and inflammation (Maines, 1997).
Unfortunately, in some instances inflammatory and immune reactions are induced against the “wrong targets” (e.g., self-antigens in the case of autoimmune diseases), or the resolution of inflammation does not occur, resulting in severe and even irreversible tissue injury (Moore, 1999). Vascular diseases such as atherosclerosis, vasculitis, graft failure, ischemia/reperfusion injury, rheumatism, restenosis, and autoimmune diseases are all associated with oxidative stress- and inflammation-induced injury (Brod, 2000). It is therefore of utmost importance to better understand the onset and resolution of inflammation in order to develop new therapies.
In this review, a historic overview, the biosynthesis and the significance of heme and heme oxygenase is presented in view of biological and pathological processes, with special reference to inflammation. In addition, the heme-HO system is discussed in relation to apoptosis and transplantation biology.
II. Heme
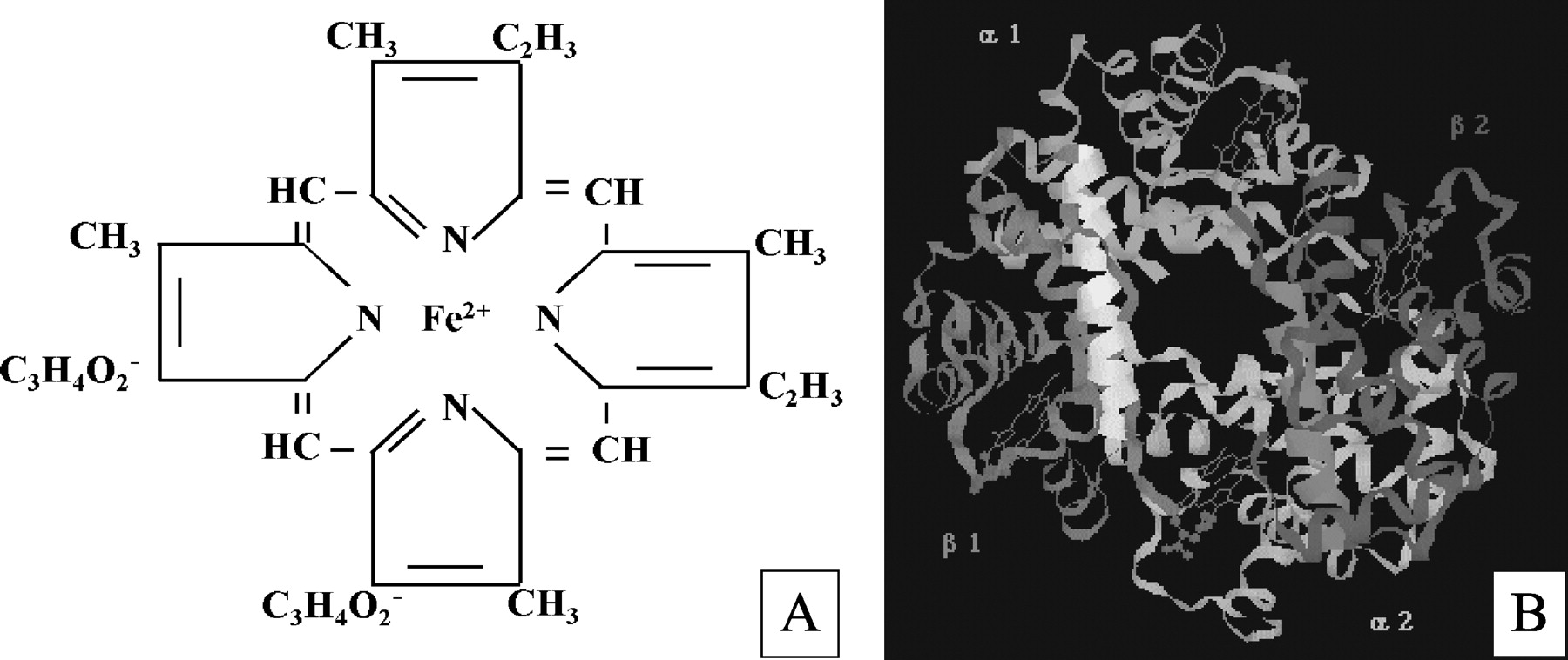
Heme (iron protoporphyrin IX) is a complex of an iron atom linked to the four ligand groups of porphyrin (Beri and Chandra, 1993). Its structure is depicted in Fig. 1A.

, structure of iron(II) protoporphyrin IX (heme); B, hemoglobin, an example of the various heme proteins (courtesy of Dr G. M. Baker, Northern Illinois University, DeKalb, IL).
A. Heme Biosynthesis
The structure of heme (Fig. 1A) was confirmed after complete organic synthesis by Hans Fischer (1881-1945) and coworkers in 1927 (Watson, 1965). In 1944, David Shemin set out experiments to determine the half-life of hemoglobin (Fig. 1B) and other blood proteins. He ingested 66 g of the simplest amino acid, glycine, containing the stable isotope 15N (nitrogen), to follow its incorporation into his own blood components. As he expected, a continuous synthesis and degradation of proteins in the blood plasma was found, and the half-life was estimated to be less than 5 days. However, to his surprise, the constituents in the red blood cells, once formed, had an average life span of 127 days and would only then be destroyed (Shemin and Rittenberg, 1946; Shemin, 1989). Indeed, when red blood cells mature both heme and hemoglobin synthesis ceases. They must therefore survive for the life of the erythrocyte (approximately 120 days). Thus, hemoglobin, in contrast to all other proteins previously examined by these techniques, is not in a dynamic state.
Interestingly, thorough analysis of these data demonstrated that glycine has to be the nitrogenous precursor of heme, the functional nonprotein component of hemoglobin (Shemin and Wittenberg, 1951; Shemin, 1970; Shemin, 1989). Additional experiments in laboratory animals confirmed the novel insight that glycine was incorporated into heme. This first observation eventually led to the unraveling of the heme molecule from glycine and another body component, succinyl coenzyme A. Shemin and colleagues subsequently unraveled the complete set of enzymes involved in the synthesis of heme from these small precursors (Fig. 2) (Shemin and Wittenberg, 1951; Shemin et al., 1953; Shemin, 1970, 1989; Kappas et al., 1995b).
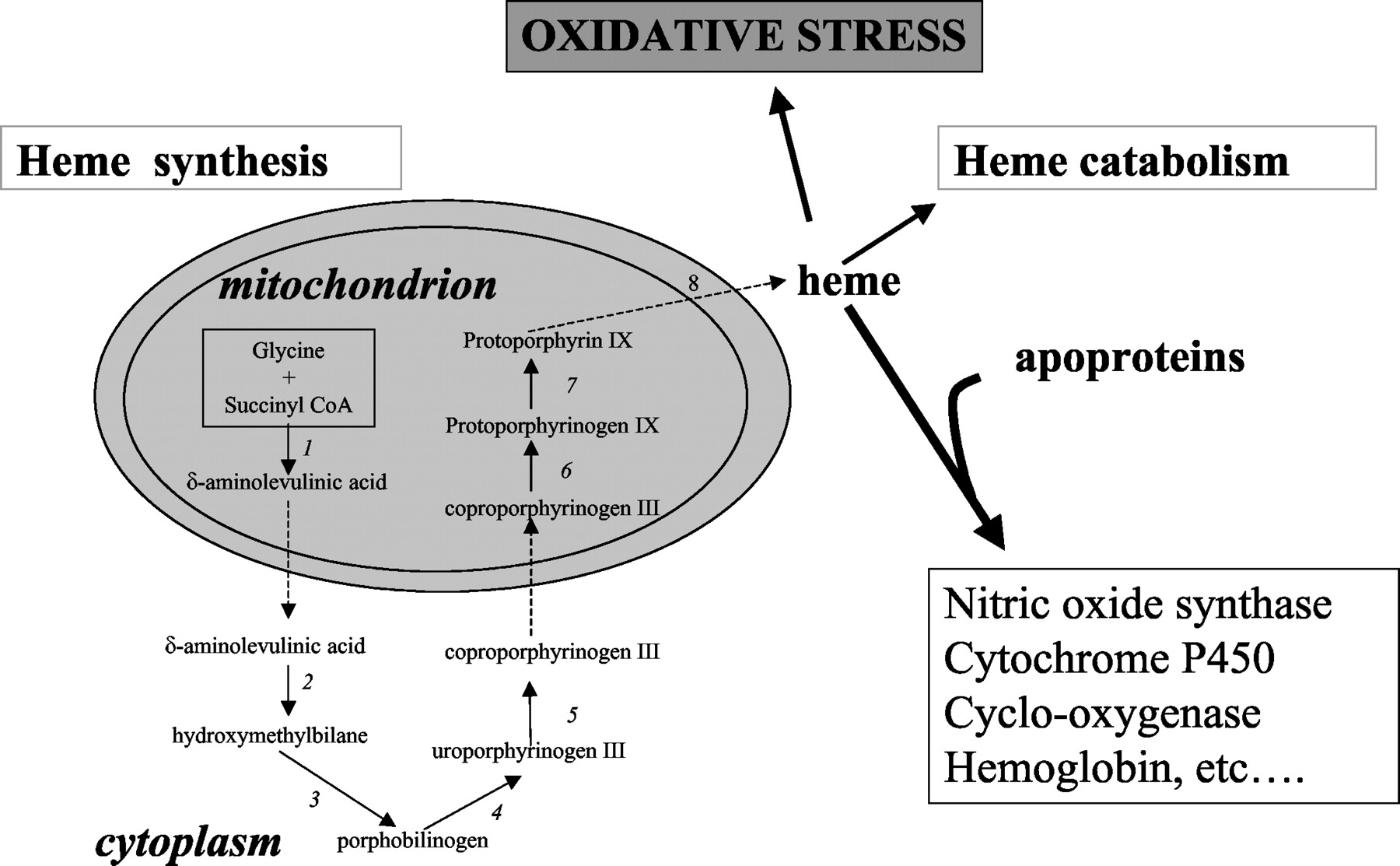
Overview of heme synthesis. The formation of heme from glycine and succinyl CoA involves the sequential participation of eight different enzymes that are named as follows: δ-aminolevulinate synthase (ALA-S) (1); δ-aminolevulinate dehydratase (ALA-D) (2); porphobilinogen deaminase (PBGD) (3); uroporphyrinogen III synthase (URO-S) (4); uroporphyrinogen III decarboxylase (URO-D) (5); coproporphyrinogen III oxidase (CPO) (6); protoporphyrinogen III oxidase (PPO) (7); and ferrochelatase (FC) (8). Newly synthesized heme can be incorporated into heme proteins or, alternatively, can be degraded. Accumulation of intracellular or exogenous (e.g., in blood clot) heme can be detrimental to cells, since heme catalyzes the formation of ROS, resulting in oxidative stress.
