


Abstract
The amphetamine derivative (±)-3,4-methylenedioxymethamphetamine (MDMA, ecstasy) is a popular recreational drug among young people, particularly those involved in the dance culture. MDMA produces an acute, rapid enhancement in the release of both serotonin (5-HT) and dopamine from nerve endings in the brains of experimental animals. It produces increased locomotor activity and the serotonin behavioral syndrome in rats. Crucially, it produces dose-dependent hyperthermia that is potentially fatal in rodents, primates, and humans. Some recovery of 5-HT stores can be seen within 24 h of MDMA administration. However, cerebral 5-HT concentrations then decline due to specific neurotoxic damage to 5-HT nerve endings in the forebrain. This neurodegeneration, which has been demonstrated both biochemically and histologically, lasts for months in rats and years in primates. In general, other neurotransmitters appear unaffected. In contrast, MDMA produces a selective long-term loss of dopamine nerve endings in mice. Studies on the mechanisms involved in the neurotoxicity in both rats and mice implicate the formation of tissue-damaging free radicals. Increased free radical formation may result from the further breakdown of MDMA metabolic products. Evidence for the occurrence of MDMA-induced neurotoxic damage in human users remains equivocal, although some biochemical and functional data suggest that damage may occur in the brains of heavy users. There is also some evidence for long-term physiological and psychological changes occurring in human recreational users. However, such evidence is complicated by the lack of knowledge of doses ingested and the fact that many subjects studied are or have been poly-drug users.
I. Introduction
3,4-Methylenedioxymethamphetamine (MDMA2; ecstasy) is a ring-substituted amphetamine derivative that is also structurally related to the hallucinogenic compound mescaline (Fig. 1). MDMA has often been said to have been originally patented for use as an appetite suppressant, but Cohen (1998) reported that it was actually first patented in Germany in 1914 as a precursor agent for therapeutically active compounds and was never intended for use as an anorectic drug. The toxicology of MDMA was first examined in the 1950s, together with other mescaline analogs, by the U.S. military, presumably as part of a chemical warfare program (Hardman et al., 1973).
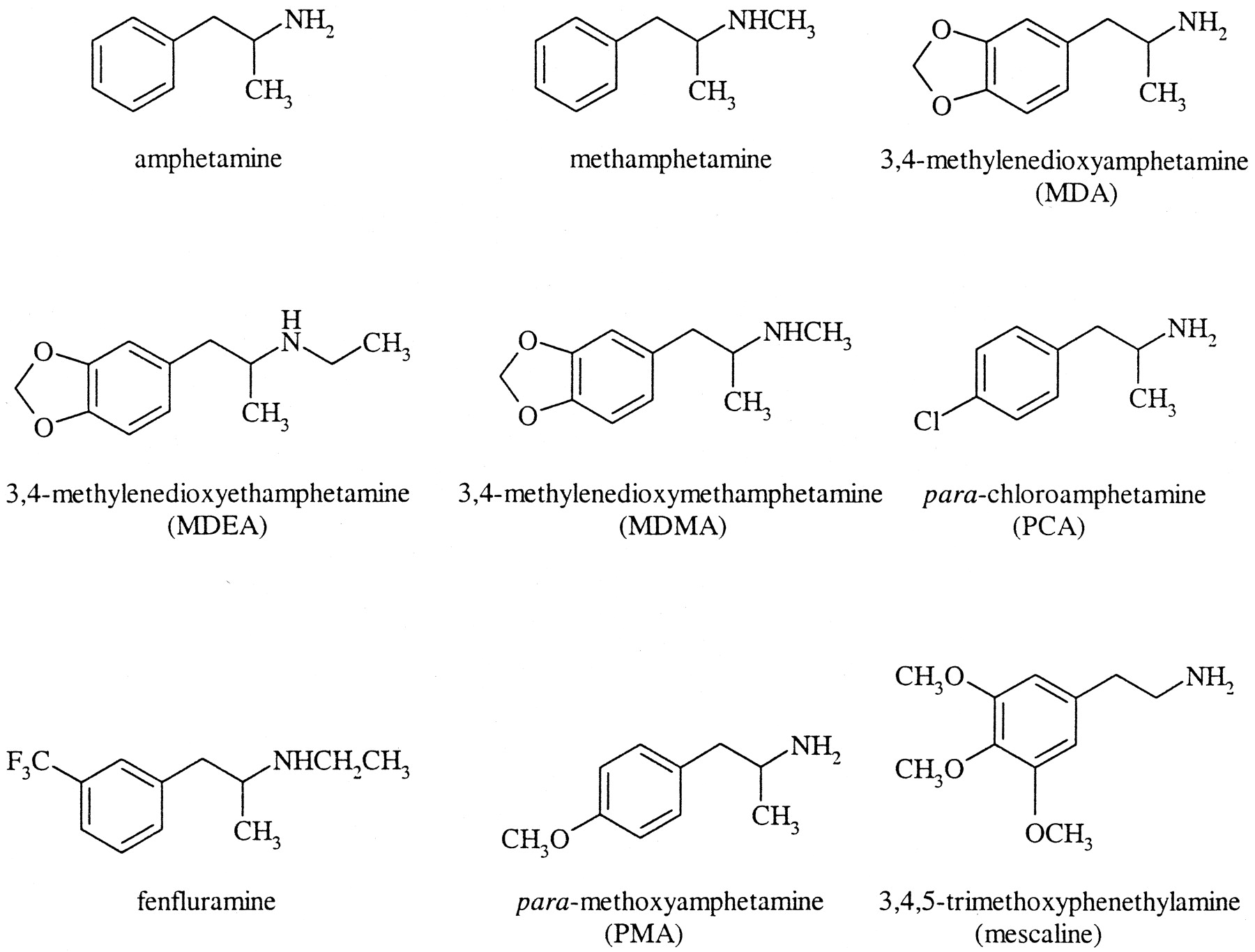
Chemical structures of amphetamine and some of its derivatives, including MDMA and mescaline.
The first report that MDMA was psychoactive in humans appears to be the report of Shulgin and Nichols (1978), although this paper does not describe the effects encountered. In the 1980s, MDMA started to be used in psychotherapy and was said to increase patient self-esteem and facilitate therapeutic communication. In such settings it was administered orally (75-175 mg) and noted to produce acute sympathomimetic effects, such as increased heart rate and blood pressure, and transient anxiety (Greer and Strassman, 1985; Grinspoon and Bakalar, 1986).
In 1985, the U.S. Drug Enforcement Administration classified MDMA as a Schedule 1 drug due to its high abuse potential, lack of clinical application, lack of accepted safety for use under medical supervision (www.usdoj.gov/dea) and evidence that 3,4-methylenedioxyamphetamine (MDA), a related compound and major MDMA metabolite, induced serotonergic nerve terminal degeneration in rat brain (Ricaurte et al., 1985). Possession of MDMA is also illegal in the United Kingdom, it being controlled as a Class A drug under the Misuse of Drugs Act (1971). Nevertheless, since the mid 1980s it has become popular as a recreational drug, often being taken at “rave” or “techno” parties, particularly in large dance clubs. “Raves” comprise heavily mixed, electronically generated sound and computer-generated video and laser light shows, where individuals are able to dance all night.
Ecstasy comes in a variety of colors, shapes, and sizes of tablet, which are decorated with a wide variety of designs or logos and may also be available in capsule form (see www.drugscope.org.uk; www.ecstasy.org; www.erowid.org; www.thesite.org). As with any illicitly prepared and obtained recreational drug, both doses and purity vary greatly (Ziporyn, 1986), but tablets have regularly been found to contain between 80 and 150 mg of MDMA.
The onset of effects can take 20 to 60 min to occur, the peak occurring 60 to 90 min after ingestion, and the primary effects last for 3 to 5 h. MDMA usually produces a relaxed, euphoric state, including emotional openness, empathy, reduction of negative thoughts, and a decrease in inhibitions (Peroutka et al., 1988; Davison and Parrott, 1997; Parrott and Stuart, 1997; Hegadoren et al., 1999; Liechti and Vollenweider, 2000b; Morgan, 2000). Sounds and colors can also appear more intense (see Davison and Parrott, 1997). Accompanying physiological changes can result in severe adverse events (see below).
II. Epidemiological Studies on the Use of MDMA
A series of studies on use patterns of MDMA have been conducted. Such studies have usually taken the form of questionnaires or interviews, and subjects may have been selected by being known drug users, being randomly selected from a particular population, or being recruited via advertisements.
Williamson et al. (1997) studied 158 known current drug users (average age 30 years, 62% male, 93% white, 76% unemployed) in the United Kingdom. Over half the subjects had used one or more illicit drugs (MDMA, cocaine, or amphetamines) during the past year, 82% of subjects using MDMA within this time, taking an average of 2 tablets on each occasion.
Solowij et al. (1992) recruited 100 subjects in Sydney, Australia, to assess the extent of recreational use of MDMA. Subjects were aged 16 to 48 years (male: 61%); 68% of subjects had used MDMA more than three times and the longest duration of use was more than 5 years. Approximately one-third of subjects reported using MDMA between once a month and once every three months, while 18% used MDMA mainly on “special occasions.”
Peroutka (1987) studied a randomly selected group of 369 U.S. university undergraduates and reported that 39% had used MDMA at least once (range: 1-38). Webb et al. (1996) performed a similar study of 3075 British 2nd-year undergraduate students (average age: 20 years) from 9 different faculties in 10 universities. Approximately equal numbers of male and female subjects from a cross-section of ethnic origins and religions took part; 5.2% of subjects had used MDMA more than once or twice, and 2.7% used MDMA at least once per week. In 1998 the National Institute of Alcohol and Drugs Research in Norway reported that 4.8% of people aged 15 to 20 years in Oslo had used MDMA at least once (Christophersen, 2000), while the estimated nationwide use of MDMA was 2.6% compared to 0.3% in 1994 (Mørland, 2000).
The U.S. National Institute on Drug Abuse publishes annual results of the “Monitoring the Future Study,” conducted at the University of Michigan's Institute for Social Research. This study examines trends in drug abuse within different populations of individuals—school children, college students, and adults aged 19 to 40. In 2001, 44,346 school children completed the survey, being recruited from 424 schools across the United States, and including 8th-, 10th-, and 12th-grade students (aged 13-14, 15-16, and 17-18 years, respectively). The use of any illicit drug, at least once during a subject's lifetime, was 26.8, 45.6, and 53.9% by 8th-, 10th-, and 12th-grade students, respectively, and the use of MDMA at least once in an individual's lifetime was reported to be 5.2, 8, and 11.7%, respectively. While the overall use of illicit drugs had marginally declined since 1999, the use of MDMA had increased in each age group; in 1999, MDMA had been used at least once by 2.7, 6, and 8% of 8th-, 10th-, and 12th-grade students, respectively.
Similar trends were observed in college students (aged 19-22) and all young adults (aged 19-28). In both of these populations there has been little change in lifetime use of any illicit drug over the past ten years. For example, illicit drug use by college students has ranged from 45.5% in 1994 and 1995 to 53.7% in 2000, while use by all young adults has ranged from 56.4% in 1996 to 62.2% in 1991 (use was reported to be 58.1% in 2001). In contrast, use of MDMA at least once in an individual's lifetime has increased dramatically from 2 and 3.2% in 1991, by college students and all young adults, respectively, to 14.7 and 13% in 2001 (NIDA, 2002).
In a recent UK study aimed to “generate information on patterns and trends among regular recreational drug consumers,” 1151 subjects were recruited via advertisements in a popular dance music magazine (60% male, average age 24 years). Ninety-six percent of subjects had used MDMA at least once, in addition to at least a single use of amphetamines (92%), cannabis (91%), cocaine (75%), and LSD (71%). The average duration of MDMA use was 4 to 5 years, 8% of users having taken the drug for over 10 years; 58% of users bought 4 or fewer tablets on each occasion. Since the subjects were self-nominating, the sample could be subject to bias and not a representative sample of drug users associated with the dance music scene in general. The majority of subjects were poly-drug users and over 70% also reported “harmful” levels of alcohol consumption (Winstock et al., 2001).
The “UK Drug Situation 2000” report to the European Monitoring Centre for Drugs and Drug Addiction was recently published by DrugScope, a government-designated body for drugs information in the United Kingdom (www.drugscope.org.uk). This reported that in England and Wales approximately one-third of adults aged 16 to 59 had used illicit drugs at least once in their lifetime. While cocaine use is on the increase, MDMA and amphetamine use has leveled off and there are indications that use is declining, particularly among individuals under age 20. MDMA use has been reported by approximately 10% of individuals in this age group. In the United States, in contrast, ecstasy use may be increasing. A very recent study on ecstasy use and related behavior in a group of over 14,000 college students found that use rose from 2.8% to 4.7% (an increase of 69%) between 1997 and 1999 (Strote et al., 2002).
All the foregoing indicates that MDMA use by young people is widespread; indeed, it has been estimated that in the United Kingdom alone 500,000 young people ingest the drug every weekend. Fatalities following ingestion of the drug are estimated to be approximately 12 persons per year.
III. Acute Effects of MDMA in Experimental Animals
A. Rats
1. Release and Depletion of Serotonin in the Brain.
MDMA administration to rats induces an acute and rapid release of 5-HT. This has been demonstrated using in vivo microdialysis (Gough et al., 1991; Yamamoto et al., 1995; Gudelsky and Nash, 1996; Sabol and Seiden, 1998; Shankaran and Gudelsky, 1999; Nixdorf et al., 2001; Mechan et al., 2002a) and is also reflected by the fact that the 5-HT concentration in brain tissue decreases markedly during the first few hours following drug administration (Schmidt et al., 1986; Stone et al., 1987a; Logan et al., 1988; McKenna and Peroutka, 1990; Gough et al., 1991; Colado and Green, 1994; Aguirre et al., 1995; Connor et al., 1998). For example, Gudelsky and Nash (1996) demonstrated a dose-related increase in extracellular 5-HT concentrations in the striatum and medial prefrontal cortex following peripheral administration of MDMA. 5-HT release in both the striatum (Gudelsky and Nash 1996) and hippocampus (Mechan et al., 2002a) is markedly attenuated by pretreatment with the serotonin uptake inhibitor, fluoxetine, indicating that MDMA-induced 5-HT release involves a carrier-mediated mechanism. Depletion of vesicular stores with reserpine also produces a significant attenuation of 5-HT release (Sabol and Seiden, 1998).
Acute 5-HT release has also been demonstrated in vitro following addition of MDMA to brain slices (Johnson et al., 1986; Schmidt et al., 1987; Schmidt, 1987b; Berger et al., 1992; Crespi et al., 1997; Koch and Galloway, 1997) or synaptosomal preparations (Berger et al., 1992; O'Loinsigh et al., 2001). Johnson et al. (1986) first demonstrated an acute release of [3H]5-HT from rat hippocampal slices by MDMA and reported that there was no significant difference in the releasing effects of the two MDMA enantiomers. Schmidt (1987b) demonstrated similar dose-dependent release of [3H]5-HT from rat striatal slices following superfusion with MDMA, MDA, or MDEA. At the highest concentration (10 μM), MDA was the most potent compound, followed by MDMA and MDEA. Berger et al. (1992) also examined the potencies of several compounds on [3H]5-HT release from synaptosomes. Dose-dependent release of [3H]5-HT was observed, with p-chloroamphetamine (PCA) and fenfluramine being the most potent (EC50 = 3 μM), MDMA slightly less so (EC50 = 8 μM), and methamphetamine being the least potent (EC50 = 23 μM). Fluoxetine significantly attenuated the [3H]5-HT-releasing actions of all four compounds (Berger et al., 1992). O'Loinsigh et al. (2001) recently reported that MDMA, MDA, and MDEA were equipotent at inducing a dose-dependent release of [3H]5-HT from frontal cortex/hippocampal synaptosomes, while 3,4-methylenedioxybutylamphetamine (MDBA) the N-butyl analog of MDMA, only induced significant release at a concentration of 100 μM.