Heme is synthesized in all human nucleated cells. It involves a series of enzymatic reactions taking place partly in the mitochondrion and partly in the cytoplasm (Fig. 2). Heme requirements vary significantly among various cells and tissues. The most rapid rates of heme synthesis occur in the erythroid cells in the bone marrow [75% of total body heme (Berk et al., 1976)] and the hepatocytes in the liver, because of the incorporation of heme into the heme proteins, hemoglobin and cytochrome P450 synthesized in very high quantities in these respective organs. Probably, most mammalian cells contain a “free” or “uncommitted” heme pool, serving both precursor and regulatory functions (Fig. 2) (Ponka, 1999).
Senescent red blood cells are removed from the circulation and degraded by the reticuloendothelial system (RES) in the spleen, liver, and bone marrow (Abraham et al., 1988). As such, heme turnover is high for erythropoiesis (∼250 mg/day) and cytochrome synthesis (∼50 mg/day). Heme derived from denatured heme proteins other than hemoglobin is probably degraded locally by HO.
As a consequence of its vital importance in biological processes, a partial defect in one of the enzymes used in heme biosynthesis is associated with a number of disease states. Examples of these inherited or acquired disorders are porphyrias, myelodysplastic syndrome, and sideroblastic anemia (Bottomley and Muller-Eberhard, 1988; Volin et al., 1988).
B. Heme As Active Site in Heme Proteins
The heme molecule provides a multitude of crucial biological functions. It can interact with various inactive apo-heme proteins giving rise to functional heme proteins. The function of the heme molecule is ultimately determined by the properties of the polypeptide bound to it (Dawson, 1988). In hemoglobin (Fig. 1B) and myoglobin, it is used for oxygen transport and storage, respectively, whereas in cytochromes it is involved in electron transport, energy generation, and chemical transformation. In catalases and peroxidases, heme functions in H2O2 inactivation or activation, respectively, and in tryptophan pyrrolase, it catalyzes the oxidation of tryptophan (Maines, 1997). Furthermore, heme is indispensable for a wide array of other important enzyme systems, such as cyclooxygenase (COX) and nitric-oxide synthase (NOS) (Seed and Willoughby, 1997).
C. Heme-Mediated Gene Modulation, Cell Differentiation, Proliferation, and Immune Stimulation
Besides its function as prosthetic moiety in heme proteins, heme itself may influence the expression of many genes. In nonerythroid cells, heme regulates its own production by down-regulating heme biosynthesis (at the level of the rate-limiting enzyme 5-aminolevulinic acid synthase (see Fig. 2) and by up-regulating heme metabolism (Yamamoto et al., 1982). In contrast, in erythroid cells, heme serves as a positive feedback regulator for heme synthesis (at the level of ferrochelatase [see Fig. 2)] and inhibits its degradation (Sassa, 1976; Rutherford and Harrison, 1979).
Heme may affect a wide spectrum of regulatory factors and can influence gene expression at almost every level by regulating transcription (Pfeifer et al., 1989; Lathrop and Timko, 1993; Zhang and Guarente, 1995), mRNA stability (Maniatis et al., 1976), protein synthesis via eIF-2α kinase (Chen et al., 1994), splicing (Ponka, 1999; Zhu et al., 1999), and post-translational modification (Swenson et al., 1991). Furthermore, heme is important in controlling the expression of numerous proteins, such as globin, heme biosynthetic enzymes, cytochromes, myeloperoxidase, heme oxygenase-1, and the transferrin receptor (Gidari and Levere, 1977). A number of these genes are regulated via heme response elements (HREs).
Using differential display, Zhu and coworkers showed that heme also strongly induces genes other than those encoding heme proteins (Sassa and Nagai, 1996; Zhu et al., 1999). These include growth-associated protein p-62 (involved in Ras signaling), chaperonin Tcp20, histone H2A.Z, and a subunit of the small nuclear ribonucleo-protein complex (involved in splicing) (Zhu et al., 1999). On the other hand, heme represses other genes, such as the H+-ATPase proton channel subunit and a cellular immediate-early response gene (Zhu et al., 1999).
It has long been hypothesized that a heme protein is involved in oxygen sensing, which is important in various molecular and cellular processes. However, in mammalians no such protein has been found yet. In yeast, heme signals are mainly mediated by the heme activator protein Hap1, which in response to heme, activates the transcription of genes necessary for respiration and control of oxidative damage (Pfeifer et al., 1987; Lee et al., 2001). Heme repression is achieved through the action of the ROX1 repressor, the expression of which is transcriptionally activated by heme (Zhang and Hach, 1999).
Recently, a mammalian transcription repressor, Bach1, was identified, which is inactivated upon heme binding (Ogawa et al., 2001). Bach1 competes with activators, including Nrf2, for the binding to Maf recognition element (MARE). HREs share a nucleotide sequence with the MARE, which is important in development and differentiation (Motohashi et al., 1997; Ogawa et al., 2001). Therefore, stress response genes bearing MARE may be up-regulated via the MARE-binding transcription factors with increased levels of heme (Ogawa et al., 2001).
In addition, heme regulates differentiation and proliferation of various cell types. It stimulates neuronal differentiation of mouse neuroblastoma cells (Ishii and Maniatis, 1978), erythroid differentiation of erythro-leukemia cells of mouse (Granick and Sassa, 1978) and man (Benz et al., 1980), formation of erythroid colonies in mouse as well as in human bone marrow cultures (Partanen et al., 1988; Abraham, 1991), differentiation of 3T3 fibroblasts into adipocytes (Chen and London, 1981), and it stimulates cell growth of cultured fibroblasts (Verger et al., 1983). Moreover, heme possesses also stimulating activity on anti-tumor immune responses (Tsuji et al., 1993).
D. Toxic Effects of Free Heme and Its Control
In contrast to the many positive functions of heme, there is accumulating evidence that an excess of free heme can cause cell damage and tissue injury since heme catalyzes the formation of reactive oxygen species (ROS), resulting in oxidative stress (Vercellotti et al., 1994; Jeney et al., 2002) (Fig. 2). Because the low-molecular weight iron chelate, heme, is lipophilic it can easily intercalate in the membrane and impair lipid bilayers and organelles, such as mitochondria and nuclei, and destabilize the cytoskeleton (Balla et al., 1991; Beri and Chandra, 1993; Ryter and Tyrrell, 2000).
Various autoimmune infectious (such as malaria) and inherited (e.g., sickle cell disease) disorders are complicated by myolysis or hemolysis, and tissues can subsequently be exposed to large amounts of free heme/heme proteins (Fig. 3) (Zager, 1996). The cellular “free heme pool” may increase after extracellular heme overload, increased heme synthesis, accelerated breakdown of heme proteins, impaired incorporation into apo-heme proteins, or because of diminished HO activity, resulting in increased levels of ROS and, subsequently, oxidative damage and cellular injury (Maines, 1992; Ryter and Tyrrell, 2000).

Following hemorrhage, huge deposits of extravascular erythrocytes are present (A), from which large amounts of heme are released (B) as visualized with benzidine staining. The free heme may then interact with the surrounding tissue resulting in oxidative stress and inflammatory reactions.
Several defense mechanisms against free heme-mediated oxidative stress and inflammation exist in mammals. They consist of intra- (e.g., heme-binding protein 23, HO-2, and HO-3) and extracellular (e.g., hemopexin, albumin) scavengers, anti-oxidative enzymes, and HO-1(Muller-Eberhard and Fraig, 1993; Immenschuh et al., 1997; Maines, 1997; Castellani et al., 2000). During hemolysis, free vascular hemoglobin is captured by its scavenger haptoglobin and transported to the RES. However, hemoglobin can be rapidly converted into methemoglobin, which liberates the incorporated heme group easily. Any free vascular heme is bound to the plasma proteins hemopexin or albumin, which transport it to the liver for degradation in the RES (Muller-Eberhard and Fraig, 1993). However, when large amounts of free heme proteins or heme (locally) accumulate, like in a blood clot or after vascular deposition, the scavengers get overwhelmed or are unable to reach them (Muller-Eberhard et al., 1968; Jacob, 1994; Wagener et al., 2001b; Jeney et al., 2002). This enables heme to exert its damaging effects.
In summary, heme exerts a dual role: in small amounts it acts by itself or as the functional group of heme proteins providing diverse and indispensable cellular functions, whereas in excessive amounts, free heme can cause severe tissue damage (Balla et al., 2000; Ryter and Tyrrell, 2000; Jeney et al., 2002). Therefore, the amount of free heme must be tightly controlled to maintain cellular homeostasis and avoid pathological conditions.
III. Heme Oxygenase: The Heme-Degrading Enzyme
HO is the rate-limiting enzyme in the catabolism of heme. It breaks down the porphyrin ring to yield equimolar amounts of biliverdin, free iron (Fe2+), and carbon monoxide (CO). In mammals, biliverdin is rapidly converted by biliverdin reductase into bilirubin (Abraham et al., 1988; Maines, 1997; Montellano, 2000) (Fig. 4).

The colorimetric actions of the heme-HO system; heme oxygenase-mediated heme degradation. Heme derived from hemoglobin in senescent erythrocytes, other heme proteins, or newly synthesized is degraded by HO into biliverdin, CO, and iron. Biliverdin gets converted into the anti-oxidant bilirubin by biliverdin reductase. Iron is directly sequestered by ferritin. CO can interact with heme proteins (e.g., guanylyl cyclase) and alter their activity. Heme and its breakdown products possess various physiological properties but are also potentially toxic (see text).
A. History of Heme Oxygenase
The first who presumed a link between red blood cell breakdown and biliverdin was probably Virchow (1847). The connection between heme and bilirubin was made by Kuster, who noted in the beginning of the 20th century that the structures were “closely related”.
Early experiments of Shemin's group demonstrated that after administration of 15N-labeled glycine, at least 20% of the heme was metabolized into 15N-labeled bile pigments as observed in the feces of volunteers within the 1st week after administration (London et al., 1950; Shemin, 1989). Furthermore, administration of labeled heme into rats showed conversion into bilirubin in the liver (Ostrow et al., 1962). In 1964, Wise and colleagues confirmed this conversion in an in vitro cell-free system (Wise and Drabkin, 1964).
In 1968, Tenhunen and coworkers presented the first evidence that in the conversion of heme to biliverdin/bilirubin, the enzyme HO is involved (Tenhunen et al., 1968). They purified HO from the microsomal fraction of the liver and spleen (Tenhunen et al., 1969). They found that HO activity had an absolute stoichiometric requirement for NADPH and molecular O2 and generated equimolar amounts of bilirubin and CO.
Tenhunen et al. (1969) reported that the microsomal HO system was not only active in heme degradation but also in the biotransformation of xenobiotics (Tenhunen et al., 1969). Thus, it was assumed that the multiple-functioned microsomal HO system was a heme protein functioning both as cytochrome P450 enzyme and terminal oxidase. However, they also demonstrated that known cytochrome P450 substrates, such as hexobarbital, aminopyrine, and cytochrome P450 inhibitors, failed to inhibit HO activity.
Maines and Kappas (1974, 1975) investigated this apparent discrepancy further, using the heavy metal cobalt, which is known to decrease both hepatic microsomal drug metabolism and the microsomal content of cytochrome P450 (Tephly et al., 1971). Rats that were fed with cobalt chloride, indeed showed a major decrease in cytochrome P450 content in microsomes of the liver, whereas, in striking contrast, the HO system increased its activity up to 8-fold. In addition, drug oxidation in microsomes was eliminated after treatment with urea, whereas HO activity levels did not change despite the absence of spectrally detectable cytochrome P450. Herewith was shown for the first time that the protein components of heme catabolism and drug metabolism were distinct and that cytochrome P450 was not required for heme oxidation.
Similarly, Yoshida and Kikuchi (1974) demonstrated that there was a high specific HO activity present in the spleen, in contrast to cytochrome P450 activity. Furthermore, they observed the absence of cytochrome P450 in their preparation of spleen microsomes but, in addition, reported the requirement of NADPH-cytochrome c (P450) reductase for enzymatic activity. Thus, the heme-degrading activity was solely attributable to HO. In addition, oxygen and hydrogen, donated by e.g., NADPH-cytochrome c (P450) reductase or NOS, are needed for the reaction (Fig. 4A).
B. Heme Oxygenase Isoforms and Gene Regulation
To date, three isoforms of heme oxygenase have been identified (HO-1, HO-2, and HO-3) (Maines, 1997). These are products of different genes, and their expression differs greatly between cell types, tissue distribution, and regulation (Table 1) (Maines, 1997; Otterbein and Choi, 2000; Maines and Panahian, 2001). They have a molecular weight of approximately 32, 36, and 33 kDa, respectively. The HO proteins are anchored to the endoplasmic reticulum by a hydrophobic sequence of amino acids at the carboxyl terminus of the protein.
Summary of the various functions of HO isoforms, their tissue distribution, and gene regulation
Maines et al. (1986) were the first to report the identification of a second form of HO from rat liver microsomes, designated HO-2. Only recently, a third isoform, HO-3, was discovered (McCoubrey et al., 1997). HO-1 and HO-2 share little similarity in amino acid sequence (40%), whereas the HO-2 and HO-3 isoform are far more homologous (90%). All HO isoforms are highly conserved among species in evolution. HO is expressed in virtually all life forms; in prokaryotic bacteria as well as in fungi, plants, and humans, regulating a wide spectrum of cellular processes (Terry et al., 2002). The homology between rat, mouse, and human is for HO-1 and HO-2 proteins higher than 80% and 90%, respectively.
Under normal physiological conditions, most cells express low or undetectable levels of HO-1 protein, whereas HO-2 proteins are constitutively expressed. HO-3 protein expression awaits further characterization. HO-2 transcription is only up-regulated by few agents, such as opiates and adrenal glucocorticoids (Li and David Clark, 2000; Liu et al., 2000).
HO-1 gene expression is highly inducible by more diverse stimuli than any other enzyme described to date (Maines, 1997) and involves a multitude of signaling pathways (Table 2) (Immenschuh and Ramadori, 2000). HO-1 expression is mainly regulated at the transcriptional level.
Selection of different inducers of HO-1 gene expression
Since HO-1 gene expression is strongly induced by agents or conditions that increase oxidative stress, redox signaling plays a crucial role in its regulation. These stimuli include heavy metals, bacterial lipopolysaccharides, hypoxia, hyperoxia, heat shock, ischemia, UV radiation, H2O2, cytokines, nitric oxide, stimuli that deplete cellular glutathione stores, and its substrate heme (Applegate et al., 1991; Shibahara, 1994; Immenschuh and Ramadori, 2000). In addition, several different redox-independent signaling pathways are involved in HO-1 gene regulation (Immenschuh and Ramadori, 2000). These include kinases [e.g., mitogen-activated protein kinases (Elbirt et al., 1998; Oguro et al., 1998), protein kinase C (Alam et al., 1995; Terry et al., 1999), cAMP-dependent protein kinase A (Immenschuh et al., 1998), and cGMP-dependent protein kinase G(Polte et al., 2000)] and protein phosphatases (Immenschuh et al., 2000). However, HO-1 gene expression is often cell type- and species-specific (Morse and Choi, 2002).
Consistent with the diversity of signaling cascades involved in HO-1 induction, the promoter region of HO-1 contains a wide variety of regulatory elements (Abraham et al., 1996). It includes DNA-binding sites for oxidative stress-responsive transcription factors, such as nuclear factor (NF)-κB, NF-E2-related factor 2 (Nrf2), and AP-1 (Lavrovsky et al., 1994; Choi and Alam, 1996).
Next to inducers of HO-1, negative regulators also exist, e.g., scavengers of ROS, such as N-acetylcysteine that reduce the magnitude of HO-1 induction by oxidative stress (Lautier et al., 1992). Furthermore, it has been reported that the HO-1 gene is further down-regulated by angiotensin II via calcium-signaling (Ishizaka and Griendling, 1997), interferon-γ (Takahashi et al., 1999), prostaglandin E2 (Tetsuka et al., 1995), transforming growth factor-β (Pellacani et al., 1998), and IL-10 (Immenschuh et al., 1999).
HO-1 is active even at low substrate concentrations, and its activity increases as the concentration of heme increases within the physiological range. In contrast, HO-2 has low activity at low substrate concentrations, and even its maximum activity is limited to less than 10% that of HO-1 (Table 1) (Maines and Panahian, 2001). However, the enzymatic activity of HO-2 can be increased in a protein kinase C-dependent manner (Dore et al., 1999).
Specific activity of HO in different organs greatly varies. The highest HO activity is found in spleen, testis, and brain. The spleen is the only organ in which under normal unstressed conditions, HO-1 is the predominant form. HO-2 is abundant in brain, testis, and liver. HO-3 has been identified in brain, kidney, liver, heart, testis, and spleen and has been reported to have only very poor heme degrading capacity (McCoubrey et al., 1997).
Because of their heme-binding capacity, HO-2 and HO-3 may function as a first buffer reservoir for sudden accumulation of free heme. Similarly, other heme proteins may not only exert their specific activity but may also have a function in restraining free heme from exerting injurious effects (Nath et al., 2000). Moreover, there are several specialized heme-binding proteins (HBP) present in a cell, such as L-FABP, glutathione S-transferase, heme-binding protein 23 (HBP23), and p22 HBP, HO-2, HO-3) (Immenschuh et al., 1995). Although, the exact function of HBPs remains unclear, they are likely involved in shielding from oxidative assaults, heme trafficking, and heme utilization for heme protein synthesis.