2. Effect on Tryptophan Hydroxylase and Monoamine Oxidase.
It has been known for some time that the activity of tryptophan hydroxylase (TPH), the rate-limiting enzyme required for 5-HT synthesis, is inhibited by MDMA administration (Stone et al., 1987a,c; 1988; Schmidt and Taylor, 1988; Johnson et al., 1992; Che et al., 1995). Stone et al. (1987c) demonstrated that TPH activity started to decline in the neostriatum, frontal cortex, hippocampus, and hypothalamus within 15 min after MDMA administration. Inhibition of the enzyme has been reported to still be detectable more than 2 weeks following a single dose of MDMA (Schmidt and Taylor, 1987).
Depletion of central dopamine content by administration of α-methyl-p-tyrosine (AMPT) or reserpine, or by selectively destroying nigrostriatal dopamine projections with 6-hydroxydopamine, provides partial blockade of the MDMA-induced reduction of TPH activity (Stone et al., 1988). Although a single, direct, central injection of MDMA did not alter cortical or striatal TPH activity, a continuous i.c.v. infusion of MDMA for 1 h resulted in a significant reduction in TPH activity (Schmidt and Taylor, 1988). These data may indicate that the peripheral generation of an active metabolite is responsible for the acute neurochemical effects of MDMA, a proposal that is supported by the observation that MDMA has no inhibitory effect on the enzyme in vitro (Schmidt and Taylor, 1987). The possible involvement of calcium influx in MDMA-induced decreases in TPH activity has been demonstrated by pretreatment with flunarizine (thereby blocking calcium influx through non-NMDA calcium channels), which significantly attenuated the inhibitory effect of MDMA (Johnson et al., 1992). The fact that MDMA can be metabolized to a quinone led Rattray (1991) to suggest that the quinone could combine with sulfhydryl groups within the enzyme molecules leading to deactivation. This proposal is supported by the observation that enzyme activity can be restored by reduction with sulfhydryl reagents under anaerobic conditions (Stone et al., 1989).
The MDMA-induced decrease in TPH activity is influenced by body temperature. Che et al. (1995) demonstrated that MDMA administration at an ambient temperature (Ta) of 25°C produced a hyperthermic response, while administration at a Ta of 6°C produced a hypothermic response. A significant reduction in TPH activity was observed in the hippocampus, striatum, and frontal cortex of hyperthermic animals, whereas TPH activity was unaltered in hypothermic animals. This observation indicates the possible involvement of free radicals in the inactivation of the enzyme, since MDMA-induced free radical formation is enhanced by hyperthermia (Colado et al., 1999b).
In common with other amphetamine analogs, MDMA inhibits the catabolic enzyme monoamine oxidase (MAO). Potency was approximately 10 times greater at MAO-A (IC50 = 44 μM) than MAO-B in a rat brain homogenate preparation (Leonardi and Azmitia, 1994). Such inhibition reduces the metabolism of 5-HT and dopamine within the nerve terminal and therefore contributes to the increased release of active neurotransmitter by MDMA.
3. Release and Depletion of Dopamine in the Brain.
MDMA also rapidly increases dopamine release from cerebral tissue, as has been shown by both in vivo microdialysis (Yamamoto and Spanos, 1988; Gough et al., 1991; Nash and Brodkin, 1991; Nash and Yamamoto, 1992; Gudelsky et al., 1994; Yamamoto et al., 1995; Koch and Galloway, 1997; Sabol and Seiden, 1998; Colado et al., 1999a; Nixdorf et al., 2001) and by in vitro studies using tissue slices (Johnson et al., 1986; Schmidt, 1987b; Crespi et al., 1997). In vivo studies have generally found the striatal tissue concentration of dopamine to be raised and the metabolite concentration lowered in the first few hours after MDMA administration (Logan et al., 1988; Yamamoto and Spanos, 1988; Gough et al., 1991; Schmidt et al., 1991; Colado and Green, 1994).
Yamamoto and Spanos (1988) placed voltammetry electrodes in the caudate and nucleus (n.) accumbens to enable measurement of dopamine release in awake-behaving rats. Following peripheral administration of MDMA there was a dose-dependent release of dopamine in both brain areas, release being significantly greater in the caudate compared to the n. accumbens at the highest dose of MDMA, but of similar magnitude at the two lower doses. The peak release occurred within 120 min after drug administration and returned toward baseline values within 180 min. Colado et al. (1999a) administered MDMA to male Dark Agouti rats and, using in vivo microdialysis, demonstrated a rapid, significant increase in extracellular dopamine concentrations in the striatum, and a sustained depletion of DOPAC and HVA.
Although there is consistent evidence that 5-HT release induced by MDMA results from an interaction of MDMA with the 5-HT uptake carrier, since fluoxetine blocks MDMA-induced 5-HT release (Gudelsky and Nash 1996; Mechan et al., 2002a), the involvement of the dopamine uptake site in MDMA-induced dopamine release is controversial. When Nash and Brodkin (1991) infused MDMA directly into the brain they observed that the dopamine uptake inhibitor GBR 12909 antagonized the enhanced dopamine release. In addition, Koch and Galloway (1997) showed that GBR 12909 prevented MDMA-induced dopamine release using an in vitro brain slice preparation. In contrast, Mechan et al. (2002a), using an in vivo microdialysis technique and peripheral MDMA administration found that GBR 12909, far from inhibiting dopamine release, in fact produced a further increase in extracellular dopamine. This suggests that MDMA enters the dopamine terminal by diffusion, not the uptake carrier, a conclusion supported both by the fact that the dopamine uptake inhibitor mazindol fails to block the dopamine releasing actions of the MDMA-related compound methamphetamine (Marek et al., 1990) and evidence that MDMA can enter nerve-ending tissue by diffusion (Zaczek et al., 1990; O'Shea et al., 2001).
Hansen et al. (2002) demonstrated that multiple doses of MDMA resulted in a 35 to 55% reduction in [3H]dopamine uptake in synaptosomes prepared from treated animals 1 h post-administration, this effect being reversed by 24 h. These data are in contrast to the effects of methamphetamine, where a 70 to 80% reduction in plasmalemmal [3H]dopamine uptake has been reported 1 h post-administration and where a 60% reduction is still apparent at 24 h (Kokoshka et al., 1998). Binding of [3H](-)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane 1,5-naphthalenedisulfonate ([3H]WIN 35,428) to the dopamine transporter was only reduced by 10% following MDMA administration and persisted for at least 24 h. In vitro, incubation of striatal synaptosomes with MDMA also resulted in a 35-55% reduction in [3H]dopamine uptake, an effect which was prevented by pretreatment with two PKC inhibitors, S-2,6-diamino-N-[[(1-oxotridecyl)-2-piperidinyl]methyl]hexanamide dihydrochloride (NPC 15437) and 2-[1-3(aminopropyl)indol-3-yl]-3(1-methylindol-3-yl)maleimide acetate (Ro 31-7549), indicating the possible involvement of PKC activation in this response (Hansen et al., 2002). These data highlight some of the differences between the effects of MDMA and methamphetamine on dopaminergic systems.
The significant attenuation of MDMA-induced striatal dopamine release by pretreatment with fluoxetine suggests an involvement of 5-HT in the response, at least in this brain region (Koch and Galloway, 1997). Gudelsky et al. (1994) demonstrated that MDMA-induced release of striatal dopamine was significantly potentiated by pretreatment with either the 5-HT2 receptor agonist 2,5-dimethoxy-4-iodoamphetamine (DOI), or the nonselective 5-HT agonist, 5-methoxy-N,N-dimethyltryptamine (5-MeODMT). These data indicate that stimulation of 5-HT2 receptors enhances MDMA-induced dopamine release. 5-HT release was unaltered by pretreatment with either the noradrenaline uptake inhibitor, desipramine, or N-(2-chloroethyl)-N-ethyl-2-bromo benzylamine (DSP4), a compound that selectively depletes brain noradrenaline (Shankaran and Gudelsky, 1998). In contrast, dopamine release from the hippocampus was inhibited by both compounds, indicating that the MDMA-induced increase in hippocampal extracellular dopamine may result from dopamine release from noradrenergic nerve terminals. MDMA may therefore be taken up by the noradrenaline transporter into noradrenergic nerve terminals and increase efflux of cytosolic dopamine (Shankaran and Gudelsky, 1998).
Yamamoto et al. (1995) demonstrated a complete blockade and significant attenuation of MDMA-induced dopamine release in the substantia nigra and striatum, respectively, following central infusion of the 5-HT2A/2C receptor antagonist, ritanserin, indicating modulation of MDMA-induced dopamine release by 5-HT2A/2C receptors. In addition, MDMA administration decreased the extracellular GABA concentration in the substantia nigra, a change that was prevented by ritanserin. The authors suggested that MDMA-induced striatal dopamine release could be modulated through an interaction between 5-HT and GABA. Administration of tetrodotoxin attenuated MDMA-induced dopamine release, indicating that release is an impulse-mediated response (Yamamoto et al., 1995).
Nixdorf et al. (2001) demonstrated a significant potentiation of MDMA-induced striatal dopamine release following co-administration of malonate and suggested that augmentation of MDMA-induced transporter-mediated dopamine release might have resulted from either malonate-induced increases in intracellular calcium or intracellular sodium accumulation due to inhibition of sodium/potassium adenosine triphosphatase (Na/K ATPase). In addition, malonate-induced inhibition of energy production might have rendered dopaminergic nerve terminals vulnerable to MDMA.
Crespi et al. (1997) demonstrated acute [3H]dopamine release in striatal synaptosomes following incubation with amphetamine, PCA, MDMA, and fenfluramine (in descending order of potency), and showed the response to be calcium-dependent. In a similar type of study, O'Loinsigh et al. (2001) found the order of potency to be MDA > MDMA > MDEA > MDBA.
4. Release and Depletion of Norepinephrine in the Brain.
In vitro MDMA has been shown to induce the release of norepinephrine (NE) from brain tissue. Induction of both basal and stimulated [3H]NE release from preloaded rat brain slices was blocked by desipramine (Fitzgerald and Reid, 1990). In a synaptosomal preparation, MDMA induced NE release with similar potency to 5-HT and greater than that for DA (Rothman et al., 2001). However the effectiveness of MDMA on NE release in vivo is unclear in the absence of microdialysis studies. MDMA depresses the firing of noradrenergic neurons in the locus ceruleus (Piercey et al., 1990), but it is unclear whether this results from the local release of NE, direct activation of α2-autoreceptors, or indirect mediation via serotonergic mechanisms. In isolated rat atrial and rabbit perfused ear preparations MDMA induced NE release, causing a positive chronotropic effect and vasoconstriction, respectively, both effects being blocked by desipramine (Fitzgerald and Reid, 1994). Although cardiovascular effects of MDMA are also seen in humans (see Section V) these are mostly inhibited by prior administration of citalopram, suggesting that they are mediated predominantly via indirect serotonergic mechanisms (Liechti and Vollenweider, 2000a).
Following administration of a neurotoxic regimen of MDMA there is generally reported to be no long-term depletion of tissue NE levels in either rat or monkey (Battaglia et al., 1987; Slikker et al., 1988; Insel et al., 1989) and no change in density of catecholamine uptake sites labeled by [3H]mazindol (Battaglia et al., 1987, 1991). Using a more intensive MDMA regimen (20 mg/kg for 10 consecutive days), Mayerhofer et al. (2001) observed a significant depletion of both 5-HT and NE, but not DA, in the n. accumbens 4 weeks after the treatment.
5. Effects on Neurotransmitter Receptors and Transporters.
MDMA binds to all three presynaptic monoamine transporters, exhibiting highest affinity (submicromolar) for the 5-HT transporter. Affinities for the noradrenaline and dopamine transporters are at least 10-fold less (Steele et al., 1987; Battaglia et al., 1988). Binding at both the 5-HT and DA transporters is stereo-selective, the S-(+) isomer being the more potent, whereas no stereoselectivity is evident at the NE transporter (Steele et al., 1987).
Binding affinities for the classical neurotransmitter receptors can be divided into three groups on the basis of KDi values: 1 to 10 μM range for 5-HT2, α2-adrenergic, M1 muscarinic, and histamine H1 receptors; 10 to 100 μM range for M2 muscarinic, α1-adrenergic, β-adrenergic and 5-HT1 receptors; and above 100 μM for dopamine D1 and D2, opioid receptors, and benzodiazepine sites (Battaglia et al., 1988). The affinities of MDA are broadly comparable (within a factor of 2) to those of MDMA at these sites. Acute administration of MDMA to rats at doses of 10 to 20 mg/kg results in brain concentrations in the micromolar range (Battaglia et al., 1990; Esteban et al., 2001), so effects at the higher-affinity group of receptors may be pertinent to the psychotropic and neurotoxic actions of MDMA.
Affinity of MDMA at 5-HT2 receptors labeled by the agonist ligand [3H]1-(4-bromo-2,5-dimethoxyphenyl)-2-aminopropane (DOB) is more than 10 times greater than that indicated by antagonist radioligands (Lyon et al., 1987), suggesting an agonist role. This has been confirmed by the demonstration that MDMA induces phosphatidylinositol turnover in cells expressing 5-HT2A or 5-HT2C receptors. These responses are highly stereospecific, the R-(-) isomer exhibiting greater potency and efficacy at the 5-HT2A receptor than the S-(+) isomer, which has negligible efficacy, whereas the opposite isomerism applies at the 5-HT2C receptor (Nash et al., 1994). Agonism at the 5-HT2A receptor is associated with the hallucinogenic effects of substituted amphetamines and ergolines (Egan et al., 1998) and, although efficacy at the 5-HT2A receptor is low (21%), this is also true for LSD (Newton et al., 1996). However, the affinity of MDMA at the human 5-HT2A receptor is slightly less than that for the rat receptor (Sadzot et al., 1989), corresponding with the low incidence of hallucinations induced by MDMA in humans.
While the presence of the 3,4-methylenedioxy substituent increases the affinity of MDMA for serotonergic sites compared to the parent amphetamine, affinity for the α2-adrenergic receptor is correspondingly decreased. Blockade of central presynaptic α2-adrenergic receptors may account for the increase in both systolic and diastolic blood pressure caused by MDMA in humans (McCann et al., 1996) and since such receptors are located on some serotonergic terminals this may also contribute to the induction of 5-HT release. In the vas deferens, however, MDMA exhibits agonist effects similar to xylazine, reducing stimulus-evoked contractions (Rajamani et al., 2001).
In addition to these classical receptors, MDMA has recently been reported to possess high affinity (EC50 = 1.7 μM) and efficacy for a novel receptor that is positively coupled to adenylyl cyclase, for which the endogenous agonist may be a trace amine such as tyramine (Bunzow et al., 2001). Unlike the normal monoamine receptors, this receptor is located within the cell cytosol, possibly on vesicular membranes (Borowsky et al., 2001). Since MDMA is rapidly transported into and concentrated within the serotonergic terminal, it may be expected to express significant intrinsic activity at this receptor. However, the level of expression of this receptor in rat brain is low, so its relevance to the psychotropic actions of MDMA is unclear.