Interestingly, expression of CD163, a hemoglobin/haptoglobin scavenger receptor on macrophages, is highly up-regulated by acute phase reactants, such as interleukin-6 (IL-6) and glucocorticoids (Buechler et al., 2000; Hogger and Sorg, 2001; Kristiansen et al., 2001). Haptoglobin, CD163, and HO-1 may be regarded as a “scavenger molecule-receptor-enzyme” system that is coherently elevated during inflammatory conditions to enhance the capacity for hemoglobin clearance (Kristiansen et al., 2001).
C. Traditional View on Heme Oxygenase and Its Toxic Breakdown Products
Traditionally, HO has been thought to be solely involved in the breakdown of heme from senescent red blood cells or denatured heme proteins. The degradation products free iron, biliverdin/bilirubin, and CO produced during this useful process were considered “toxic waste” materials (Johnson et al., 1999).
Free iron(II) (Fe2+) is capable of causing severe oxidative stress by the generation of reactive molecules, such as the highly reactive hydroxyl radical, since it can participate in Fenton reaction, which involves H2O2 and iron reacting with a variety of organic molecules, resulting in a series of radical reactions (Tyrrell, 1999; Alayash et al., 2001). This oxidative stress might then result in membrane damage and consequently tissue injury. Iron is thus generally considered a powerful prooxidant and injurious to cells.
The heme breakdown product biliverdin is rapidly reduced to bilirubin by the cytosolic enzyme biliverdin reductase. Reduction of biliverdin to bilirubin occurs in all mammals, but in some non-mammals, such as birds, amphibians, and reptiles, biliverdin is the end product of heme catabolism and is excreted in e.g., egg shells and manure (Lane, 1998). Since bilirubin is highly lipophilic, it is excreted as the glucuronide conjugate. The increased water solubility of bilirubin diglucuronide facilitates its excretion with bile as the bile pigments.
Bilirubin production is two to three times elevated in newborns compared with normal adults, because of the switching from fetal to adult hemoglobin (Maines and Trakshel, 1992). Approximately 5% of newborns suffer from neonatal jaundice, or hyperbilirubinemia. If the bilirubin levels become dangerously high, bilirubin passes through the blood-brain barrier and can cause neuronal damage associated with kernicterus (Gourley, 1997). The most common treatment for hyperbilirubinemia is phototherapy, in which the jaundiced infant is exposed to blue light. The therapeutic effect is mediated by photoisomerization of unconjugated bilirubin, resulting in more polar and readily excretable photoisomers (McDonagh and Lightner, 1985). Recently, alternative treatment with competitive inhibitors of HO activity, such as stannic mesoporphyrin (single dose of 6 μmol/kg), has shown to prevent and to reverse bilirubinemia (Kappas et al., 1995a, 2001).
CO has been considered a dangerous poisonous gas for a long time (Johnson et al., 1999). It is usually generated during the incomplete burning of organic materials. Bernard reported already in 1857 that CO could bind to hemoglobin. In fact, this odorless, colorless, tasteless gas binds about 200 times better to hemoglobin than oxygen, resulting in the formation of carboxyhemoglobin. This subsequently results in decreased oxygen release to the metabolizing tissues and ultimately death (Johnson et al., 1999). CO was shown to be an endogenous metabolic product in man by Sjoestrand (1949), who also observed that the rate of CO production was elevated in patients with increased destruction of erythrocytes (Sjoestrand, 1949a,b). The hypothesis that heme is the source of endogenous CO production in mammals was confirmed in vivo by investigators who used radioactive tracers to show that the α-meso carbon of heme is oxidized to CO and bilirubin (Coburn et al., 1967). The rate of CO production in the human body is normally approximately 16 μmol/h (Marks, 1994), but it can get 10-fold higher in pathological circumstances, such as hemolysis (Coburn et al., 1966).
D. Recent Insights in Heme Oxygenase and a Novel Role for Heme-Derived Metabolites
In addition to the role of HO in recycling iron for heme synthesis, current understanding of HO has demonstrated that HO is important in a wide variety of other physiological and pathological processes. To date, no other known enzyme is induced by so many stimuli of diverse origin as HO-1 (Maines, 1997; Otterbein and Choi, 2000) (Table 2). It is this diversity of non-heme inducers and the existence of different HO isoforms that has led to the hypothesis that HO-1 plays a vital function in maintaining cellular homeostasis in addition to heme degradation (Maines, 2000).
Elevated HO-1 expression levels are detected in a variety of pathological conditions (Otterbein and Choi, 2000) (Table 3). HO-1 induction protects against ischemia/reperfusion injury, oxidative stress, inflammation, transplant rejection, apoptosis, and many more conditions (Amersi et al., 1999; Yang et al., 1999; Brouard et al., 2000, 2002; Willoughby et al., 2000; Inguaggiato et al., 2001; Sato et al., 2001; Ke et al., 2002; Melo et al., 2002). Although the precise mechanism is poorly understood and requires further investigation, consensus has been reached about the potent cytoprotective properties of HO-1.
Selection of diseases/conditions associated with HO-1 expression
Previous studies demonstrated that the induction of HO-1 induction is accompanied by increased ferritin synthesis, whereas inhibition of HO activity causes a decrease in ferritin levels (Eisenstein et al., 1991; Vile et al., 1994). Chelating iron prevents ferritin synthesis and fails to protect against iron-mediated oxidative stress (Otterbein et al., 1997). In contrast, HO-dependent release of iron results in up-regulation of ferritin, scavenging of the free iron, and subsequently protection from the adverse effects of iron. Each apo-ferritin molecule of 450 kDa can sequester up to approximately 4,500 iron atoms (Harrison and Arosio, 1996). Maintenance of low iron pools by increased ferritin levels appears to play a central role in cellular anti-oxidant defense and cytoprotection (Torti and Torti, 2002).
In addition, HO-1 is closely associated with cellular iron extrusion mechanisms, which may constitute an alternative cytoprotective mechanism of HO-1 (Snyder and Baranano, 2001). Attenuated HO-1 activity results in decreased cellular iron efflux and subsequently iron overload, whereas HO-1 overexpression accelerates efflux (Poss and Tonegawa, 1997a; Ferris et al., 1999). Baranano et al. (2000) demonstrated that HO-1 decreases intracellular levels of free (unbound) iron by up-regulating an iron ATPase present in the endoplasmic reticulum that can pump iron out. Interestingly, iron regulates nuclear transcription of several other genes as well, such as inducible NOS (iNOS), transferrin, and HO-1 (Abraham et al., 1988; Siow et al., 1999; Baranano et al., 2002).
Bilirubin was only recently recognized as a potentially important anti-oxidant of physiological significance (Stocker et al., 1987), although this property had already been known for some time (Bernard et al., 1954). Administration of bilirubin demonstrated to be cytoprotective in models of ischemic heart injury and oxidative damage (Dore et al., 1999; Clark et al., 2000). It was shown in vitro that at micromolar concentrations both biliverdin and bilirubin efficiently scavenge peroxyl radicals, thereby inhibiting lipid peroxidation (Stocker et al., 1987). In liposomes, bilirubin suppressed oxidation even more effectively than α-tocopherol or vitamin E, which are regarded as excellent anti-oxidants (Stocker et al., 1987). These results indicate that bilirubin functions as a very prominent anti-oxidant in human serum and provides potent protection against oxidative injury and inflammation and decreases risk for familial coronary artery disease (Gopinathan et al., 1994; Hopkins et al., 1996).
CO had no known physiological function until Marks and coworkers proposed that CO may play a role similar to nitric oxide (NO) in signal transduction (Marks et al., 1991). During the last decade, NO research has taken a prominent position into the pathogenesis of virtually all diseases. NO signaling prevents hypertension, ameliorates inflammation, and functions as an important messenger molecule (Ignarro, 1996).
However, NO has not been able to fulfill the high expectations (Lane, 1998). The efficacy is hampered by the fact that NO is a reactive nitrogen species (RNS). Under oxidative conditions, NO reacts with other ROS resulting in the formation of the highly reactive ONOO- (peroxynitrite) (Wolin et al., 1998). Peroxynitrite does not prevent or ameliorate disease states like NO but, in contrast, exacerbates oxidative and inflammatory stress.
CO, in contrast to NO, does not contain free electrons and is therefore relatively inert. Moreover, this simple molecule shares the ability of NO to activate the heme protein guanylyl cyclase by binding to its active site, the heme molecule, resulting in enhanced conversion of guanosine triphosphate (GTP) to guanosine 3,5-cyclic monophosphate (cGMP) and subsequently vasodilation (Marks et al., 1991; Morita and Kourembanas, 1995; Maines, 1997). CO can activate this second messenger system and thereby mediate numerous physiological processes, such as activities of protein kinases, ion channels, and phosphodiesterases (Maines, 1997). Interestingly, NO and peroxynitrite induce HO-1 activity and thus form a feedback loop, where CO takes over the roles of NO under conditions of oxidative stress (Maines, 1997). Therefore, HO activity is important in controlling the activity of heme proteins, such as cytochromes P450, COX, guanylyl cyclase, and NOS, not only at the level of the intracellular heme pool available for the apo-heme proteins (Kappas and Drummond, 1986) but possibly also at the level of HO's downstream effector molecule CO by interactions with the heme group (Haider et al., 2002).