6. Induction of Immediate Early Genes.
The regional expression of immediate early genes (IEGs) such as Fos, in response to neurochemical stimulation, provides a means of mapping neuronal activation (Hughes and Dragunow, 1995). MDMA induces localized but widespread induction of c-fos mRNA and Fos protein in rat brain, particularly throughout the cerebral cortex, striatum, lateral septum, n. accumbens, amygdala, and paraventricular nucleus of the thalamus (Hashimoto et al., 1997; Erdtmann-Vourliotis et al., 1999; Stephenson et al., 1999). Colocalization studies indicated that in the striatum no neurons were double-labeled with Fos and parvalbumin or neuropeptide Y following MDMA (Dragunow et al., 1991) and in the raphe nuclei very few of the Fos-positive cells were serotonergic neurons as labeled with a 5-HT antibody (Stephenson et al., 1999). Similar patterns of Fos expression were seen following administration of fenfluramine or PCA but a differentially stronger effect of MDMA was noted in the n. accumbens, supraoptic hypothalamic nucleus, and dorsal raphe (Moorman and Leslie, 1996; Rouillard et al., 1996). Fos expression in the striatum and n. accumbens was inhibited by pretreatment with the NMDA antagonist MK-801 and the dopamine D1 antagonist SCH 23390, but not by fluoxetine (Dragunow et al., 1991; Hashimoto et al., 1997).
Induction of egr-1 mRNA, which is constitutively expressed at higher levels than c-fos in several brain regions, resulted in a similar pattern of expression by MDMA in prefrontal cortex and striatum but additionally in the dentate gyrus of the hippocampus. This response was inhibited by pretreatment with MK-801, SCH 23390 or paroxetine but not by the 5-HT2A receptor antagonist SR46349B (Shirayama et al., 2000). Arc mRNA, which is implicated in the development of synaptic plasticity (Steward and Worley, 2001), again generated a broadly similar pattern of expression by MDMA, but notably included the hippocampal CA1 region but not the dentate gyrus (Aston et al., 2002). Pretreatment with paroxetine inhibited arc mRNA expression in the frontal but not parietal cortex (Aston and Elliott, 2002).
The localized expression of IEGs induced by MDMA may be particularly useful in mapping brain areas associated with specific functional or behavioral effects, such as Fos induction in the pontine reticular nucleus oralis, an area concerned with the control of masticatory muscles, corresponding with the frequent observation of bruxism reported in subjects taking ecstasy (Stephenson et al., 1999). The differences in expression revealed by individual IEGs may suggest important differences associated with the functional role of the corresponding proteins. Pharmacological studies of the neurotransmitter control of IEG expression by MDMA implicate glutamate acting at NMDA receptors and dopamine at 5-HT receptors in the striatum and n. accumbens and serotonin at some, but not all, cortical sites. Further analysis of this type using more specific receptor antagonists should lead to a clearer understanding of the biochemical mechanisms and neuronal circuitry underlying both the acute and the neurotoxic effects of MDMA.
7. Effects on Free Radical Production in the Brain.
The first indication that neurotoxic damage produced by amphetamines results from free radical formation was the paper of Steranka and Rhind (1987), which reported that the free radical scavenger cysteine attenuated brain damage produced by administration of PCA and amphetamine. Sprague and Nichols (1995) subsequently showed that MDMA administration increased lipid peroxidation, a marker of free radical-induced damage. This finding was confirmed by Colado et al. (1997a), although they found that increased lipid peroxidation occurred much earlier following MDMA than Sprague and Nichols showed (1995). In 1995 it was also reported that administration of the nitrone radical trap α-phenyl-N-tert-butyl nitrone (PBN) attenuated MDMA-induced damage to cerebral 5-HT nerve endings (Colado and Green, 1995). PBN was further shown to lessen the damage produced by PCA, but not fenfluramine (Murray et al., 1996).
Direct evidence for MDMA administration increasing free radical formation in the brain was provided by Colado et al. (1997a). This group perfused salicylic acid through a microdialysis probe implanted in the hippocampus and demonstrated that peripheral MDMA injection increased the conversion of salicylate to 2,3-di-hydroxybenzoic acid (2,3-DHBA). Since this reaction only occurs in the presence of free radicals (Halliwell et al., 1991; Halliwell and Kaur, 1997), these data provided the first direct evidence for MDMA increasing free radical formation in the brain. This study also showed a similar increase in free radical formation following injection of PCA, but not fenfluramine. Administration of PBN inhibited free radical formation and attenuated neurotoxic damage and was shown to do so without altering MDMA-induced hyperthermia. The protective effect of PBN against MDMA-induced damage was confirmed by Yeh (1999).
The failure of fenfluramine administration to increase free radical formation is supported by the fact that PBN injection fails to provide protection against fenfluramine-induced damage to 5-HT nerve endings (Murray et al., 1996). It was suggested that fenfluramine, in contrast to MDMA and PCA was not metabolized to catechol or quinone compounds, which are capable of forming free radicals on further degradation. Although the mechanism of fenfluramine-induced neurotoxicity still appears uncertain, these data do indicate that one cannot extrapolate from the apparent clinical safety profile of fenfluramine to a projected safely profile for MDMA, as has sometimes been done (Saunders, 1996). In any event, the weakness of this argument is emphasized by the fact that fenfluramine has never been ingested in high recreational doses.
Other free radical scavenging drugs have also been found to protect against MDMA-induced damage. Gudelsky (1996) reported that administration of large doses of sodium ascorbate or L-cysteine prevented the long-term depletion of 5-HT induced by MDMA injection, and subsequently Shankaran et al. (2001) found that ascorbic acid administration suppressed the MDMA-induced formation of hydroxyl radicals, as indicated by the inhibition of 2,3-DHBA formation from salicylic acid in the striatum. MDMA also produced a significant reduction in vitamin E and ascorbate in the striatum and hippocampus. Aguirre et al. (1999) administered a high dose of the metabolic antioxidant α-lipoic acid before MDMA injection and found that it fully protected against damage to 5-HT nerve endings, again supporting the suggestion that free radical formation is responsible for MDMA-induced neurotoxicity. Shankaran et al. (1999b) using in vivo microdialysis observed that mazindol suppressed the MDMA-induced increase of both 2,3-DHBA and dopamine in the striatum and stated that this result supported the suggested role of extracellular dopamine in producing free radicals and neurotoxic damage. However, other recent data fail to support this contention (see below). Finally, Yeh (1997) reported that salicylate administration did not produce neuroprotection and suggested that MDMA-induced neurotoxicity might occur more through production of superoxides than hydroxyl radicals. However, these data are somewhat at variance with the other data presented above.
In a study demonstrating that clomethiazole did not act as a neuroprotective agent by a free radical scavenging action, Colado et al. (1999b) also observed that free radical formation was markedly inhibited when the MDMA-induced hyperthermic response was prevented. This result provides a plausible explanation as to why hypothermia or normothermia is neuroprotective against MDMA-induced damage and is perhaps analogous to the observation that hypothermia is neuroprotective against ischemia-induced damage and also attenuates free radical production (Globus et al., 1995; Kil et al., 1996).
Finally, the fact that a prior 5-HT lesion (produced by pretreatment with fenfluramine) prevented the MDMA-induced rise in free radical formation, as measured by the conversion of salicylate to 2,3-DHBA in a hippocampal probe, suggests that the 5-HT nerve endings are the site of the enhanced free radical formation (Colado et al., 1997a). This proposal was supported by the subsequent study by Shankaran et al. (1999a), who found that MDMA-induced free radical production was attenuated by fluoxetine, which indicates that free radical production occurs following activation of the 5-HT transporter.
8. Neuroendocrine and Immune Responses.
Administration of MDMA produces a significant elevation of rat serum corticosterone and prolactin concentrations 30 min post-injection (Nash et al., 1988). Serum corticosterone concentration remains elevated for over 4 h, whereas the peak prolactin response occurs at 60 min and concentrations return to control values by 4 h. Although the increase in serum corticosterone concentration was dose-dependent, such a relationship was not apparent with the prolactin response. Ketanserin, mianserin, or fluoxetine administration all attenuated the MDMA-induced increase in corticosterone but not prolactin, which indicates that MDMA-induced corticosterone secretion, at least, is mediated by serotonergic systems.
Aldosterone and renin secretion have also been shown to increase following MDMA administration to rats, and in vitro studies using adrenal capsules suggested that this effect was the result of MDMA increasing aldosterone secretion by potentiating the action of 5-HT on secretion (Burns et al., 1996). In vitro studies using isolated hypothalamic tissue have demonstrated that MDMA and some of its metabolites can stimulate release of both oxytocin and vasopressin, the response being dose-dependent (Forsling et al., 2001; 2002).
An MDMA-induced alteration in immune function has been reported by Connor et al. (1998), who measured brain monoamine concentrations, serum corticosterone levels, total leukocyte counts, and concanavalin A-induced lymphocyte proliferation, 30 min and 6 h following MDMA administration. Serum corticosterone levels were significantly increased 30 min post-injection and had returned to control levels within 6 h. Total leukocyte counts were reduced by approximately 50% at 30 min and 6 h post-treatment, as was concanavalin A-induced lymphocyte proliferation. Thus, acute MDMA administration produced a rapid, sustained suppression of mitogen-stimulated lymphocyte proliferation and total leukocyte count and, therefore, a suppression of immune function. These changes suggest that recreational users of MDMA may be subject to a reduced immunocompetence. A subsequent study (Connor et al., 1999) demonstrated that mitogen-induced lymphocyte proliferation was suppressed at doses of MDMA that did not alter serotonergic function. However, MDMA-induced reductions in circulating lymphocyte numbers were only apparent at doses that caused an increase in serotonergic activity and plasma corticosterone levels. MDMA-induced alterations in lymphocyte functional activity might therefore be occurring via a glucocorticoid-independent mechanism, while reductions in circulating lymphocytes could be a glucocorticoid-mediated event.
9. Cardiovascular and Sympathetic Effects.
While clinical reports have linked MDMA use with cardiovascular toxicity, cardiovascular and sympathetic nerve responses in rats are still being characterized. MDMA was shown to produce a range of effects on cardiovascular function in the rat some time ago when Gordon et al. (1991) reported that the compound had cardiac stimulant effects, resulting in tachycardia and arrhythmia. The compound also facilitates vasoconstriction (Fitzgerald and Reid, 1994).
O'Cain et al. (2000) recently reported that MDMA (0.01-3 mg/kg i.v.) produced a dose-dependent increase in mean arterial pressure, significant bradycardia following administration of the highest dose of drug, and a significant decrease in renal sympathetic nerve activity. The increases in mean arterial pressure are consistent with reported increases in arterial pressure in humans following MDMA ingestion (e.g., Vollenweider et al., 1998), while bradycardia may have been due to pressor-mediated baroreceptor reflex activation, and the observed inhibition of sympathetic nerve activity could have been due to an action on medullary α2-adrenergic receptors (O'Cain et al., 2000). Repeated frequent administration of MDMA to rats followed by a period of abstinence (binge administration) appears to be particularly effective in altering cardiovascular function and inducing cardiac toxicity (Badon et al., 2002).
MDMA can displace noradrenaline from adrenergic nerve terminals (Fitzgerald and Reid, 1993; Lavelle et al., 1999) and appears to have direct α2-adrenoceptor-mediated actions both in the periphery (Lavelle et al., 1999) and at central α2-adrenoceptors mediating depressor responses (McDaid and Docherty, 2001). Rajamani et al. (2001) provided further evidence for the drug having potency at α2-, α2AD-, and α2C-adrenoceptor subtypes. Data suggest that MDMA may competitively block the noradrenaline transporter (Al-Sahli et al., 2001). In contrast, MDMA does not appear to significantly alter 5-HT-induced aortic contraction (Cannon et al., 2001; Murphy et al., 2002). However both 4-methylthioamphetamine and 4-methylthiomethamphetamine are potent inhibitors of 5-HT-mediated vascular contraction (Murphy et al., 2002).
The first study on the effect of MDMA on glucose utilization was that of Wilkerson and London (1989) who observed significant effects in several brain regions within 5 min of drug administration. Marked stimulation was seen in areas of the extrapyramidal motor system, while parts of the limbic system showed decrements. Some of these effects on glucose utilization in the brain resembled changes seen after cocaine, amphetamine, and phencyclidine administration. Recently, Quate et al. (2003) examined the effect of MDMA on intracerebral blood flow and intracerebral glucose utilization in Dark Agouti rats and obtained similar results. MDMA resulted in an increase in glucose utilization in many brain regions, particularly areas concerned with the motor system, together with decreases in blood flow in regions such as the limbic and primary sensory nuclei, thereby indicating an uncoupling of blood flow from metabolic demand. Darvesh et al. (2002) found that the glucose concentration increased following MDMA administration and demonstrated that this increase was linked to an increase in glycogenolysis, which in turn appeared to be linked to the MDMA-induced hyperthermia. The authors speculated that the altered cellular bioenergetics might be associated with the oxidative stress and subsequent neurotoxicity.
10. Body Temperature.
a. Effect on Body Temperature.
Under “normal” Ta conditions (20-22°C), MDMA administration to rats has generally been reported to produce a marked hyperthermic response of approximately +1-2°C, with a peak at about 40 to 60 min post-injection (Nash et al., 1988; Schmidt et al., 1990a; Colado et al., 1993; Dafters, 1994; Broening et al., 1995; Che et al., 1995; Malberg et al., 1996; O'Shea et al., 1998). However, an acute decrease in temperature has also been reported in a few studies. Marston et al. (1999) reported a hypothermic response in Hooded Lister rats, and Malberg and Seiden (1998) demonstrated a hypothermic response in Holtzman rats at a Ta of 20-22°C, no change from control animals at a Ta of 24-26°C, and a hyperthermic response at a Ta of 28-30°C.
The influence of ambient temperature on the effect of MDMA on body temperature seen by Malberg and Seiden (1998) has been observed by others. For example, Broening et al. (1995) administered MDMA to female Sprague-Dawley (SD) rats under Ta conditions of 10, 25, and 33°C on postnatal days (PND) 10, 40, and 70. There was no clear temperature response to MDMA administration in the PND-10 group under any of the temperature conditions. However, both PND -40 and -70 animals demonstrated a hypothermic response at a Ta of 10°C, and an acute hyperthermia following MDMA administered at a Ta of 25°C or 33°C. Dafters (1994) administered MDMA to male Wistar rats housed under Ta conditions of either 11 or 24°C. At a Ta of 11°C there was a dose-dependent hypothermic response, while at a Ta of 24°C a dose-dependent hyperthermic response was seen. When rats were administered MDMA under Ta conditions of 24°C, and subsequently transferred to a “cool” room (Ta = 11°C), their hyperthermic response was significantly attenuated. In a subsequent study, Dafters and Lynch (1998) found that MDMA produced hyperthermia when given to rats in a 22°C environment and a hypothermic response when they were in a 17°C environment, indicating a high sensitivity to small changes in Ta.
Gordon et al. (1991) investigated the effects of MDMA on the thermoregulatory mechanisms of rats by monitoring metabolic rate (MR), evaporative water loss (EWL), and rectal temperature under three Ta conditions (10, 20, and 30°C). MR was significantly increased, compared to control animals, under Ta conditions of 20 and 30°C and was unchanged at 10°C. MDMA-treated rats demonstrated an increasing EWL with increasing Ta; EWL values in MDMA-treated rats were approximately 275% above control values at a Ta of 30°C. Rectal temperature increased with increasing Ta: hypothermia (-2°C) occurred at 10°C, while at 20°C there was no difference between MDMA- and saline-treated animals, and at 30°C hyperthermia was seen (+2°C). It therefore appears that MDMA administration has profound effects on the thermoregulatory system of the rat, involving increases in MR, EWL, and rectal temperature, and that such effects are apparently dependent on Ta. A recent study has further shown that the tail temperature of rats was unchanged following a hyperthermic dose of MDMA (Mechan et al., 2002a). Since vasodilation of the tail blood vessels is a major heat loss mechanism in rats (Grant, 1963), these data suggest that MDMA interferes with normal heat loss mechanisms, a proposal also advanced to explain the hyperthermic action of methamphetamine (Mohaghegh et al., 1997). Presumably, when the animal is kept in a low-temperature environment the loss of this mechanism is of little consequence and hyperthermia no longer occurs. Finally, Dafters (1995) also showed that 14-day administration of MDMA at a presumed non-neurotoxic dose resulted in an increase in peak temperature responses across the test days, indicating a sensitization effect.
b. Pharmacology of the Hyperthermic Response.