It has been shown that exogenous CO (ca. 300 parts per million) reduces inflammatory responses in several models of oxidant injury, which is consistent with results observed with HO-1 overexpression (Abraham et al., 1995a; Amersi et al., 1999). Although CO acts in many ways that are similar to NO, CO possesses additional functions on signal transduction pathways. CO inhibits pro-inflammatory genes while augmenting antiinflammatory cytokine production by selective activation of several p38 mitogen-activated protein kinase (MAPK) signaling pathways in a guanylyl cyclase-independent manner (Brouard et al., 2000; Otterbein et al., 2000; Sarady et al., 2002; Song et al., 2003).
Other important effects of CO involve inhibition of proliferation of vascular smooth muscle cells, inhibition of platelet aggregation, protection against apoptosis, and acting as a neurotransmitter (Brune and Ullrich, 1987; Durante and Schafer, 1998; Brouard et al., 2000; Togane et al., 2000; Baranano and Snyder, 2001). Therefore, although high amounts of CO are lethal, low concentrations of CO mediate many important physiological functions, such as maintenance of vasomotor tone and mediating neurotransmission (Johnson et al., 1999).
In summary, although the mechanisms underlying the anti-inflammatory actions of HO-1 remain unclear, the heme-degradation products CO, biliverdin/bilirubin, and iron-induced ferritin seem to be mediators of the beneficial effects of HO-1 activation. CO causes vasodilation, inhibits platelets aggregation, and suppresses the production of cytokines, whereas alternative actions could be exerted by the potent anti-oxidant properties of biliverdin/bilirubin and the iron-capturing actions of ferritin (Maines, 1997; Otterbein and Choi, 2000).
IV. Inflammation and the Role of Adhesion Molecules
Inflammation is a defense system employed by organisms to protect them from pathogenic invaders, to clean up damaged cells after injury and to prevent further damage. The main purpose of inflammation is to identify and eliminate injurious agents and to repair the surrounding tissue. The inflammatory response involves several stages: 1) dilation of capillaries to increase blood flow; 2) microvascular structural changes and escape of plasma proteins from the bloodstream; 3) the so-called “leukocyte-adhesion cascade”; 4) elimination of possible pathogens; and 5) the resolution of inflammation.
An essential step of the leukocyte-adhesion cascade involves a series of sequential activation and adhesion events. This cascade requires the activation of endothelial cells resulting in highly increased surface expression of specific inflammatory adhesion molecules (Table 4), such as selectins, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule (VCAM-1), enabling circulating activated leukocytes to specifically interact with their ligands on the endothelium. Next, capture, rolling, and firm adhesion of leukocytes is followed by transmigration through the endothelium toward the site of injury by chemotaxis. Here they exert their effector functions and eradicate the source of inflammation (Fig. 5).
Some important adhesion molecules involved in the interaction between leukocytes, endothelium, and the extracellular matrix

The “leukocyte cascade” involves sequential events, starting with adhesion molecule expression on activated endothelium. Activated leukocytes interact with the endothelium via adhesion molecules (see inset) and start rolling, firmly adhere, and finally transmigrate through the endothelium toward the site of injury by chemotaxis (Bevilacqua, 1993).
Although inflammation is an essential and beneficial process in the protection against pathogens, occasionally the inflammatory process is directed against autologous antigens, or the process does not alleviate after its onset and escalates into long-term persistence of inflammation, i.e., chronic inflammation (Robbins et al., 1999). The excessive release of proteases and ROS by the activated leukocytes and endothelial cells in these conditions can ultimately result in severe tissue damage (Conner and Grisham, 1996). In the pathogenesis of several diseases, such as rheumatoid arthritis, inflammatory bowel disease, atherosclerosis, multiple sclerosis, Alzheimer disease, and transplant rejection, inflammation-mediated tissue injury plays an important role (Cotran and Mayadas-Norton, 1998).
In many inflammatory diseases, currently available intervention strategies fail or are of limited success. Therefore, there is a great need for novel strategies to treat chronic inflammatory conditions. One approach could be to control adhesion molecule expression, which is pivotal in the outcome of inflammatory processes (Behrend, 2000).
A. The Role of Heme in Inflammation
Large amounts of free heme and heme proteins are found after hemolysis or rhabdomyolysis in various pathological conditions, including ischemia/reperfusion, hemoglobinopathies, hematoma, hemorrhage, and muscle injury (Letarte et al., 1993; Nath et al., 1998, 2001a; Alayash, 2000) (Fig. 3).
Heme-mediated oxidative insults and inflammation are likely important in a wide variety of pathophysiological processes (Balla et al., 2000). Heme and heme proteins have been implicated in vasospasm, vasoconstriction, oxidative stress, and endothelial cell adhesiveness, whereas a variety of toxic effects are attributed to oxidized lipids (Letarte et al., 1993; Wagener et al., 1997; Balla et al., 2000; Lavrovsky et al., 2000).
We previously demonstrated that elevated levels of heme can act pro-inflammatory. We observed that heme induces the expression of pro-inflammatory adhesion molecules both in vitro (Wagener et al., 1997) and in vivo (Wagener et al., 2001b). Moreover, vascular heme was also found to promote an increase in vascular permeability and the infiltration of leukocytes into a variety of tissues in a mouse model (Wagener et al., 2001b).
Nath et al. (2001b) demonstrated in a kidney model that heme proteins elicit inflammatory processes. Heme proteins, such as hemoglobin, get stuck in the glomeruli and accumulate in the kidney. Based on our observations, subsequent release of heme may then result in local inflammatory reactions and ultimately lead to renal failure. In fact, they observed an increased expression of monocyte chemoattractant protein (MCP-1) upon exposure to heme proteins. This finding adds significantly to our understanding of the observed heme-induced leukocyte recruitment.
Several groups have reported that the efficacy of artificial blood substitutes, such as modified hemoglobin solutions, are often hampered by oxidative and inflammatory complications (Simoni et al., 1997; Goldman et al., 1998; Baldwin, 1999). This phenomenon may be related to a difference in heme-releasing capacity of the different hemoglobin solutions. Upon release from hemoglobin, heme interacts with the vascular wall and induces oxidative stress, adhesion molecule expression, and vasopermeabilization (Wagener et al., 1997, 2001b).
Furthermore, the demonstrated vaso-occlusion and vascular inflammation present in several hemoglobinopathies are thought to be caused by increased adhesive interactions between the vascular endothelium and circulating blood cells (Moore et al., 1996; Hebbel, 1997; Solovey et al., 2001). In sickle cell disease, accumulation of heme, adhesion molecule expression, and an increased risk of renal failure have recently been demonstrated (Shiu et al., 2000; Nath et al., 2001a; Bonaventura et al., 2002; Frenette, 2002), strengthening the idea that heme-induced oxidative and inflammatory insults relate to its etiology (Wagener et al., 2001a).
B. Consequences of Heme-Induced Oxidative Stress and Inflammation
These recent observations may have widespread implications for the understanding of various inflammatory complications. It implicates, on the one hand, that at sites of injury, the released heme may be a physiological response that is necessary to recruit inflammatory cells, to initiate inflammatory processes, and to function as a first “danger signal”. However, on the other hand, excess of free heme may cause oxidative and inflammatory injury. In that case, it is of utmost importance to shield the oxidative and inflammatory properties of free heme; e.g., prevention of the formation of methemoglobin, which easily releases its heme, may be helpful in the design of safe “blood substitutes”.
This adds to the hypothesis that heme-induced inflammation is involved in the pathology of diverse conditions, such as renal failure, atherosclerosis, complications after artificial blood transfusion, peritoneal endometriosis, and heart transplant failure (Jacob, 1994; Nath et al., 1995; Alayash, 2000; Sato et al., 2001; Van Langendonckt et al., 2002).
Based on our findings, it seems important that packed red blood cells should be first washed before administration to a patient to prevent inflammatory complications during blood transfusions, since during storage a significant amount of erythrocytes will have been lysed, resulting in large amounts of free heme that may initiate inflammation. Alternatively, patients with increased vascular free heme, e.g., after intravascular hemolysis, may benefit from plasmapheresis or dialysis to dilute its potential damaging activities.
Furthermore, appearance of functional relevant polymorphisms for haptoglobin, hemopexin, and HO could be important for people with increased risk of heme release or people with already compromised redox balance, such as diabetes patients, sports people, or people undergoing transplantation or surgery. In case these individuals have a compromised protection against heme and heme protein-mediated inflammation priming of HO-1 expression or administration of heme/hemoglobin scavengers may protect them from injury.
For the hemoglobin scavenger, haptoglobin, there have been three major polymorphisms described that are associated with different prevalence of many inflammatory diseases, including infections, atherosclerosis, and autoimmune disorders (Langlois and Delanghe, 1996). Interestingly, these different polymorphisms possess also different hemoglobin-scavenging activity (Wuyts et al., 2000). Moreover, the carriers of the polymorphisms that are severely impaired in hemoglobin binding show to be more susceptible to cardiovascular diseases, such as atherosclerosis (Braeckman et al., 1999). This may possibly be explained by our findings of heme-mediated oxidative and inflammatory effects, such as foam cell formation, adhesion molecule expression, and leukocyte recruitment (Wagener et al., 2001b, 2003b). Interestingly, both polymorphisms in haptoglobin and HO-1 are considered risk factors for restenosis after percutaneous transluminal angioplasty (Exner et al., 2001; Roguin et al., 2001).
C. The Role of Heme Oxygenase in Inflammation
In 1916, Suzuki postulated the idea that prior exposure to toxins induces resistance to secondary toxic insults, although the mechanism for this protection remained unclear (Platt and Nath, 1998). Keyse and Tyrrell (1989) were the first to assume that up-regulation of HO-1 by stress-causing agents could mediate this protection against subsequent noxious stimuli (Keyse and Tyrrell, 1989). The stress protein HO-1 is up-regulated by a wide range of stress signals, including cytokines, heavy metals, and also by its substrate heme (Maines, 1997). This illustrates the different faces of the heme molecule in inflammation; small concentrations of heme act cytoprotective via the swift up-regulation of HO-1 (Hayashi et al., 1999), whereas large amounts of heme may act deleterious on tissue via its pro-oxidative and pro-inflammatory functions (Nath et al., 2001b; Wagener et al., 2001b), which cannot be neutralized anymore by the anti-oxidative and anti-inflammatory properties of the HO-1 end-products. Moreover, the time kinetics of HO-1 expression in relation to the inflammatory insult is important for the pro-inflammatory potency of heme and other mediators.
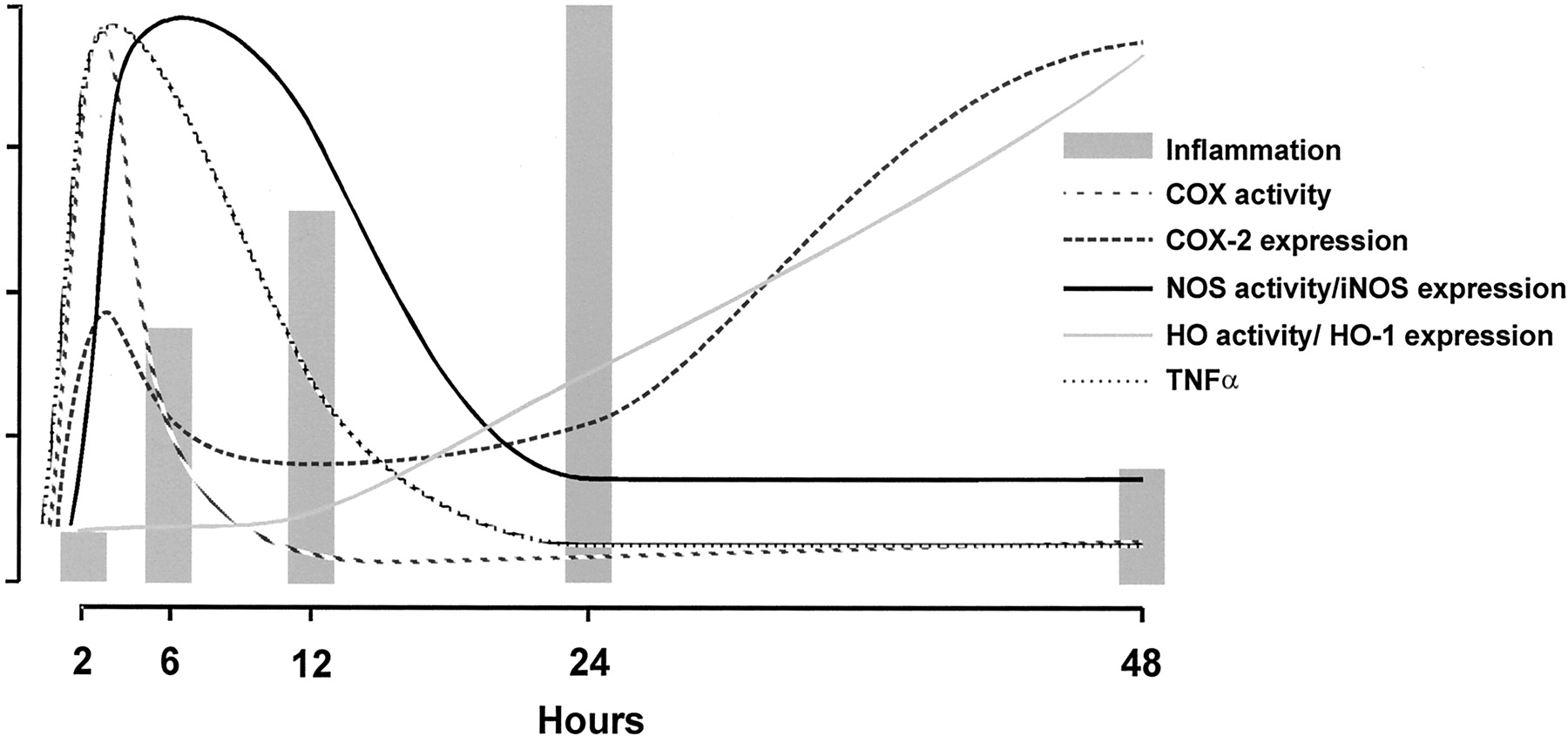
The evolving paradigm of HO-mediated protection of cells and tissues is supported by several animal models of oxidant injury (endotoxic shock, ischemia, hyperoxia, etc.) and acute inflammation (Hancock et al., 1998; Otterbein et al., 1999a; Li et al., 2000; Tamion et al., 2001). In these models, HO-1 elevation confers potent resistance to stress, cell injury, and lipopolysaccharide-induced death whereas blocking of HO activity abrogates cytoprotection, resulting in severe tissue damage. In addition, increased HO-1 expression levels have clinically been demonstrated in a wide variety of inflammatory conditions, such as ischemia/reperfusion injury, atherosclerosis, asthma, Alzheimer disease, and acute renal failure (Amersi et al., 1999; Siow et al., 1999; Agarwal and Nick, 2000; Maines, 2000; Tullius et al., 2001). Moreover, HO-1 production is greatly elevated in inflammatory cells during the resolution phase of inflammation (Fig. 6) (Willis et al., 1996). Very recently, it has been suggested that IL-10, a key molecule for controlling inflammation, mediates many of its anti-inflammatory effects via up-regulation of HO-1 (Lee and Chau, 2002). Induction of HO-1 corresponds to a significant suppression of inflammation, whereas inhibition of the enzyme potentiates the inflammatory response in several models of inflammation (Amersi et al., 1999; Nath, 1999; Vogt et al., 1996; Agarwal and Nick, 2000; Willis et al., 2000; Wagener et al., 2001b). These include corneal, lung, and renal inflammatory models, and in transplantation biology (Laniado-Schwartzman et al., 1997; Platt and Nath, 1998; Otterbein et al., 1999a; Agarwal and Nick, 2000).

This diagram demonstrates the intricate relationships of HO-1, TNF-α, and the heme proteins iNOS and COX during inflammation in a pleurisy model. During inflammation, TNF-α, COX, and iNOS get induced, adhesion molecule expression increases, and leukocytes are recruited (Willis et al., 1996, 2000; Wagener et al., 2001b). HO-1 is up-regulated during the resolution of inflammation and controls the activity of the heme proteins COX and NOS both on the level of the prosthetic group heme, as well as by the generation of CO, which can bind to the heme group and alter the activity of the enzymes.
D. Mechanism of Heme Oxygenase-Mediated Down-Modulation of Inflammation
To date, the mechanism by which HO-1 functions as a cytoprotective and anti-inflammatory protein remains poorly understood. Based on our data, which is in line with observations of others, we postulate that one mechanism by which HO exerts its cytoprotective effects is mediated by down-modulation of adhesion molecule expression. It is evident that adhesion molecules are important to recruit inflammatory cells to sites of acute and chronic inflammation and are therefore crucial in determining the outcome of the inflammatory process (Behrend, 2000). This hypothesis is supported by our observations that HO-1 overexpression reduces heme-induced ICAM-1 expression, whereas inhibition of HO activity increases heme-induced ICAM-1 expression and leukocyte influx (Wagener et al., 1999, 2001b).
Moreover, it has now been confirmed by several other laboratories that HO-1 up-regulation ameliorates adhesion molecule expression and leukocyte adhesion in several other models of inflammation, whereas inhibition of HO activity exacerbates adhesion molecule expression both in vitro and in vivo (Hayashi et al., 1999; Vachharajani et al., 2000; Rucker et al., 2001).
Hayashi et al. (1999) demonstrated that HO-1 induction down-modulates H2O2-mediated induction of P-selectin and subsequently decreases leukocyte binding in vivo, whereas Rucker et al. (2001) observed in vivo decreased ICAM-1 expression in a model of arterial injury when HO-1 was up-regulated prior to injury. This model may thus provide a plausible molecular basis for the previously reported anti-inflammatory properties of HO-1.
The HO-2 and HO-3 isoforms probably function normally as first defense against oxidative/inflammatory insults, whereas upon stress HO-1 is swiftly up-regulated and strongly protects against further injurious signals. HO-2, which is highly expressed in brains and testis, protects, e.g., neurons against oxidative stress. HO-2 may function as a physiologic regulator of cellular function, whereas HO-1 plays a role in modulating tissue responses to injury in pathophysiologic states (Wagener et al., 1999).
Adhesion molecule expression can be down-regulated by anti-oxidants and the gaseous mediator NO (De Caterina et al., 1995; Berendji-Grun et al., 2001). HO activity may decrease adhesion by depleting heme and generating anti-oxidants biliverdin/bilirubin and the gaseous signaling molecule CO, which shares many properties with NO. Unfortunately, under oxidative circumstances, NO is converted into peroxynitrite, which possesses pro-inflammatory properties (van Der Vliet et al., 2000). However, both NO as peroxynitrite up-regulate HO-1 expression (Foresti et al., 1999; Liang et al., 2000). The hereby generated CO has now been postulated to take over the role of NO under oxidative conditions (Foresti and Motterlini, 1999).