It is well established that hyperthermia can be produced by increasing 5-HT function by administering L-tryptophan plus an MAO inhibitor (Grahame-Smith, 1971a) or various 5-HT agonists such as 5-MeODMT (Grahame-Smith, 1971b), 6-chloro-2-(1-piperazinyl)pyrazine (MK212) (Yamawaki et al., 1983), or the 5-HT-releasing drug PCA (Colado et al., 1993). There has been an assumption, therefore, that the hyperthermia that follows MDMA administration is also 5-HT receptor-mediated (Shankaran and Gudelsky, 1999). However, methamphetamine-induced hyperthermia has been shown to involve dopamine release (Bronstein and Hong, 1995), which implies that dopamine could also be involved in MDMA-induced hyperthermia, given the fact that MDMA and methamphetamine release both 5-HT and dopamine.
A recent study has strongly supported the contention that MDMA-induced hyperthermia is a consequence of dopamine release. Methysergide, ritanserin, and selective 5-HT2A and 5-HT2C antagonists all failed to block MDMA-induced hyperthermia (Mechan et al., 2002a), and while MDL 11,939 did antagonize the hyperthermic effect, confirming an earlier report (Schmidt, 1987b), the authors suggested that this might be due to lack of receptor selectivity of this compound or its active metabolites. Crucially, it was shown that administration of the selective 5-HT uptake inhibitor fluoxetine almost totally inhibited the increase in extracellular 5-HT, as measured by in vivo microdialysis, but had no effect on the hyperthermic response in the same animals. This finding confirmed earlier studies that measured these two parameters in separate groups of animals (Schmidt et al., 1990a; Berger et al., 1992; Malberg et al., 1996). The separation of 5-HT release and hyperthermia strongly indicated that neurotransmitters other than 5-HT might be involved in the hyperthermic response. Furthermore, the observation that the dopamine D1 receptor antagonist SCH 23390 dose-dependently inhibited MDMA-induced hyperthermia leads to the conclusion that MDMA might be producing hyperthermia by enhancing the release of dopamine, which then acts on D1 receptors (Mechan et al., 2002a). Support for this proposal was supplied by another study published almost simultaneously which found that PCA-induced hyperthermia was also unaltered by fluoxetine or the 5-HT-depleting drug p-chlorophenylalanine (PCPA), but was antagonized by SCH 23390 (Sugimoto et al., 2001).
c. Aggregation Toxicity.
Over 60 years ago Gunn and Gurd (1940) reported that when mice were grouped or “aggregated” (as opposed to being housed singly), both the behavioral and toxic effects of amphetamine were enhanced. This observation was confirmed and extended by Chance, who also noted that toxicity was enhanced if mice were grouped even if each mouse was given the area allocated to a singly housed animal. He also noted that toxicity was increased by elevated ambient temperature, poor hydration, and loud noise (Chance, 1946, 1947; Morton et al., 2001).
Although the mechanism of toxicity has generally been assumed to be directly related to raised body temperature (Askew, 1961; Craig and Kupferberg, 1972), acute toxicity can occur without marked hyperthermia (Wolf and Bunce, 1973). However, the mechanism for the increased toxicity on exposure to loud noise is unknown.
While specific studies on aggregation toxicity have not been performed with MDMA, there is clear evidence that the phenomenon occurs when using this amphetamine derivative and indications are that the toxicity primarily relates to hyperthermia. Rats kept at elevated temperatures display a greater hyperthermic and neurotoxic response to MDMA (Dafters, 1995; Malberg and Seiden, 1998). Water deprivation also enhances these effects (Dafters, 1995), and Gordon and Fogelson (1994) demonstrated an enhanced hyperthermic response to MDMA when the cage construction failed to assist body heat loss (an acrylic floor rather than a grid). Such data suggest that the conditions at dance parties, where people are grouped and there is loud music, high ambient temperatures, and sometimes lack of availability of drinking water, could result in increased acute MDMA-induced adverse effects in comparison to ingestion in quiet surroundings.
11. Acute Behavioral Effects—The Serotonin Syndrome and Hyperactivity.
The “serotonin behavioral syndrome” was first described by Grahame-Smith (1971a) following administration to rats of an MAO inhibitor and L-tryptophan. Subsequent studies showed that the syndrome could also be produced by nonselective 5-HT agonists (Grahame-Smith, 1971b; Green and Grahame-Smith, 1976), the selective 5-HT1A agonist 8-OH-DPAT (Tricklebank et al., 1984; Goodwin and Green, 1985) and 5-HT releasing compounds such as PCA (Green and Kelly, 1976). The syndrome included hyperactivity, accompanied by head-weaving, piloerection, fore-paw treading, proptosis, penile erection, ejaculation, salivation, and defecation. Not surprisingly, therefore, given the evidence that MDMA administration results in a major release of 5-HT in several brain regions, this compound also produces an acute, dose-dependent, hyperlocomotor response (Slikker et al., 1989; Spanos and Yamamoto, 1989; Callaway et al., 1990; Colado et al., 1993; McNamara et al., 1995; De Souza et al., 1997) together with the appearance of all the major behavioral features of the serotonin syndrome (Slikker et al., 1989; Spanos and Yamamoto, 1989; Colado et al., 1993; De Souza et al., 1997; Marston et al., 1999; Shankaran and Gudelsky, 1999). Callaway et al. (1990) reported that MDMA produced a dose-related increase in locomotor activity that was prevented by pretreatment with fluoxetine, indicating that 5-HT release plays a key role in the behavioral effects of MDMA.
Kehne et al. (1996a) demonstrated a reduction of the MDMA-induced locomotor response following pretreatment with the 5-HT2A receptor antagonist MDL 100,907, while the increase in rearing behavior was unaffected. These data indicate the importance of 5-HT2A receptors in expression of MDMA-induced locomotor responses. McCreary et al. (1999) further showed that MDMA-induced hyperactivity was also blocked by pretreatment with the 5-HT1B/1D receptor antagonist GR 127935, but not the 5-HT1A antagonist N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-(2-pyridinyl) cyclohexane carboxamide (WAY 100,635), implicating the 5-HT1B receptor in the locomotor component of the behavior. More recently, Bankson and Cunningham (2002) provided evidence that MDMA-induced hyperactivity was potentiated by 5-HT2B/2C antagonism by use of 5-methyl-1-(3-pyridylcarbamoyl)-1,2,3,5-tetrahydropyrrol[2,3-f]indole (SB 206553), which indicates that the 5-HT2B/2C receptor might normally have an inhibiting influence.
Gold and Koob (1988) reported that MDMA-induced hyperactivity was enhanced by the nonselective 5-HT1/2 antagonist methysergide. This is not surprising given the earlier observation that the hyperactivity induced by administration of tranylcypromine and L-tryptophan is similarly enhanced (Green et al., 1981). It again indicates that 5-HT2 (probably 5-HT2C) receptors inhibit hyperactivity mediated through 5-HT and, probably, dopaminergic mechanisms.
The hyperactivity induced by MDMA is complex in neurochemical terms as there are undoubtedly both 5-HT and dopamine components. While amphetamine administration increased activity over the whole of an activity chamber, MDMA increased activity predominantly in the periphery of the apparatus (Gold et al., 1989; Callaway et al., 1990; McCreary et al., 1999).
Slikker et al. (1989) administered MDMA once daily for 4 days and assessed subsequent behavior. On the 1st day the serotonin motor syndrome score was significantly higher in MDMA-treated animals compared to controls. However, over the following 3 days the scores progressively decreased and were no different from control values by day 4. Although the mean locomotor activity score was greater in MDMA-treated animals on the first test day, this did not reach statistical significance and there was no difference between the groups on the subsequent test days.
12. Effects on Motor Function Tests.
Marston et al. (1999) assessed skilled motor function in male rats via a skilled paw reach (“staircase”) task. The test box comprised two staircases with six steps in each, situated opposite to one another, and the task involved the retrieval of food pellets from each step. MDMA was administered on three consecutive days and behavior monitored on these days and up to 15 days postdrug administration. Skilled paw-reaching behavior was significantly reduced in MDMA-treated rats during the drug administration period compared to control animals, indicating a perturbation of the motor control.
13. Anxiety-Related Behaviors.
Little work has been done on the acute effects of MDMA on the responses of rats in tests of anxiety-like behavior. Morley and McGregor (2000) examined rat behavior on the elevated plus maze and reported a dose-related decrease in the time spent on the open arms and the total number of arm entries, indicating an anxiogenic effect at the doses chosen (1.25-5 mg/kg). However, in the social interaction test MDMA (5 mg/kg) produced an apparent anxiolytic response. Bhattacharya et al. (1998) similarly found an anxiogenic effect of MDMA when using the plus maze test but also observed an anxiogenic effect of the drug when using the social interaction test.
14. Effects on Reinforcement and Self-Stimulation Behavior.
Hubner et al. (1988) used intracranial medial forebrain bundle self-stimulation, an animal model used to assess the abuse liability of drugs in humans, to test the effects of MDMA. MDMA administration resulted in a dose-dependent lowering of the reward threshold and self-stimulation, indicating that MDMA has effects on reinforcement behavior mediated by this brain region. This increase in self-stimulation has been shown to be blocked by pretreatment with naltrindole, which indicates that δ-opioidergic processes may be involved in the effect (Reid et al., 1996).
Lin et al. (1997) examined the acute effects of MDMA on n. accumbens self-stimulation in male Wistar rats. MDMA decreased total and maximal rate responding and frequency threshold, indicating an inhibitory effect of MDMA on operant behavior. Pretreatment with the 5-HT antagonist methysergide reversed the effects of MDMA, resulting in an increase in both total responding and maximal rate, without altering the threshold-lowering effects of MDMA. These results indicate a role for serotonin in the mediation of MDMA-induced effects on performance, but not the reinforcing effect of self-stimulation.
Byrne et al. (2000) tested the effects of MDMA administration on the acquisition of lever-pressing (reinforcement) behavior in SD rats. Animals demonstrated increasing rates of reinforcement lever-pressing over time, indicating response-acquisition, while increased delay of reinforcement led to decreased pressing of the reinforcement lever. MDMA treatment 15 min before the response-acquisition session resulted in a delayed response acquisition and increased the number of presses of the reinforcement lever under conditions of immediate reinforcement. Recently, Braida and Sala (2002) found that administration of a cannabinoid agonist reduced the number of MDMA-associated lever pressings in a self-administration test, suggesting a synergistic action of cannabinoids and MDMA.
15. Effects on Cognitive Behavior.
A more intricate version of the lever-pressing test is the delayed nonmatch to place (DNMTP) test, which provides a measure of cognitive ability. Marston et al. (1999) investigated DNMTP performance in rats administered different doses of MDMA. During the first 3 days of the testing period MDMA or saline was administered twice daily, the dose of MDMA being increased on each successive day. DNMTP performance was assessed during a 40-min period on each drug administration day, and then up to 18 days after the first drug administration. The total number of completed trials was significantly reduced in MDMA-treated rats on the first drug treatment day and the number of food responses was significantly lower on the first two drug treatment days. However, the accuracy of response could not be analyzed in MDMA-treated animals during the drug treatment period due to the low number of completed trials. The progressive improvement in DNMTP performance seen in control animals was not observed in the MDMA-treated group at the longer delay periods. The authors suggested that the behavioral effects observed could be primarily attributed to serotonergic nerve terminal dysfunction.
16. Effects on Startle Reflexes and Prepulse Inhibition.
Kehne et al. (1992) measured startle reflexes elicited by either acoustic or tactile stimulation. Rats were given MDMA and then exposed to 315 acoustic and 315 tactile stimuli over approximately 3.5 h. MDMA treatment resulted in enhanced acoustic and tactile startle reflexes, the peak excitatory effects occurring between 1 and 3 h post-injection. The 5-HT uptake blockers MDL 27,777A (2,3-dihydro-N-methyl-1-[4-(trifluoromethyl)phenoxyl]-7H-indene-2-methanamine hydrochloride) and fluoxetine significantly attenuated the excitatory effects of MDMA, as did 5,7-DHT-induced 5-HT depletion, leading the authors to conclude that the excitatory effects of MDMA on this behavioral phenomenon are mediated by the release of central 5-HT, particularly involving pathways arising from the dorsal raphe nuclei.
Prepulse inhibition (PPI), where the startle reflex is significantly reduced when the pulse is preceded by a weaker prepulse, has also been shown to be affected by MDMA administration. PPI provides a measure of sensorimotor gating and has been used in investigation of attentional deficits characteristic of schizophrenia and obsessive-compulsive disorder (see Vollenweider et al., 1999). Mansbach et al. (1989) demonstrated an attenuation of PPI following MDEA, while the effect of MDMA was similar but failed to reach statistical significance. Vollenweider et al. (1999) compared the effects of MDMA administration on PPI responses in rats and humans. MDMA had no effect on habituation of the startle response (reduction in response magnitude with successive trials) in either species. In rats, MDMA significantly reduced the percentage of PPI, whereas in humans the percentage of PPI was increased following MDMA administration. Several possible explanations were provided for this result, including potential experimental procedural differences, interspecies differences in the mechanism of action of MDMA, or different behavioral expression of a similar pharmacological effect.
B. Mice
1. Effects on Monoamine Biochemistry in the Brain.
In contrast to the substantial numbers of investigations on the pharmacological effects of MDMA in rats, rather few studies have been conducted into the effects of this compound in mice. Some early studies on the neurotoxic actions of MDMA in mouse brain demonstrated a very different profile to that seen in rats, namely long-term neurotoxic loss of striatal dopamine (Stone et al., 1987a; Logan et al., 1988; O'Callaghan and Miller, 1994). However, few further investigations were made until recently.
Three hours after the last of three doses of MDMA (given 3 h apart), O'Shea et al. (2001) observed a small decrease in 5-HT and 5-HIAA in cortex and hippocampus with little effect in the striatum. Stone et al. (1987a) had previously reported a slight and reversible depletion of both indoles in the striatum. This, of course, contrasts strongly with the marked acute effects of MDMA on 5-HT concentration in the rat.
MDMA also appears to have little effect on tryptophan hydroxylase activity in mouse brain. Stone et al. (1987a) found no inhibition following MDMA administration unless multiple doses of the drug were given.
With regard to dopamine biochemistry there is good evidence that MDMA administration produces an acute release of dopamine. The striatal content of both dopamine and its metabolites HVA and DOPAC is reduced 3 h after the last of three injections of MDMA (O'Shea et al., 2001). Furthermore, a recent study provided direct evidence for MDMA-induced dopamine release by using in vivo microdialysis, which confirmed that the extracellular dopamine concentration in the striatum increased after MDMA administration (Colado et al., 2001; Camarero et al., 2002). A single injection of MDMA only produced a modest rise in the extracellular dopamine concentration, but the rise was magnified and sustained by the two subsequent doses of MDMA (Colado et al., 2001; Camarero et al., 2002). Administration of the dopamine uptake inhibitor GBR 12909 enhanced the MDMA-induced increase in the extracellular dopamine concentration. This observation is identical to that seen by Mechan et al. (2002a) in rats and indicates that MDMA may enter the nerve terminal by diffusion and not via the dopamine uptake site (Camarero et al., 2002).