Pro-inflammatory mediators, like TNF-α and heme, activate the transcription factors NF-κB, AP-1, and SP-1 signaling pathways via the formation of ROS, which may result in the transcriptional activation of several genes involved in inflammation, such as ICAM-1, VCAM-1, and selectins (Lavrovsky et al., 1994; Shono et al., 1996).
Interestingly, the HO effector molecules biliverdin/bilirubin and CO have also been implicated in the modulation of a range of signaling pathways, including the cGMP and MAPK system, by which they may modulate inflammatory processes and also be involved in ROS-mediated signaling (Adeagbo, 1997; Siow et al., 1999; Otterbein et al., 2000; Nath et al., 2001b). It is likely that HO effector molecules bilirubin and CO, similar to anti-oxidants and NO (De Caterina et al., 1995), play a role in the regulation of ROS-mediated pathways, since HO-1 knockout mice show increased nuclear staining of NF-κB after exposure to hemoglobin, compared with control mice (Nath et al., 2001b).
E. Effects of Heme Oxygenase Inhibition versus Heme Oxygenase-1 Overexpression
The observation that both large amounts of free vascular heme as well as oxidative and chronic inflammatory complications and a remarkable sensitivity to stressful assaults in the first known human case of HO-1 deficiency (Yachie et al., 1999; Kawashima et al., 2002) is consistent with data from our mouse model (Wagener et al., 2001b). This boy who died at age 6 suffered from growth retardation, anemia, leukocytosis, thrombocytosis, coagulation abnormality, elevated serum levels of haptoglobin, ferritin, and heme, a low serum bilirubin concentration, and hyperlipidemia (Yachie et al., 1999; Kawashima et al., 2002). Autopsy revealed the presence of amyloid deposits, foamy macrophages, fatty streaks, and fibrous plaques. Interestingly, it was observed that endothelial ICAM-1 and Von Willebrand factor were highly increased in this patient, which supports our findings in mice (Wagener et al., 2001b; Kawashima et al., 2002).
These findings in human are paralleled by similar phenotypic data obtained from HO-1 targeted (HO-1 -/-) mice (Nath et al., 2001b; Poss and Tonegawa, 1997a,b). These knockout mice are characterized by anemia, tissue iron depositions, and severe inflammatory conditions, such as enlarged spleen and lymph nodes, hepatic inflammatory cell infiltrates, vasculitis, and glomerulonephritis (Poss and Tonegawa, 1997a). These mice are also extremely sensitive to oxidative injury and to endotoxin-mediated cell death (Poss and Tonegawa, 1997b). Furthermore, they express growth retardation and are prone to abortions (Poss and Tonegawa, 1997a).
HO-2-derived CO plays an important role in the neurotransmitter control and in activation of guanylyl cyclases, in conjunction with NO. Mice targeted in HO-2 demonstrate important abnormalities in the central and peripheral nervous systems as exemplified by interference with ejaculation (Burnett et al., 1998).
Overexpression of HO-1 can lead to hyperbilirubinemia in humans with certain hepatic disorders especially in patients in whom bilirubin disposition is impaired for developmental or genetic reasons, e.g., in newborns and in patients with the Crigler-Najjar Type I syndrome (Kappas et al., 1993). Drummond and colleagues have shown that pharmacological agents such as the inhibitor of HO, stannic mesoporphyrin, can inhibit HO activity significantly and is highly effective in a single dose in controlling hyperbilirubinemia (Martinez et al., 2001) (personal communication). However, such agents can exert only transient control of the activity of HO-1. Furthermore, the long-term overexpression or inhibition of different HO isoforms would have considerable experimental value in elucidating the role of the enzyme in physiological and pathological processes.
Quan et al. (2001) examined the feasibility of utilizing the retrovirus-mediated transfer of a human HO-1 (hHO-1) sense (S) and antisense (AS) orientation sequence under the control of the hHO-1 promoter to regulate endogenous HO-1 expression and function and thus permit development of gene transfer technology to regulate the rate of heme catabolism over the long term (Quan et al., 2001). Their data demonstrate for the first time that selective delivery of the hHO-1 gene in antisense orientation into human endothelial cells results in an attenuation of hHO-1 protein leading to a decrease in the rate of catabolism of cellular heme and that this effect is brought about without altering endogenous HO-2 protein.
Thus by using retroviral HO-1 antisense methodology, it is possible to envisage prolonged down-regulation of the rate of heme catabolism to its degradation product bilirubin in clinical or experimental circumstances where this might prove useful. Alternatively, inhibition of HO isoforms may be established using RNA interference techniques.
In contrast, the development of gene transfer techniques has provided the opportunity to deliver a functional HO-1 gene and to evaluate the direct effects of this gene on, e.g., vascular functions.
Using a retroviral vector, Sabaawy et al. (2001) established chimeric rats expressing the human HO-1 gene to study hypertension. Surprisingly, they observed that these rats, in addition to developing decreased blood pressure, grew significantly faster than rats lacking the hHO-1 gene, particularly during the first 12 weeks. Importantly, the increase in somatic growth associated with hHO-1 expression in rats was both proportionate and not associated with an increase in food intake. Interestingly, both human (Yachie et al., 1999) and mice (Poss and Tonegawa, 1997a) lacking the HO-1 gene display severe growth retardation.
An alternative means of establishing HO isoform overexpression is the use of TAT-HO proteins. The fusion protein of HO-1 to an 11-amino acid cell-penetrating peptide from the human immunodeficiency virus TAT protein enables efficient HO-1 protein entry and proved to protect pancreatic β-cells against TNF-α-mediated injury (Ribeiro et al., 2003).
The importance of a fine balance of the heme-HO system is further underscored by accumulating data demonstrating that induction of HO-1 by gene transfer protects cells from hemoglobin/heme-mediated oxidative stress, leukocyte infiltration, and against a range of inflammatory complications, such as ischemia/reperfusion injury and hyperoxia-induced injury (Abraham et al., 1995b; Amersi et al., 1999; Yang et al., 1999). Moreover, it has recently been found that HO-1 gene transfer protects blood vessels from pathological vasoconstriction and excessive smooth muscle cell proliferation in animal models of arterial injury (Duckers et al., 2001). Furthermore, HO-1 transgenic mice are protected from hypoxia-induced inflammation and vascular dysfunction (Minamino et al., 2001).
Interestingly, HO-1 has been shown to interact with cellular and synthetic proteins, which may modulate the activity of the enzyme. Interactions with amyloid precursor proteins, which are implicated in the pathogenesis of Alzheimer disease, have been demonstrated to inhibit HO-1 activity, resulting in a compromised stress defense (Takahashi et al., 2000; Takahashi and Snyder, 2000; Wagener et al., 2003b). On the other hand, Buelow and colleagues have demonstrated that the synthetic peptide RDP1258 can bind HO-1 and cause increased HO-1 expression and activity in vivo (Cuturi et al., 1999; Magee et al., 1999). This protein has further shown to be highly effective in ameliorating a range of inflammatory conditions (Cuturi et al., 1999; Oberyszyn et al., 2001).
Thus, HO seems to be cytoprotective by limiting the influx of activated inflammatory cells and by ameliorating the extent of cellular necrosis and apoptosis in injured tissues. On the other hand, too high HO-1 expression may also cause tissue injury by the generation of high levels of iron or result in bilirubinemia (Suttner and Dennery, 1999).
V. The Heme-Heme Oxygenase System
The heme-heme oxygenase system is tightly involved in both physiological as well as pathophysiological processes, such as cytoprotection, apoptosis, and inflammation. A better understanding of the complex heme-heme oxygenase system may provide us with novel tools to combat diverse conditions, such as inflammation, wound healing, transplant rejection, sickle cell disease, and cancer (Lane, 1998; Maines, 2000; Wagener et al., 2003a).
A. The Heme-Heme Oxygenase-1 System and Apoptosis
Under oxidative and inflammatory conditions necrosis and apoptosis may occur, which result in the release of large amounts of heme and subsequently exacerbation of the inflammatory process and cellular injury. However, under these conditions the stress-responsive protein HO-1 will be immediately induced as well.
A growing body of evidence suggests that expression of HO-1 acts in an anti-apoptotic manner in several cells types. This was originally demonstrated by the observation that overexpression of HO-1 in endothelial cells protected these cells from TNF-α-mediated apoptosis (Soares et al., 1998). In a similar manner, overexpression of HO-1 has also been shown to protect fibroblasts from TNF-α-mediated apoptosis (Petrache et al., 2000). Subsequently, Ferris et al. (1999) showed that the antiapoptotic action of HO-1 was associated with the expression of an iron pump, which is responsible for the decrease of intracellular iron content. This may relate to the anti-apoptotic effect of HO-1 because by decreasing iron content cells would become less likely to generate ROS through the Fenton reaction. Given the critical role of ROS generation in the induction of signal transduction pathways that lead to apoptosis, cells that have decreased levels of ROS would become protected from undergoing apoptosis. These authors also suggested that CO does not mediate the anti-apoptotic effects of HO-1 (Ferris et al., 1999). This assumption was largely based on the observation that activation of guanylyl cyclase and/or generation of cGMP (which occur when CO is generated) does not protect cells from undergoing apoptosis (Ferris et al., 1999). Although this observation is also true in other cell types such as endothelial cells, recent work from Brouard et al. (2000) have shown that whereas the cGMP pathway is not involved in the antiapoptotic action of HO-1, CO can mediate the anti-apoptotic effect of HO-1 via the activation of the p38 MAPK signal transduction pathway in endothelial cells. It should be noted that, as for endothelial cells, CO can also mediate anti-apoptotic effects in fibroblasts. However, in fibroblasts the protective effect of CO occurs through a mechanism that is dependent on the activation of guanylyl cyclase and the generation of cGMP but independent of the activation of the p38 MAPK signal transduction pathway (Petrache et al., 2000). In other cells types, such as in β-cells of pancreatic islets, CO exerts anti-apoptotic effects through a mechanism that requires both the activation of the cGMP and the p38 MAPK signal transduction pathways.
These observations indicate that the anti-apoptotic effect of CO can be mediated through the modulation of different signal transduction pathways depending on the cell type. The mechanism by which HO-1 and/or CO act to protect cells from undergoing apoptosis is not yet clear. Recently, Brouard et al. (2002) have shown that the ability of HO-1 and/or CO to protect endothelial cells from TNF-α-mediated apoptosis is dependent on the activation of NF-κB. Activation of this transcription factor promotes the expression of a series of anti-apoptotic genes, e.g., c-IAP2 and A1, that interact functionally with CO to prevent endothelial cell apoptosis (Brouard et al., 2002). More recently Duckers et al. (2001) have shown that expression of HO-1 induces the expression of the cyclin-dependent kinase inhibitor p21Cip1/Waf in smooth muscle cells whereas Inguaggiato et al. (2001) have shown that the same phenomenon occurs in renal epithelial cells. Interestingly p21Cip1/Waf expression confers resistance to apoptosis in several cell types and promotes the activation of NF-κB (Polyak et al., 1996; Megyesi et al., 1998). This would indicate that the antiapoptotic effect of HO-1 and/or CO might be mediated through the expression of p21Cip1/Waf.
B. The Heme-Heme Oxygenase System and Transplantation Biology
The heme-HO system plays also a vital role in transplantation biology (Hancock et al., 1998; Sato et al., 2001). Graft rejection is a complex phenomenon that is associated with the induction of adhesion molecules in the graft endothelium, influx of immune cells, ischemia/reperfusion injury, and cell injury (Fuggle and Koo, 1998). Vascular dysfunction, immune cell influx, and arteriosclerosis are evident and ultimately result in organ rejection. It is tempting to speculate that the elevated release of denatured heme proteins, derived from injured cells, is of major importance in the initiation and progression of inflammation during the process of graft rejection.
Interestingly, in several transplantation models, HO-1 has been demonstrated to play an indispensable role in terms of promoting graft survival. Elevated HO activity prevents the development of vascular lesions, intragraft apoptosis, ischemia/reperfusion injury, and significantly prolongs allograft survival (Hancock et al., 1998; Soares et al., 1998; Pileggi et al., 2001; Tullius et al., 2001; Katori et al., 2002). Moreover, in a model of xeno-transplantation, expression of HO-1 (Soares et al., 1998) and generation of CO (Sato et al., 2001) have been shown to suppress graft rejection and promote long-term graft survival. Under a given immunosuppressive regimen that allows the survival of wild-type mouse hearts transplanted into rat, mouse hearts lacking HO-1 expression (HO-1 -/-) are rejected (Soares et al., 1998). Given the above, it is tempting to postulate that injury-derived heme recruits leukocytes and promotes inflammation during transplant rejection, whereas HO activity prevents heme-induced inflammatory and oxidative actions. Presumably this effect of HO-1 acts through a complex mechanism that involves the down-modulation of adhesion molecules, subsequently preventing leukocyte recruitment and organ failure.
The increasing usage of “marginal” organs for transplantation because of the organ shortage brings new problems into transplant medicine. Organs from elderly donors suffering from diseases such as hypertony, hyperlipidemia, stroke, etc. are very sensitive to ischemia/reperfusion injury resulting in a higher risk for developing chronic graft injury. Very recently we could show that donor pretreatment with cobalt protoporphyrin 6 to 24 h before organ harvesting up-regulates HO-1 in the grafts and prevents both short-term and long-term ischemia/reperfusion injury of kidney allografts in normal donors as well as marginal donors. The kidney allograft recipients showed less proteinuria and improved histology and graft function even 6 months post-treatment. Interestingly, monitoring of intragraft gene expression suggests that the resistance of parenchymal graft cells to inflammation might be more important for the observed protection than down-regulation of inflammation by intragraft HO-1 up-regulation (Tullius et al., 2002).
C. The Heme-Heme Oxygenase System As Target for Inflammatory Control
The ultimate goal of research aimed at understanding inflammation is to develop methods to control inflammation in situations that are harmful to patients. In patients with increased inflammatory activity (e.g., chronic inflammation, rheumatoid arthritis, graft rejection), amelioration of inflammation is required, whereas in patients with a compromised immune system (e.g., cancer, AIDS), stimulation of the immune system is needed. An important role for the heme-HO system in the control of inflammation by modulation of cell adhesion can be envisioned (Fig. 6).
D. Some Considerations Concerning Modulation of the Heme-Heme Oxygenase System
Novel strategies in the control of inflammation may involve systemic administration of antisense oligonucleotides or antibodies against adhesion molecules, or antioxidants (Haller et al., 1996; Menger and Vollmar, 1996). Targeted expression of the inflamed tissue remains, however, difficult. A better understanding of the heme-HO system may result in novel therapeutic strategies to inflammatory diseases. HO-1 induction using gene therapy or pharmacological modulation show promising results both in vitro and in vivo (Yang et al., 1999; Duckers et al., 2001). The use of tissue-specific promoters would enable the generation of local vasorelaxants and anti-oxidants at places prone to inflammatory or oxidative insults. Alternatively, administration of solely the HO effector molecules bilirubin or CO seem to possess therapeutical value as well (Otterbein et al., 1999b; Chapman et al., 2001; Dennery et al., 2001). Bilirubin administration has been demonstrated to alleviate adhesion molecule expression, leukocyte binding, and to provide neuronal protection (Vachharajani et al., 2000). Furthermore, exposure to CO, or a synthetic precursor molecule of CO such as methylene chloride, prolongs allograft survival significantly.
It is important to take into account that the heme-HO system has regulatory functions in a wide variety of processes. Heme is the crucial part of many major enzymatic systems, such as nitric-oxide synthase, cytochrome P450, and COX (Maines, 1997). HO activity controls the activity of these heme proteins in at least two ways: directly, by regulating the availability of heme for the synthesis of heme proteins, or indirectly, via the generation of CO, which can bind to the heme moiety of heme proteins and thereby affecting their enzymatic activity (Fig. 6) (Haider et al., 2002).
Although HO-1 and its effector molecules, CO, biliverdin/bilirubin, and iron-ferritin, are nowadays recognized as cytoprotective agents, many of the effects of HO-1 are highly dependent on the redox state of the cell. Data from several laboratories demonstrated that directly after inducing HO-1, cells are sensitized to oxidative stress, whereas after prolonged time, ferritin is upregulated, and these cells are desensitized and significantly protected against a wide range of stresses (Balla et al., 1993; da Silva et al., 1996; Ryter and Tyrrell, 2000). It is evident that each of the heme breakdown products, when present in high quantities can mediate cell injury instead of cytoprotection (Fig. 3A) as described under Section III.C. (Suttner and Dennery, 1999). Whether the fine balance of the heme-HO-1 system mediates either cytoprotection or cytotoxicity is largely dependent on the microenvironmental circumstances. The sum of the anti-oxidative, pro-oxidative, and inflammatory signals within the surrounding tissue will probably determine the outcome. The understanding that the heme-HO system exerts a wide variety of functions, besides inflammatory control, is important when modulation of the system is considered as a potential therapeutic strategy.
VI. Conclusion
The heme-HO system plays a crucial role in the control of inflammatory processes, probably in part through its effects on cellular adhesion and migration. Besides the observation that the heme-HO system provides a promising target for new treatment possibilities, it offers a novel view on the etiology of diverse physiological as well as pathophysiological processes.
Acknowledgments
Acknowledgments. Preparation of this review and the work from the authors' laboratories were supported in part by European Commission Grant HO-1 QLK3-CT-2001-00422, the Roche Organ Transplantation Research Foundation (998521355), National Institutes of Health Grant HL67040, and the Vanderes Foundation.
Footnotes
-
↵1 Heme is ferroprotoporphyrin IX; hemin is ferric protoporphyrin IX. In this review, the term heme is used as a generic expression denoting no particular iron valence state.
-
↵2 Abbreviations: HO, heme oxygenase; AP-1, activator protein-1; COX, cyclooxygenase; HBP, heme-binding protein; hHO-1, human HO-1; HRE, heme-responsive element; ICAM, intercellular adhesion molecule; iNOS, inducible nitric-oxide synthase; MAPK, mitogen-activated protein kinase; MARE, Maf recognition element; MCP-1, monocyte chemoattractant protein 1; NF-κB, nuclear factor-κB; NO, nitric oxide; NOS, nitric-oxide synthase; RES, reticuloendothelial system; RNS, reactive nitrogen species; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; VCAM, vascular cell adhesion molecule.
-
Article, publication date, and citation information can be found at http://pharmrev.aspetjournals.org.
-
DOI: 10.1124/pr.55.3.5.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}