2. Effects on Free Radical Production in the Brain.
An indication that MDMA administration to mice increases free radical formation was given by the observation that transgenic mice with high superoxide dismutase (SOD) activity were resistant to the neurotoxic actions of MDMA (Cadet et al., 1995) and the fact that MDMA administration decreased glutathione peroxidase activity and increased lipid peroxidation in several brain regions (Jayanthi et al., 1999). Recently, direct evidence for an increase in free radical production in the brain following MDMA administration has been obtained. Two studies (Colado et al., 2001; Camarero et al., 2002) have shown an increase in 2,3-DHBA formation from salicylic acid perfused through a dialysis probe implanted in the striatum. The putative neuronal NOS inhibitor AR-R17477AR inhibited the MDMA-induced rise in free radical formation in vivo, indicating that MDMA or dopamine metabolite breakdown products were producing radicals that combine with nitric oxide to produce peroxynitrites (Colado et al., 2001; Camarero et al., 2002). Such data are in accord with the evidence that peroxynitrites are formed following neurotoxic doses of methamphetamine (Imam and Ali, 2000; Imam et al., 2001).
MDMA not only facilitates free radical generation, but also impairs endogenous antioxidant resources in the mouse brain. A reduction in vitamin E, total antioxidant reserve, and protein thiols is evident 72 h after MDMA dosing, a time coincident with the maximal neuronal damage (Johnson et al., 2002a). As a consequence, vitamin E-deficient mice show a greater susceptibility to MDMA-induced neurotoxicity to dopamine neurons than normal mice (Johnson et al., 2002a).
3. Effects on Body Temperature.
In general, MDMA administration produces a similar body temperature response in mice to that seen in other species, namely hyperthermia. However, the changes in body temperature seen by mice after MDMA administration at a room temperature of 20-22°C are much more variable than those observed in rats. Although MDMA has been reported to produce a hyperthermic response, this does not always occur and the response is dependent on both dose administered and the strain studied. Several groups have examined the response on temperature of female C57BL/6J mice after administration of MDMA (20 mg/kg s.c., 4 times, every 2 h) and found that MDMA causes an elevation of body temperature of approximately 2°C over the 8 h dosage period (Johnson et al., 2000, 2002b; Miller and O'Callaghan, 1994). In contrast, the same laboratory (Johnson et al., 2002a) using male BALB/c mice and lower doses of MDMA (5 and 10 mg/kg s.c. every 2 h for 4 doses) observed a dose-dependent hypothermic response that was still evident 24 h after administration of the higher dose studied. Carvalho et al. (2002) measured the subcutaneous temperature of male Charles River mice and reported that a single administration of MDMA (5, 10, and 20 mg/kg i.p.) produced an increase in body temperature that reached its maximum (2°C) at approximately 30 min and remained elevated for more than 4 h. Using Swiss-Webster mice, O'Shea et al. (2001) reported that repeated administration of MDMA (3 times at 3-h intervals i.p.) altered the body temperature biphasically in such a way that hypothermia was the predominant effect following MDMA at the dose of 10 mg/kg, while a higher dose (30 mg/kg) induced hyperthermia followed by hypothermia.
In contrast, the same group using male NIH/Swiss mice and given a similar protocol of MDMA (20-25 mg/kg i.p., 3 times at 3-h intervals) observed a pronounced hyperthermic response immediately after each injection lasting over 2 h, the magnitude of hyperthermic response being more pronounced after the first and second injection (Colado et al., 2001). Similar results have been observed in male C57BL/6J mice after receiving 15 mg/kg MDMA (3 times, once every 3 h) (Sanchez et al., 2003).
4. Effects on Locomotor Activity.
MDMA-induced locomotor responses in mice have been shown to be mediated, at least in part, by the 5-HT1B receptor. Scearce-Levie et al. (1999) administered MDMA (3.3-30 mg/kg) to wild-type and 5-HT1B-knockout mice before analysis of locomotor behavior in an open field arena. The lowest dose had no effect on locomotor activity in either group, whereas higher doses resulted in increased locomotor activity in wild-type mice. Only the highest dose produced an increase in locomotor activity in the knockout mice, although this response was delayed. The alterations in MDMA-induced locomotor behavior were confirmed to be due to the absence of the 5-HT1B receptor, since administration of the 5-HT1B/1D antagonist, GR 127935, blocked MDMA-induced locomotor stimulation in wild-type mice in a similar manner to that observed in the knockout mice.
5. Effect on Behavioral Tests.
Lin et al. (1999) reported dose-dependent effects of MDMA when mice were tested on an elevated plus maze 30 min later. Anxiogenic effects were observed at lower doses and anxiolytic effects at higher doses, as shown by changes in the percentage number of open arm entries and time spent on the open arms. Maldonado and Navarro (2001) conducted a study on social interaction behaviors between male mice 30 min after MDMA injection and found that MDMA-treated animals performed significantly less grooming, digging, social investigation, threat, and attack behaviors compared to control animals. Nonsocial exploration, defense/submission, stretched attend posture, and avoidance/flee behaviors were all increased in MDMA-treated mice. These behavioral changes are all indicative of anxiogenic-like activity being produced by MDMA in mice.
C. Nonhuman Primates
1. Effects in Psychological Tests.
Frederick and Paule (1997) reported on a series of behavioral tests performed on male rhesus monkeys commencing 30 min after MDMA (0.1-1 mg/kg i.m.). The behaviors assessed comprised performance in a monkey operant test battery. In the time estimation task, where the animals depress a lever for a defined time to receive food reward, MDMA administration prevented correct performance, the monkeys tending to press the lever rapidly rather than hold the lever down for the required time period. In a delayed-match-to-sample paradigm measuring short-term memory, MDMA administration was without significant effect. However, motivation to work for a food reward was highly sensitive to disruption by MDMA administration. In a learning test, again involving delivery of a food reward, MDMA significantly decreased response accuracy but did not affect the response rate, indicating that MDMA could disrupt processes associated with learning/acquisition of new information, but that retention of newly acquired information (short-term memory) was less sensitive to drug effects. Finally, color and position discrimination were not affected by MDMA. Thus, operant schedules, where correct performance is believed to be dependent on learning and time estimation capabilities, appeared to be more sensitive to the acute effects of MDMA than to tasks involving short-term memory and visual discrimination.
The reinforcing effect of MDMA has recently been investigated in rhesus monkeys by examining whether the animals would self-administer the drug. MDMA and its stereoisomers did serve as reinforcers, but resulted in a bell-shaped dose-response curve and the effect of MDMA was weaker than cocaine or methamphetamine. It was also antagonized by the 5-HT2A antagonists ketanserin and MDL 100,907, suggesting an integral role of this receptor in the response (Fantegrossi et al., 2002).
IV. Long-Term Effects (Neurotoxicity) in Experimental Animals
A. Rats
1. Evidence for Long-Term Serotonin Loss in Brain.
a. Biochemical Mechanisms.
There are more than 60 published reports on the fact that administration of single or multiple doses of MDMA to rats results in a long-term depletion of 5-HT and 5-HIAA (for example, Battaglia et al., 1987; Commins et al., 1987; Schmidt, 1987a; Stone et al., 1987c; Slikker et al., 1988; Laverty and Logan, 1990; McKenna and Peroutka, 1990; Nash and Yamamoto, 1992; Colado et al., 1993; Farfel and Seiden, 1995; Malberg et al., 1996; O'Shea et al., 1998; Shankaran and Gudelsky, 1998; Wallace et al., 2001). One factor that has to be borne in mind in evaluating these reports is that different strains of rats have been used by different investigators and the strains have different sensitivities to both the acute (Malpass et al., 1999) and the long-term neurotoxic effects of MDMA. That is, the dose required to induce neurotoxicity is strain-dependent. The most obvious example is the Dark Agouti strain, which requires a single dose (10-15 mg/kg) of MDMA to produce a clear 30 to 50% or greater loss in cerebral 5-HT content (Colado et al., 1995; O'Shea et al., 1998). This contrasts with the several doses of MDMA, often of 20 mg/kg or more, that are usually required to produce a similar loss in Sprague-Dawley, Hooded Lister, and Wistar rats (Colado et al., 1993; Aguirre et al., 1998a; Shankaran and Gudelsky, 1999).
Following the initial decrease in 5-HT content resulting from MDMA-induced release, concentrations return toward pretreatment levels within 24 h. Schmidt (1987a) monitored the time course of cortical 5-HT depletion following a single dose of MDMA (10 mg/kg) and showed two clearly distinguishable phases of the response. 5-HT was significantly depleted within 3 h of drug treatment, concentrations being 16% of control values between 3 and 6 h post-drug administration. Between 6 h and 24 h, however, a sharp recovery was observed and the 5-HT concentration had returned to control values 1 day later. The second phase of depletion was apparent 1 week post-treatment, 5-HT levels gradually declining during the period between 1 and 7 days, being reduced to 74% of control values at 1 week. Stone et al. (1986) and Battaglia et al. (1988) demonstrated a dose-dependent reduction in the concentration of 5-HT and 5-HIAA in the frontal cortex during the subacute phase, 18 h after multiple doses of MDMA. Similar reductions were observed following four 20 mg/kg doses administered to guinea pigs (Commins et al., 1987).
O'Shea et al. (1998) conducted a comprehensive study in Dark Agouti rats on MDMA-induced long-term serotonergic depletion, assessing the extent of neurotoxicity produced by single doses of MDMA (4, 10, and 15 mg/kg i.p.), multiple low doses (4 mg/kg) administered once or twice daily for four consecutive days, and multiple low doses (4 mg/kg) administered twice weekly for eight consecutive weeks. Single doses produced dose-dependent decreases in hippocampal, cortical, and striatal 5-HT and 5-HIAA measures 1 week post-treatment, with the lowest dose (4 mg/kg) having no significant depleting effect. Administration of 4 mg/kg MDMA daily for 4 days also had no effect on regional brain concentrations of 5-HT or 5-HIAA, while twice-daily administration resulted in a substantial depletion in all brain areas examined (40% loss of cortical 5-HT). In contrast, twice-weekly administration of low-dose MDMA had no effect on brain 5-HT or 5-HIAA content. The data thus indicate that high or frequent doses of MDMA are required to produce neurotoxic damage. Although these results may have significant implications for human recreational users of MDMA, the authors specifically point out that rat data provide no indication of doses or frequency regimes that may put human users at risk (O'Shea et al., 1998).
An early study examined the effect of route of administration on MDMA-induced neurotoxicity. Finnegan et al. (1988) compared oral with subcutaneous dosing and reported comparable effects. However, a recent study on the acute temperature effect of MDMA suggested that in rats, oral administration of MDMA was less effective than intraperitoneal (De Souza et al., 1997), and this probably also applies to the doses required for neurotoxicity (see also Slikker et al., 1989).
Since MDMA administration results in an inhibition of tryptophan hydroxylase, decreased cerebral tissue concentrations of 5-HT and 5-HIAA may indicate inhibition in indole synthesis rate rather than neurotoxic damage to the presynaptic nerve ending. However, there are other data supporting the contention that serotonergic neuronal damage occurs after MDMA, such as the use of [3H]paroxetine binding to the presynaptic 5-HT transporter. There are again many papers reporting that [3H]paroxetine binding is reduced following MDMA administration (Battaglia et al., 1987; Scanzello et al., 1993; Hewitt and Green, 1994; Broening et al., 1995; Colado et al., 1995; Obradovic et al., 1998; O'Shea et al., 1998). For example, 7 days after a single dose of MDMA Aguirre et al. (1995) reported a 35% reduction in [3H] paroxetine binding in the frontal cortex, while multiple doses resulted in an approximately 45% reduction. In contrast, [3H]mazindol binding to dopamine and noradrenaline uptake sites is unaffected by MDMA administration (Battaglia et al., 1987; 1991).
While Battaglia et al. (1987) demonstrated a significant loss of 5-HT content in the cortex and hypothalamus, much smaller effects were seen in the striatum and hippocampus. These decreases contrasted with the significant reduction of [3H]paroxetine binding observed in all brain regions examined. These results possibly indicate that the loss of 5-HT content may underestimate the full magnitude of MDMA-induced neurotoxicity (Battaglia et al., 1987). Hewitt and Green (1994) also showed that the loss of high-affinity [3H]5-HT uptake in cerebral tissue taken from MDMA-pretreated rats correlated more closely with the [3H]paroxetine binding measures than the indole concentration. The fact that the indole concentration can be influenced by tryptophan hydroxylase activity, this being modified by MDMA administration (see Section III.A.2), does suggest that [3H]paroxetine binding might be a more accurate indication of 5-HT nerve ending loss. This view is supported by evidence that the MDMA-induced loss of [3H]paroxetine binding is not due to a neuroadaptive response (Boot et al., 2002).
b. Histology.
It is reasonable to argue that all biochemical measures of neurodegeneration are indirect and that absolute identification of neurotoxic damage can only be made with histological/histochemical analysis and several substantial studies exist to support the contention that MDMA can cause long term neurodegeneration in the brain.
Silver-staining of rat striatal slices 13 to 16 h after several high doses of MDMA demonstrated the presence of argyrophilic deposits in MDMA-treated rats, which were absent in control animals. Primary somatosensory cortex slices contained shrunken, argyrophilic neuronal cell bodies and what appeared to be fragmented dendrites and degenerating axon terminals (Commins et al., 1987). However, the Fink-Heimer staining method used does not enable identification of the specific neurotransmitter contained in the damaged nerve terminals. O'Hearn et al. (1988) performed immunocytochemical analysis of regional brain sections 2 weeks after MDMA administration and reported on the presence of gross changes. There was reduced intensity of staining in MDMA-treated brain slices, reflective of a marked reduction in serotonergic axonal density, and these changes were particularly apparent in the neocortex, striatum, and thalamus, with smaller reductions occurring in the hippocampus, septum, and amygdala. The terminal portions of axons were shown to be selectively vulnerable to MDMA-induced damage, as indicated by the reduced density of fine, arborized 5-HT axons and sparing of smooth, straight preterminal fibers, while fibers of passage and raphe cell bodies were unaffected. Morphological evidence of damage to axon terminals is consistent with the observed reductions in 5-HT uptake sites (O'Hearn et al., 1988; Molliver et al., 1990).
The time course of the lesion has been shown to be region-specific. For example, 2 weeks after drug administration, neurodegenerative processes in the dorsal cau-date region are only just fully expressed, while a maximal and persistent deficit in 5-HT innervation is apparent in the cortex, and some regeneration is beginning to occur in the substantia nigra. Furthermore, brain regions containing 5-HT pathways or perikarya are little affected by MDMA, the predominant effects being mediated on axons and terminals (Battaglia et al., 1991). Such data are consistent with the lack of 5-HT depletion in the dorsal raphe region of the brain stem (Aguirre et al., 1995), which includes serotonergic cell bodies.
Measurement of anterograde axonal transport provides an additional method for assessment of serotonergic neurotoxicity. In the study of Callahan et al. (2001), rats were administered several doses of MDMA 3 weeks before injection of [3H]proline into the rostral raphe nuclei. Two days after injection of the labeled amino acid regional brain radioactivity levels were measured, enabling study of ascending 5-HT axonal projections to be made by tracing the transport of radioactive material. MDMA pretreatment resulted in significant decreases in anterograde axonal transport of labeled material, which paralleled (but were less severe than) decreases in 5-HT and 5-HIAA content. These effects of MDMA were similar to those observed following administration of the neurotoxin 5,7-DHT.
Astrocyte hypertrophy can occur as a result of neuronal injury and can lead to the enhanced expression of glial fibrillary acidic protein (GFAP). This marker of neuronal damage has been used in several studies assessing MDMA-induced toxicity in mice. Several studies have shown a correlation between MDMA-induced dopamine damage in mouse striatum and an increase in GFAP expression 3 days after drug treatment (Miller and O'Callaghan, 1995; Johnson et al., 2002a,2002b). Thus, although the increase in GFAP expression produced by MDMA is greater than the corresponding decrease in dopamine levels, it exhibits a similar dose dependence (Johnson et al., 2002a). The changes in both parameters are also prevented by either mechanical or pharmacological prevention of MDMA-induced hyperthermia (Miller and O'Callaghan, 1995) and augmented by a vitamin E-deficient diet (Johnson et al., 2002a) or treatment with supraphysiological levels of corticosterone (Johnson et al., 2002b). These results are similar to those obtained with other amphetamines, such as methamphetamine and MDA (Miller and O'Callaghan, 1994; O'Callaghan and Miller, 1994), both of which also produce hyperthermia. However, fenfluramine, which causes 5-HT toxicity but a decrease in temperature, failed to produce an increase in GFAP expression (O'Callaghan and Miller, 1994). Studies using this technique to examine MDMA-induced damage in rats are few but Aguirre et al. (1999) did report an increase in GFAP in the hippocampus of MDMA-pretreated rats that paralleled 5-HT damage and was prevented in the same way by α-lipoic acid administration. However, in general, data referring to 5-HT terminal damage are not consistent, thus administration of other neurotoxicants of the 5-HT system such as para-chloroamphetamine (Wilson and Molliver, 1994) and fenfluramine (Rowland et al., 1993; Bendotti at al., 1994) do not produce increases in GFAP expression despite profound losses of 5-HT levels and “abnormal” 5-HT-immunoreactivity, whereas administration of 5,7-DHT has been reported to produce both an increase in GFAP (Bendotti et al., 1994) and also no effect (Rowland et al., 1993). Thus, it has been suggested that a lack of GFAP expression increase may be due to an insufficiently strong signal and that the use of this parameter for detecting selective degeneration of serotonergic axons may have limitations (Bendotti at al., 1994).
2. Recovery of Serotonin Neurochemical Markers.
The rate of neuronal recovery in the rat frontal cortex following a neurotoxic dose of MDMA has been followed by measuring [3H]paroxetine binding to 5-HT uptake sites. Battaglia et al. (1988) administered MDMA and analyzed [3H]paroxetine binding 18 h and 1, 2, 4, 8, 26, and 52 weeks later. Eight weeks post-treatment 5-HT uptake sites were approximately 40% of control values. At 26 weeks post-treatment this value had increased to approximately 75% of control values, and by 52 weeks there was no difference between MDMA-treated and control animals. There appeared to be a faster rate of recovery between 18 h and 4 weeks, after which a slower recovery rate was observed.
Scanzello et al. (1993) measured regional brain content of 5-HT and 5-HIAA and [3H]paroxetine-labeled 5-HT uptake sites, and performed immunocytochemical analysis of 5-HT-containing nerve fibers for up to 1 year post-treatment. The earliest recovery of 5-HT content was observed in the hypothalamus 8 weeks after drug administration, while hippocampal and striatal levels had recovered by 16 weeks. All brain regions examined showed complete recovery of 5-HT within 1 year of drug treatment, and similar patterns were observed in the recovery of 5-HIAA content. [3H]Paroxetine binding values in the cortex and striatum had returned to control levels within 32 weeks, while hippocampal binding was still 29% below control values at 52 weeks post-treatment. With regard to morphological changes, all animals demonstrated a significant reduction in 5-HT axon density in the parietal cortex 2 weeks post-treatment. Only one of three animals demonstrated recovery 52 weeks post-treatment.
Sabol et al. (1996) investigated the extent of recovery of both regional brain 5-HT content and [3H]5-HT uptake in striatal and hippocampal synaptosomes up to 1 year after MDMA administration. MDMA administration resulted in a significant decrease in [3H]5-HT uptake 2 and 8 weeks after treatment, but there was no difference between MDMA- and saline-pretreated animals at any of the later time points. Brain 5-HT concentrations were significantly reduced in all regions examined, apart from the septum, and different rates of recovery were observed in different regions. For example, frontal cortex levels were depleted by approximately 70% 2 weeks post-treatment and showed complete recovery by 52 weeks post-treatment, while striatal levels were depleted by approximately 30% at 2 weeks and showed complete recovery by 16 weeks. In contrast, significant depletion of 5-HT was still apparent in both the frontal-parietal and occipital-temporal cortex 1 year post-treatment, while some hyperinnervation was observed in the hypothalamus at 52 weeks. These data indicate that MDMA-induced 5-HT depletion and rate of recovery are region-dependent. Furthermore, 5-HT innervation of nerve terminal regions after MDMA-induced damage may represent growth from raphe cell bodies, and the time course of innervation could reflect the distance from 5-HT cell bodies or fiber bundles (Sabol et al., 1996).
In an accompanying study, Lew et al. (1996) used radioligand binding and autoradiography to assess the extent of serotonergic recovery over a 1-year period. Rats administered MDMA were sacrificed 2, 8, 16, 32, or 52 weeks later for tissue analysis. Dopamine uptake sites in striatal homogenates were unaffected following multiple doses of MDMA up to 1 year previously while, in contrast, the densities of 5-HT uptake sites were altered. The density of hippocampal sites was significantly reduced (by 66% compared to control values) 2 weeks post-treatment, partial recovery being apparent by 16 weeks, and full recovery by 52 weeks post-treatment. In the frontal-parietal cortex, 5-HT uptake site density was reduced by 75% 2 weeks postdrug administration and partial recovery was observed at 16 weeks. However, recovery did not continue, the uptake site density at 32 and 52 weeks being the same as that at 16 weeks. Incubation of regional brain slices with 125I-RTI-55 and [125I]3β-(4-iodophenyl)tropane-2β-carboxylic acid isopropyl ester hydrochloride (125I-RTI-121) enabled visualization of the distribution of 5-HT uptake sites. Two weeks after drug administration, 125I-RTI-55 binding in terminal field regions was decreased or abolished in the brains of MDMA-treated animals, while binding in the substantia nigra, ventral tegmental area, dorsal and median raphe, and lateral hypothalamus was unaffected. Binding in the ventromedial hypothalamus had recovered by 16 weeks and remained unaltered at 32 and 52 weeks post-treatment. These data were consistent with previous studies with regard to the regional specificity of MDMA-induced serotonergic damage. Although these data contrast with those of Scanzello et al. (1993), who reported complete serotonergic recovery in all brain regions by 52 weeks, that study used lower doses of MDMA, which might explain the differing results.
3. Effect of Central Administration of MDMA.
Several years ago two studies reported that direct administration of MDMA into the brain failed to induce neurotoxicity. However, one communication was an abstract, so detailed methodology was sparse (Molliver et al., 1986), while the other reported on the lack of effect of intraraphe injection of MDMA on neurotoxicity in terminal regions (Paris and Cunningham, 1991). However, this result was perhaps unsurprising given the fact that systemic injection of MDMA leaves cell bodies in the raphe region intact (Battaglia et al., 1991; Lew et al., 1996).
A recent comprehensive study on the effects of central injection of MDMA confirmed its lack of neurotoxicity when administered by intracerebral injection. Even when MDMA was infused into the hippocampus at a dose producing a drug tissue concentration 4 times greater than that observed following a peripherally injected neurotoxic dose, it failed to induce neurodegenerative loss of 5-HT. The centrally administered dose nevertheless induced an acute release of 5-HT (Esteban et al., 2001). These data do suggest strongly that the MDMA molecule increases 5-HT release, while it is a metabolite or other breakdown product, initially produced peripherally, that is responsible for the neurotoxicity.
4. Effects of Preventing Acute MDMA-Induced Hyperthermia.
Prevention of the MDMA-induced hyperthermic response tends to provide protection against the subsequent neurotoxic loss of 5-HT, and a significant number of compounds that were initially reported to be neuroprotective have been subsequently demonstrated to have this property not because of a specific neurochemical action, but because of an effect on body temperature. For example, Colado et al. (1993) and Hewitt and Green (1994) reported that the NMDA antagonist MK-801 (dizocilpine) was neuroprotective against MDMA-induced damage to 5-HT nerve endings. This was confirmed by Farfel and Seiden (1995). However, this group also showed that co-administration of MK-801 and MDMA produced a hypothermic response. When the temperatures of MK-801 + MDMA animals were kept elevated, the neuroprotective effect of MK-801 was completely abolished. The finding that administration of another NMDA antagonist S-(+)-α-phenyl-2-pyridine ethanamide dihydrochloride (AR-R15896AR; Colado et al., 1998), a compound that does not attenuate MDMA-induced hyperthermia, produced no neuroprotection, strengthened the contention that NMDA antagonists have no intrinsic protective effect and that neuroprotection had only occurred in some earlier studies because of a body temperature-lowering action of the NMDA antagonist being examined.
Malberg and Seiden (1998) demonstrated the apparent importance of Ta in the long-term depletion of 5-HT and 5-HIAA following MDMA administration. No significant depletions were observed in any of the brain regions examined when MDMA had been administered at a Ta of 20, 22, or 24°C. However, under Ta conditions of 26, 28, or 30°C, significant depletion was observed. Significant negative correlations between core temperature and serotonergic depletion were observed. Thus, small changes in Ta were shown to produce marked changes in the degree of serotonergic neurotoxicity.
Although the Ta required for MDMA-induced neurotoxicity may vary with the strain of rat it is now clear that little long-term loss of cerebral 5-HT occurs unless the rats have a hyperthermic response to the drug. This may be so when a single dose of MDMA is injected. However, repeated administration of low doses of MDMA (4 mg/kg, twice daily, for 4 days) did not produce hyperthermia but did induce a long-term depletion of 5-HT parameters (O'Shea et al., 1998). Nevertheless, in general, administration of a drug that prevents hyperthermia will produce neuroprotection (Colado et al., 1998). Consequently, most early studies that have claimed a specific neurochemical mechanism of protection as the result of administering a drug that also attenuated the acute hyperthermic response of MDMA must be viewed with suspicion. A prime example is the involvement of dopamine in the neurotoxic mechanism of MDMA, since neuroleptics such as haloperidol and the dopamine synthesis inhibitor α-methyl p-tyrosine produced normothermia or hypothermia (Hewitt and Green, 1994; Malberg et al., 1996).
If we assume increased free radical formation is a key element in MDMA-induced neurotoxicity, then the role of body temperature in the neurodegenerative process is more easily explained. Free radical formation in the brain following MDMA administration is markedly enhanced in hyperthermic animals (Colado et al., 1998). This is consistent with other reports studying ischemia-induced neurodegeneration where it was found that free radical formation is influenced by body temperature (Globus et al., 1995; Kil et al., 1996).
Finally, it appears that the protective effect of hypothermia can be overcome to some extent if the dose of MDMA is high enough (Broening et al., 1995). Presumably, this indicates that it is the rate of free radical formation that is key to the neurodegenerative process.
5. Studies on Neuroprotection.
There are compounds that provide protection against MDMA-induced neurotoxicity in rats by a mechanism not related to changes in body temperature. The 5-HT uptake inhibitors fluoxetine and fluvoxamine, co-administered with MDMA, completely prevented the long-term loss of 5-HT concentration without altering MDMA-induced hyperthermia (Malberg et al., 1996; Sanchez et al., 2001). Fluoxetine continued to provide total protection when given up to 4 days before MDMA. This long-lasting neuroprotective effect might be due to the maintained presence of fluoxetine and its main active metabolite norfluoxetine in the brain. Both compounds inhibit the 5-HT transporter and could be blocking the entry of a toxic metabolite of MDMA into the 5-HT nerve terminal (Sanchez et al., 2001).
PBN is a radical trapping agent that partially prevents the neuronal damage induced by MDMA, presumably as a result of its free radical trapping activity. PBN, at a dose that did not modify hyperthermia, attenuated the MDMA-induced neuronal damage and prevented hydroxyl radical formation (Colado et al., 1997a; Yeh, 1999). Supporting the existence of an oxidative stress process is the fact that the antioxidant ascorbic acid, administered 1 h before each dose of MDMA, also prevented the long-term loss of striatal 5-HT depletion and suppressed the generation of hydroxyl radicals (Shankaran et al., 2001). Repeated administration of the metabolic antioxidant α-lipoic acid before MDMA also prevented the serotonergic deficits and the changes in the glial response induced by MDMA without affecting the hyperthermic response (Aguirre et al., 1999). Nitrogen-reactive species could also be involved in MDMA damage. N-nitro-L-arginine (L-NOARG) inhibits brain NOS activity and provides protection against MDMA-induced indole depletion. Nevertheless, this protection is not complete and does not affect all the lesioned brain areas (Zheng and Laverty, 1998).
Although it can be shown that part of the neuroprotective action of clomethiazole involves attenuation of MDMA-induced hyperthermia, it also has an additional neuroprotective effect (Colado et al., 1998). What remains uncertain is the mechanism by which clomethiazole provides protection against MDMA neurotoxicity since it is not a radical trapping agent (Colado et al., 1999b) and, while it is a GABAmimetic compound (Green, 1998), other GABAmimetics are not protective (Colado et al., 1999c).
The dopamine uptake inhibitor mazindol, administered concomitantly with MDMA, has also been reported to attenuate the long-term depletion of 5-HT in the striatum without altering the acute hyperthermic response to MDMA. Mazindol also partially prevented the MDMA-induced increase in the extracellular concentration of dopamine and 2,3-DHBA (Shankaran et al., 1999b). A problem in interpreting these data, however, is the fact that mazindol does have some serotonin reuptake inhibitory activity, although its activity at dopamine and noradrenaline sites is undoubtedly higher (Heikkila et al., 1981; Angel et al., 1988; Shimizu et al., 1992). Other compounds, such as 5-HT2 receptor antagonists (MDL 11,939 or ritanserin) and MAO-B inhibitors (l-deprenyl or MDL-72974), have shown efficacy in preventing neuronal damage when administered within 1 h of MDMA (Schmidt et al., 1990b; Sprague and Nichols, 1995), but their effect on rectal temperature was not evaluated.
6. Role of Dopamine in the Neurodegenerative Process.
The role of dopamine in MDMA-induced damage to 5-HT nerve endings remains controversial. Several early studies on MDMA indicated that altering dopamine function could also alter the degree of neurodegeneration. For example, Stone et al. (1988) proposed a role for dopamine based on their study showing that damage to 5-HT nerve endings was attenuated in animals given the dopamine synthesis inhibitor α-methyl-p-tyrosine or the depleting agent reserpine. Stone et al. (1989) reported that selective lesioning of dopamine nerve endings with 6-OH-dopamine blocked the neurotoxic effects of MDMA in several brain regions. Both Stone et al. (1988) and Shankaran et al. (1999b) have reported that GBR 12909 is neuroprotective and there are also reports that haloperidol is neuroprotective (Schmidt et al., 1990c; Hewitt and Green, 1994). Based partly on these data an integrated hypothesis linking dopamine with MDMA-induced neurotoxicity has been proposed (Sprague et al., 1998).
One problem with all of these studies, however, is the fact that body temperature was not controlled and several of the compounds (reserpine, α-methyl-p-tyrosine, haloperidol) can and do attenuate MDMA-induced hyperthermia. The hypothesis that dopamine is involved in the neurotoxicity is associated with the fact that MDMA induces dopamine release (Johnson et al., 1986; Schmidt et al., 1987; Nash, 1990; Nash and Brodkin, 1991), and that L-DOPA administration potentiates MDMA-induced damage (Schmidt et al., 1991). More recently, Shankaran et al. (1999b) showed that repeated MDMA administration produced a sustained increase in the extracellular dopamine concentration in the striatum that was suppressed by mazindol. MDMA also increased the conversion of salicylic acid to 2,3-DHBA, suggesting an increase in free radical formation, a change that was also attenuated by mazindol. The authors concluded that enhanced dopamine release and hence enhanced free radical formation in the striatum could contribute to the mechanism by which MDMA induced damage to 5-HT nerve endings. Also supporting the notion of the involvement of dopamine metabolism in MDMA-induced neurotoxicity is the fact that prior administration of an antisense oligonucleotide targeted at MAO-B attenuated both MAO-B activity and the loss in 5-HT and 5-HIAA concentration induced by MDMA (Falk et al., 2002). This work extends an earlier observation by this group that MAO-B inhibitors are neuroprotective (Sprague and Nichols, 1995).
Rather different conclusions on the role of dopamine in MDMA-induced neurotoxicity were reached by Colado et al. (1999a) in their study. Like Shankaran et al. (1999b) they also observed that MDMA increased extracellular dopamine concentrations in the striatum and they also showed that L-DOPA administration produced an enhancement in extracellular dopamine concentration. However, this enhancement did not increase free radical formation (measured by 2,3-DHBA concentration in the dialysate), at least in the hippocampus; the striatum was unfortunately not measured. It did, however, extend the period of MDMA-induced hyperthermia. This group, therefore, suggested that the enhancement of damage following MDMA in L-DOPA-treated rats was temperature-related rather than a result of increased dopamine release. This conclusion was supported by data showing that the neuroprotective effect of haloperidol was marginal when the temperature of the MDMA + haloperidol group was kept elevated relative to that of MDMA-treated rats. Similarly, the apparent protective effect of reserpine is lost when studies are made 24 h after its administration, when its hypothermic action is largely lost, despite the fact that dopamine stores were still low (Hekmatpanah et al., 1989). In addition, Shankaran and Gudelsky (1998) noted that suppression of dopamine release from noradrenergic neurons in the hippocampus failed to attenuate MDMA-induced damage to 5-HT neurons.
Support for the conclusion that dopamine is not involved in the mechanism of MDMA-induced neurotoxicity has now also been provided by Yuan et al. (2002), who demonstrated that no protection to 5-HT nerve endings could be detected when reserpine or α-methyl-p-tyrosine had been given and the body temperature of the rats was kept elevated relative to that of the MDMA-alone group. These data complement those of their earlier study on methamphetamine-induced neurotoxicity, which similarly suggested that previous evidence for a role of dopamine had been confounded by body temperature changes (Yuan et al., 2001).
In conclusion, therefore, earlier evidence for a major role of dopamine release in the neurotoxic changes induced in the striatum by MDMA has been complicated by changes in body temperature produced by many of the putative neuroprotective compounds, and recent data appear to deny a role for dopamine. The role of dopamine in other brain regions where dopamine content is very low can also be questioned, and it perhaps seems unnecessarily complicated to propose different mechanisms of damage in different brain regions (see Shankaran and Gudelsky, 1998).
7. Perinatal and Early Postnatal Sensitivity to MDMA.
Broening et al. (1994) demonstrated that MDMA administration fails to produce long-term 5-HT loss in immature rats (PND 10) while damage does occur in rats injected with MDMA at PND 40 and PND 70. This finding was further explored (Broening et al., 1995) with regard to the involvement of Ta and MDMA-induced acute hyperthermia in serotonergic neurotoxicity. One week after MDMA administration to rats in Ta conditions of 10, 25, or 33°C, no 5-HT depletion was observed in PND 10 rats under any Ta condition. In PND 40 animals no loss was observed at 10°C, but dose-dependent reductions in 5-HT content were observed at Ta = 25°C and 33°C. However, PND 70 animals also demonstrated a dose-dependent loss of 5-HT at 10°C. Since acute hyperthermia was prevented under low Ta conditions, but significant loss of 5-HT was still observed in PND 70 rats, these data indicate that either mechanisms in addition to hyperthermia are responsible for serotonergic damage or, more probably, that large doses of MDMA can overcome any neuroprotective effects of low Ta (Broening et al., 1995).
Colado et al. (1997b) administered MDMA to pregnant female Wistar rats on days 14-17 of the gestational period. On PND 7 (11 days after cessation of drug administration) both dams and pups were sacrificed for measurement of regional brain monoamine concentrations. Striatal and hippocampal 5-HT concentrations were markedly depleted in the brains of the dams, while no loss of 5-HT, 5-HIAA, or dopamine was observed in the dorsal telencephalon of the pups. These results support other studies (St. Omer et al., 1991; Broening et al., 1994, 1995) and demonstrate an apparent lack of vulnerability of the fetal or neonatal rat brain to MDMA-induced serotonergic neurotoxicity. It was suggested by Colado et al. (1997b) that the young rat brain has high endogenous radical trapping activity and is thus resistant to the neurotoxic effects of MDMA. A very recent study on the lack of MDMA-induced neurotoxicity in neonates has observed that serotonin transporter site density is much higher in the neonatal brain than in the adult (Kelly et al., 2002). Since availability of these sites is a major requirement for neurotoxicity (Shankaran et al., 1999; Sanchez et al., 2001) the resistance to MDMA-induced neurotoxicity does not appear to involve the density of the uptake sites in neonates.
Aguirre et al. (1998b) administered MDMA to pregnant female Wistar rats on alternate days, from embryonic day 6 to 20. The rat pups were sacrificed on PND 15 for analysis of 5-HT content and 5-HT transporter density. There was neither a loss of 5-HT content nor a reduction in 5-HT transporter density in rat pups whose mothers had been administered MDMA. In contrast, pups that were administered MDMA on PND 35 exhibited significant 5-HT reductions in the cortex, striatum, hippocampus, and hypothalamus, and decreased frontal cortex 5-HT transporter density 7 days later. In addition, MDMA-induced neurotoxicity was apparent at an earlier postnatal age in pups that were co-administered MDMA and L-DOPA. The authors suggested that the lack of neurotoxicity at early postnatal ages could be due to low dopamine concentrations, and that a particular threshold of dopamine release is required to produce a serotonergic deficit. Equally plausibly, it could be argued that the high endogenous radical trapping ability of the young brain can be overwhelmed when L-DOPA is also administered. Functional behavioral changes have been observed in young rats administered MDMA even in the absence of overt biochemical changes in the brain and these are discussed elsewhere.
8. Neuronal Firing.
Gartside et al. (1996) investigated 5-HT neuronal activity in the dorsal raphe nuclei of rats administered repeated doses of MDMA. There were no observable differences in the mean firing rate or regularity of firing of 5-HT neurons between MDMA-treated and control animals. Furthermore, electrical stimulation of the dorsal raphe nucleus evoked a threefold increase in cortical and hippocampal dialysate 5-HT levels in both treatment groups. The apparent lack of effect of MDMA administration on electrical activity in the dorsal raphe nucleus is consistent with the observed lack of damage to dorsal raphe nuclei 5-HT cell bodies.
Obradovic et al. (1998) also administered repeated doses of MDMA and then performed neuronal recording on days 1-4 and days 9-15 after the last drug injection. Neuronal recording was performed in the n. accumbens, a brain region believed to be involved in the rewarding properties of abused drugs, cells in the core region being sustained at stable, low firing rates by the application of glutamate. Glutamate-evoked firing was dose-dependently inhibited by the application of either 5-HT or dopamine, these inhibitory effects being markedly attenuated by MDMA pretreatment. There was no difference in the effects of treatment on 5-HT- and dopamine-mediated inhibition between animals tested 1 to 4 days and 9 to 15 days post-treatment, which indicates persistent changes in neuronal excitability following repeated exposure to MDMA.
9. Alterations in Serotonin Receptor Density.
In vitro binding of [3H]8-OH-DPAT in rat brain cortical and hypothalamic homogenates has been examined to assess the effects of MDMA administration on 5-HT1A receptor density. Both a single dose and multiple doses of MDMA resulted in a significant increase in [3H]8-OH-DPAT binding in both the frontal cortex (Aguirre et al., 1995) and hypothalamus (Aguirre et al., 1998a). Although a single dose had no effect on [3H]8-OH-DPAT binding in the dorsal raphe region, multiple doses resulted in a significant decrease in binding, indicating a reduction of 5-HT1A inhibitory autoreceptors in this region (Aguirre et al., 1995). A decrease in [3H]paroxetine binding in the frontal cortex correlated with the increase in 5-HT1A receptors, which could indicate adaptive changes to compensate for the loss of serotonergic nerve terminals (Aguirre et al., 1995). Pretreatment with fluoxetine, haloperidol, or ketanserin prevented MDMA-induced increases in [3H]8-OH-DPAT binding in the frontal cortex (Aguirre et al., 1998a). However, the fact that changes in [3H]8-OH-DPAT binding occurred after a single dose of MDMA argues strongly against the change being associated with any neurodegenerative change. Using functional tests to examine 5-HT1A receptor density, namely the 8-OH-DPAT-induced hypothermic or stereotyped behavioral responses, neither Mechan et al. (2001), Granoff and Ashby (2001), nor McNamara et al. (1995) found any long-term alteration of the response in rats administered a neurotoxic dose of MDMA, again arguing against a 5-HT1A receptor change in terminal regions produced by neurodegeneration. However, the results contrast with those of Aguirre et al. (1998a), who observed an enhanced response.
A transient down-regulation of 5-HT2 receptors following MDMA administration has been reported by Scheffel et al. (1992), who performed in vivo and in vitro labeling of 5-HT2A and 5-HT2C receptors in rat brain using the radioligand N-1-methyl-2-[125I]lysergic acid diethylamide ([125I]MIL). In vivo [125I]MIL binding was unaffected by acute MDMA administration, whereas chronic administration resulted in a 55 to 80% decrease in binding 24 h post-treatment. However, this change had disappeared after a further 6 days. Acutely, treatment with MDMA (20 mg/kg) reduced specific in vivo binding of [125I]MIL in all regions of the brain studied. For example, in the frontal cortex, specific binding of [125I]MIL was decreased by 80% at 6 h and by 62% at 24 h after cessation of treatment with MDMA. Twenty-one days after administration of MDMA, however, the number of binding sites for [125I]MIL had returned to control levels. Similar results have been obtained by Reneman et al. (2002a) measuring 5-HT2A postsynaptic receptor densities using [123I]R91150 SPECT. Rats showed an immediate decrease followed by a time-dependent recovery of cortical 5-HT2A receptor densities that coincides with the 5-HT neurotoxic damage, and probably are reflecting a compensatory up-regulation of postsynaptic 5-HT2A receptors due to 5-HT depletion. Functional evidence for a lack of 5-HT2A/2C change has been provided by Granoff and Ashby (1998), who reported that neither DOI-induced locomotor activity nor head-twitch response of rats was altered by an earlier neurotoxic dose regime of MDMA.
10. Long-Term Functional Changes.
a. Behavior.
Spanos and Yamamoto (1989) showed that chronic administration (over 24 days) of MDMA resulted in an increase in the intensity of locomotion and serotonin syndrome behaviors during chronic drug administration, suggesting sensitization to the effects of the amphetamine. McNamara et al. (1995) measured locomotor activity in an open field arena on each day of MDMA administration (5, 10, or 20 mg/kg twice daily for 4 days) and on the 4 days following the treatment period. Although total locomotor activity was significantly higher in MDMA-treated rats compared to control animals during the drug treatment period, activity had returned to baseline/control values within 48 h after the last drug administration. Thus, using this treatment regimen, the MDMA-induced increase in locomotor activity was dose- and time-dependent and returned to normal following cessation of drug treatment.
In contrast, Wallace et al. (2001) reported reductions in locomotor activity 1 week after multiple doses of MDMA administered during 1 day. Spontaneous locomotor activity was measured during diurnal and nocturnal cycles for seven consecutive days, and MDMA-treated animals demonstrated significant reductions in activity compared to control animals during both cycles. There was no difference in the activity of either treatment group between diurnal and nocturnal values. Such alterations were accompanied by significant reductions in striatal 5-HT levels, although a connection between the two findings was not established. Biello and Dafters (2001) examined the effect of MDMA pretreatment on the in vitro response of the circadian clock to 8-OH-DPAT administration and reported that the MDMA-lesioned rats had an impaired response to the phase-shifting action of this 5-HT1A agonist, speculating that this might account for some of the reported sleep disorders in human recreational ecstasy users.
In addition to measurement of the acute behavioral effects of MDMA administration, Marston et al. (1999) monitored behavior every 1 or 2 days following the treatment period, up to 18 days after the initial MDMA exposure. While skilled paw-reaching was significantly attenuated in MDMA-treated rats during the treatment period, the performance of this group did not differ from that of the control group during the post-treatment period.
b. Temperature.
Since 5-HT has long been associated as a neurotransmitter involved in thermoregulatory mechanisms (Milton, 1977; Jacob and Girault, 1979; Myers, 1981; Salmi and Ahlenius, 1988) the possibility existed that MDMA-induced neurotoxicity would lead to alterations in the ability of rats to thermoregulate.
Studies on the influence of a prior exposure to the drug on the size of the temperature response following a second dose have produced conflicting data. Dafters (1995) reported sensitization to the second dose, Shankaran and Gudelsky (1999) observed an attenuation, while other studies (T. Beveridge and J. M. Elliott, unpublished) found no change. However, Colado et al. (1997a) reported an attenuation in the MDMA-induced hyperthermic response following an earlier neurotoxic dose of fenfluramine.
Dafters and Lynch (1998) provided evidence that prior administration of several doses of MDMA altered the ability of rats to thermoregulate when exposed to a high-temperature environment. This work was confirmed and extended by Mechan et al. (2001), who observed that when rats that had been pretreated with MDMA 33 days earlier were exposed to a high ambient temperature (30°C) they displayed both a faster rise in rectal temperature in the high-temperature conditions and a sustained hyperthermia when returned to normal (20°C) conditions. No difference was observed in these rats in their hypothermic response to the 5-HT1A agonist 8-OH-DPAT (Mechan et al., 2001). This agrees with McNamara et al. (1995), who studied this response in MDMA-pretreated rats, but not Aguirre et al. (1998a), who observed an enhanced 8-OH-DPAT-induced hypothermia in rats given MDMA 1 week earlier. However, since this group also observed this response following acute MDMA treatment, it seems unlikely that the effect they saw was associated with neurodegeneration. Since the rectal temperature of the MDMA-pretreated rats was the same as control animals in normal ambient temperature conditions (Mechan et al., 2001), it seems that the defect in thermoregulation only becomes apparent in “challenging” situations such as high ambient temperature.
c. Effects on Cognitive Behavior.
Marston et al. (1999) showed that following a neurotoxic dose of MDMA cognitive behavior, as measured by the accuracy of DNMTP performance, did not differ between treatment groups at the shorter delay period of 3 s (between pressing the “sample” lever and being presented with a “choice”). However, at the longer delay period of 30 s the accuracy of control animals progressively improved on successive test days, while the accuracy of MDMA-treated rats remained the same across all post-treatment test days up to day 16. MDMA treatment therefore appeared to have resulted in cognitive impairment. The authors suggested that the behavioral effects observed could be primarily attributed to serotonergic nerve terminal dysfunction (Marston et al., 1999). Broening et al. (2001) used a multiple-T water maze and Morris water maze to assess sequential learning and cued and spatial learning in neonatal rats that had been administered MDMA during the periods PND 1-10 or PND 11-20. Rats were tested in the multiple-T maze at an average age of PND 63, and the Morris water maze at an average age of PND 77. The PND 1-10 treatment group demonstrated no significant deficits in any of the parameters tested, whereas the PND 11-20 group demonstrated dose-related impairments of sequential learning and spatial learning and memory. These data thus indicated that MDMA exposure during brain development resulted in a disruption of sequential and spatial memory-based learning, and that such deficits were developmentally specific. In addition, these effects were not related to any long-term changes in 5-HT, dopamine, or noradrenaline. However, Kelly et al. (2002) have now shown that exposure to MDMA in utero did lead to increased cerebral glucose utilization in the locus ceruleus and areas receiving ascending norepinephrinergic projections such as the thalamus of neonates, indicating that some long-term cerebral neurochemical changes do occur.
d. Anxiety Models.
Given the number of clinical studies that have suggested a possible association between psychiatric disorders and ecstasy ingestion, it is surprising that relatively few controlled studies have been performed using animal models to examine a possible relationship.
Studies on the effects of prior MDMA exposure on the behavior of rats in models of anxiety have produced conflicting data. Morley et al. (2001) found that rats treated 3 months earlier with MDMA showed greater anxiety-like behaviors than controls in emergence, elevated plus maze, and social interaction tests. Somewhat similar results have been reported by Fone et al. (2002) following administration of MDMA to adolescent rats and subsequent testing of open field behavior and social interaction up to 29 days later; although in this study the increased anxiety response was not accompanied by any measurable neurotoxic loss of 5-HT. Prior administration of MDA has also been reported to produce a decrease in open field behavior (Harkin et al., 2001). In contrast, Mechan et al. (2002b) reported both an increase in open field behavior and an apparent anxiolytic response on the elevated plus maze when rats were tested 73 to 80 days after a neurotoxic dose of MDMA.
These conflicting data may be explained by the strain differences in the rats used. The Dark Agouti strain used by Mechan et al. (2002b) display a high level of endogenous anxiety compared to Sprague-Dawley rats (Mechan et al., 2002c). Comparison of percentage of time spent on the open arm by control animals suggests that the same is true when the response of Dark Agouti rats (Mechan et al., 2002b) is compared to Wistar rats (Morley et al., 2001). A further complication remains in associating the change seen with a change in cerebral 5-HT content or function. Neither Mechan et al. (2002b) nor Morley et al. (2001) measured cerebral 5-HT content (although neurotoxic doses of MDMA were given). No significant change in 5-HT content was observed by Fone et al. (2002) and clear anxiolytic effects were seen by Morley et al. (2001) following administration of low doses of MDMA. Thus, prior MDMA administration could conceivably change long-term behavioral function without having caused a neurotoxic lesion. Such a proposal is not unreasonable given the observed alteration of memory performance in young animals, which also occurred without overt neurotoxic damage (Broening et al., 2001). In an attempt to clarify this point Gurtman et al. (2002) repeated the Morley et al. (2001) study, but measured cerebral 5-HT depletion. The results seen in the earlier study were replicated in animals with clear evidence of 5-HT loss. These data add to considerable other evidence that a decrease in cerebral 5-HT function can result in an anxiolytic or anxiogenic effect (see Soubrie, 1986; Griebel, 1995; Green and McGregor, 2002). It has recently been suggested that an MDMA-induced lesion may produce an anxiolytic effect in rats with a normally high basal anxiety state but an anxiogenic response in rats with low basal anxiety (Green and McGregor, 2002; Fig. 2.).

Plot of the mean time on the open arms, as a percentage of total arm time, spent by Wistar and Dark Agouti (DA) rats on the plus maze both before and approximately 9 weeks after an MDMA-induced neurotoxic lesion. Data recalculated from results published by Morley et al. (2001) and Mechan et al. (2002) and published in Green and McGregor (2002). Reprinted by permission of Springer-Verlag.
In their study Mechan et al. (2002b) raised the possibility that it was impulsivity, or risk-taking behavior, that was being examined rather than anxiety. This proposal has been taken further by Harro (2002), who presented further evidence that impulsivity following 5-HT depletion might present as an anxiolytic effect in the plus maze. It has also recently been demonstrated that lowering cerebral serotonin levels by rapid depletion of tryptophan in normal human individuals increases impulsiveness (Walderhaug et al., 2002), which provides some support for the Mechan et al. (2002b) interpretation of their data.
e. Dopamine.
Shankaran and Gudelsky (1999) demonstrated that MDMA-induced striatal 5-HT release was inhibited by pretreatment with a neurotoxic dose regimen of MDMA, while dopamine release was unaltered. These data support the reports from many other groups that MDMA produces selective neurotoxic damage to serotonergic nerve terminals, leaving dopaminergic neurons unaffected (Stone et al., 1986; Battaglia et al., 1987; Schmidt and Kehne, 1990; Lew et al., 1996; Sabol et al., 1996; Colado et al., 1997a, 1999a). Depleting the antioxidant activity of rat brain by providing a selenium-deficient diet also failed to induce MDMA-induced damage to striatal dopamine (Sanchez et al., 2003).
B. Mice
1. Long-Term Dopamine Depletion.
The fact that MDMA has a different pharmacology in the mouse compared to the rat is well established. Several groups have reported that MDMA is a relatively selective dopamine neurotoxin in mice, leaving 5-HT concentrations intact, in contrast to its selective 5-HT neurotoxicity in rats (Stone et al., 1987a; Logan et al., 1988; O'Callaghan and Miller, 1994).
In a recent study O'Shea et al. (2001) confirmed these earlier findings in mice and showed that fluoxetine failed to alter the MDMA-induced long-term neurotoxic damage. In contrast, administration of the dopamine uptake inhibitor GBR 12909 proved to be neuroprotective. This compound did not inhibit the acute release of dopamine induced by MDMA (as measured by the tissue concentration of dopamine), but rather enhanced it, suggesting that its neuroprotective action was not because it inhibited carrier-mediated uptake of MDMA. The neuroprotective effect of GBR 12909 was confirmed in a subsequent study (Camarero et al., 2002), which also showed, using in vivo microdialysis, that GBR 12909 enhanced the rise in extracellular concentration of dopamine that follows MDMA injection. However, GBR 12909 did inhibit the MDMA-induced increase in free radical formation in the striatum as measured by in vivo microdialysis. The data therefore suggest that free radical formation in mice is not associated with dopamine release and indicate that free radical-producing neurotoxic metabolites may enter the dopamine nerve ending via the dopamine uptake site.
In a study on the mechanisms involved in MDMA-induced neurotoxicity in mice Colado et al. (2001) found that NMDA antagonists did not prevent long-term dopamine loss in mice and that clomethiazole, a compound that was effective in preventing neurotoxic damage to 5-HT neurons in rats (Colado et al., 1993) was without neuroprotective efficacy in mice. Despite the evidence for a role of free radicals in the damage using in vivo microdialysis techniques (Colado et al., 2001; Camarero et al., 2002), a clear protective action of the nitrone radical trap PBN was not observed. However, this was primarily due to the investigators being unable to separate neuroprotection from a hypothermic action of PBN, and the involvement of free radicals in the long-term damage to dopamine neurons seen in mice does seem clear since studies using CuZn-superoxide dismutase-transgenic mice have demonstrated their resistance to MDMA-induced neurotoxicity (Cadet et al., 1994, 1995, 2001).
Two nitric oxide synthase inhibitors (S-methyl-thiocitrulline and AR-R17477AR) were found to be effective neuroprotective agents and had little effect on MDMA-induced hyperthermia. Both of these compounds are suggested n-NOS inhibitors. Interestingly, ARR17477AR inhibited the MDMA-induced rise in free radical formation in vivo, suggesting that it was either MDMA or dopamine metabolic breakdown products that were producing radicals that combine with nitric oxide to produce tissue-damaging peroxynitrites (Colado et al., 2001; Camarero et al., 2002). However, since GBR 12909 increased MDMA-induced dopamine release, but attenuated neurotoxicity, one may suggest that MDMA metabolites rather than dopamine metabolites are involved in the damage process. The results further indicate that the neurotoxin uses the dopamine transporter to enter the terminal. These recent studies also complement the available data indicating the involvement of NOS and peroxynitrites in methamphetamine-induced neurotoxicity (Ali and Itzhak, 1998; Callahan and Ricaurte, 1998; Itzhak et al., 1998; 2000; Imam et al., 1999).
Administration of MDMA to selenium-deficient mice resulted not only in the appearance of a greater dopamine loss than seen in mice fed a normal diet, but also in the occurrence of 5-HT loss in specific brain regions (Sanchez et al., 2003). In contrast, selenium depletion in rats did not alter either the size or selectivity of the 5-HT loss induced by MDMA administration, suggesting that the antioxidant capacity of rats and mice differs (Sanchez et al., 2003). This interpretation of the effect of depleting endogenous defenses against toxic radicals reported by Sanchez et al. (2003) is supported by another investigation that examined the effect of producing vitamin E deficiency in MDMA-induced neurotoxicity. Johnson et al. (2002a) reported that a low dose of d-MDMA (5 mg/kg) produced a substantial loss of striatal dopamine in vitamin E-deficient mice but was without effect in animals with a sufficient diet.
C. Primates
1. Long-Term Serotonin Depletion and Neuronal Damage.
Serotonergic depletion and neuronal damage has also been demonstrated in nonhuman primates (Ricaurte et al., 1988a,b, 1992; Slikker et al., 1988, 1989; Insel et al., 1989; Wilson et al., 1989; Ricaurte and McCann, 1992; Fischer et al., 1995; Scheffel et al., 1998; Hatzidimitriou et al., 1999), the effects being more pronounced than those observed in rodents. For example, a study administering MDMA to rats and squirrel monkeys (twice daily, for four consecutive days), illustrated the significantly greater sensitivity of monkeys to the 5-HT-depleting effects of MDMA. The dose-response curve was considerably steeper in monkeys and the maximal effect was greater in the monkey than the rat (Ricaurte et al., 1988b; Ricaurte and McCann, 1992). Serotonergic depletion in squirrel monkeys has also been shown to be dose-dependent; 2.5 mg/kg (twice daily for 4 days) resulted in a 44% depletion of 5-HT in the somatosensory cortex, while 5 mg/kg resulted in a 90% depletion (Ricaurte et al., 1988b).
The degree of 5-HT depletion may be dependent upon the route of administration, although data at this point are conflicting. Oral administration has been reported to be less effective than subcutaneous injection; Ricaurte et al. (1988c) found that MDMA (5 mg/kg, twice daily for four consecutive days) produced an 86% depletion of frontal cortex 5-HT when given s.c. compared with a 42% depletion when given orally. In contrast, Kleven et al. (1989) obtained a more marked effect of the drug when it was given to rhesus monkeys by the intragastric route rather than subcutaneously. While administration of multiple doses of MDMA to squirrel monkeys resulted in significant losses in 5-HT content in all brain areas examined, a single dose only produced significant depletion of 5-HT in the thalamus and hypothalamus (Ricaurte et al., 1988c). Since a single 5 mg/kg oral dose has been proposed to be equivalent to a 1.4 mg/kg dose in a 70-kg human based on interspecies dose scaling (see below), these data may indicate a possible risk of serotonergic damage in humans even after a single dose (see Ricaurte et al., 1988c).
Even when MDMA had been given at a dose producing a 5-HT depletion of approximately 90% in the caudate nucleus of squirrel monkeys, dopamine and noradrenaline levels were unaffected. 5-HT depletion was accompanied by a significant reduction (60%) in the CSF concentration of 5-HIAA, while HVA and MHPG concentrations were unchanged (Ricaurte et al., 1988a). These data demonstrated that measurement of CSF 5-HIAA might be used for evaluating whether recreational MDMA was producing damage to serotonergic neurons in humans, particularly since similar reductions in brain 5-HT and 5-HIAA content and CSF 5-HIAA concentration have been reported to occur in rhesus monkeys (Insel et al., 1989). A recent study confirmed the decrease in CSF 5-HIAA and found that the effect persisted for approximately 3 months (Taffe et al., 2001). Some equally persistent abnormalities in evoked potentials were also reported but some cognitive/behavioral abnormalities that were seen after the drug administration normalized within a week (Taffe et al., 2001).
Ricaurte et al. (1992) has also demonstrated a reduction in the maximal number of [3H]paroxetine-labeled 5-HT uptake sites in primate brain 2 weeks after MDMA administration. Ten weeks post-treatment several brain regions (such as the caudate nucleus head and hippocampus) exhibited partial recovery of 5-HT and 5-HIAA content and [3H]paroxetine binding, while the frontal cortex only demonstrated significant recovery of 5-HIAA content. Only partial recovery was observed in the hippocampus 8 months post-treatment, but almost complete recovery was seen in the hypothalamus. However, with the exception of the thalamus and hypothalamus, recovery had not continued when measured 18 months postdrug administration. Possible hyperinnervation had occurred in the hypothalamus 18 months post-treatment, since 5-HT and 5-HIAA levels had risen to 140% and 187% of control values, respectively. The authors suggested several possible reasons for the lack of recovery in primates compared to rats, including the fact that initial damage to serotonergic neurons is greater in the primate than the rodent, serotonergic neurons in the primate may have less regenerative potential, and axonal recovery is likely to be inversely proportional to the distance involved for a damaged axon to re-establish synaptic contact (Ricaurte et al., 1992).
Immunocytochemical analysis has also shown structural damage to serotonergic nerve fibers in nonhuman primates. Following MDMA administration to macaque monkeys there is a marked reduction in the density of serotonin-immunoreactive axons throughout the forebrain and some axons appeared swollen and misshapen (Ricaurte et al., 1988b). MDMA did not cause any obvious cell loss in the raphe nuclei, but did result in the presence of numerous, shrunken nerve cells containing cytoplasmic inclusions in the dorsal raphe nucleus. These inclusions were demonstrated to contain lipofuscin, which could be due to lipid peroxidation of cell components and phagolysosomal activity. Inclusion-containing cells were not observed in the median raphe nucleus. These data indicate that serotonergic depletion almost certainly results from actual damage to nerve fibers. Wilson et al. (1989) demonstrated a marked reduction of 5-HT immunoreactive axons throughout the cortex of macaque monkeys 2 weeks after MDMA administration and forebrain regions exhibiting a 60 to 90% loss of 5-HT axons. MDMA-treated animals showed a profound loss of fine axons, while beaded axons, with large, spherical varicosities, tended to be spared.
Thus MDMA administration results in a lasting reorganization of ascending 5-HT axonal projections—those to distal forebrain targets, such as the dorsal neocortex, demonstrate minimal recovery while projections to proximal targets, such as the hypothalamus, recover fully or are subject to hyperinnervation (Lew et al., 1996; Sabol et al., 1996). It appears that initial lesion severity is likely to be an important factor in the extent of neuronal recovery, since axons in brain areas such as the hypothalamus that demonstrated the least severe injury showed the greatest recovery, while areas such as the dorsal neocortex, which were the most severely injured, showed the least recovery (Fischer et al., 1995).
Abnormal brain innervation patterns are long-lasting after MDMA administration. Hatzidimitriou et al. (1999) reported that 5-HT axon density was only 50 to 65% of control values in neocortical regions 7 years post-treatment. Some recovery was apparent in the CA3 region of the hippocampus, while significant reductions in axonal density were still seen in the CA1 and CA2 regions. The caudate and putamen demonstrated partial recovery, while the globus pallidus showed some evidence of hyperinnervation, possibly due to its innervation from both the dorsal and median raphe, the former not being susceptible to neurotoxicity. Variables associated with the presence or absence of regrowth included the severity of the initial damage and the distance of the terminal region from the cell bodies, although no simple pattern of explanation could be ascertained by the investigators. There was no apparent loss of cell bodies and catecholaminergic axons appeared unchanged.
Scheffel et al. (1998) used positron emission tomography (PET) and a 5-HT transporter ligand, [11C](+)McN-5652, to measure the effects of chronic MDMA treatment in the baboon (Papio anubis). Significant reductions in regional radioactivity were apparent in the hypothalamus and frontal cortex of MDMA-treated animals 13 days post-treatment, and decreases in [11C](+)McN-5652 accumulation occurred in all regions examined during the first 40 days post-treatment. Nine months after MDMA administration, significant recovery of [11C](+)McN-5652 binding was observed in the midbrain and hypothalamus, while persistent reductions were seen in all cortical regions. At 13 months, [11C](+)McN-5652 concentrations were greater than control levels in the pons (+34%), midbrain (+37%), and hypothalamus (+37%), whereas tracer concentrations remained diminished in all cortical areas. Thus a time-dependent redistribution of 5-HT transporter sites was demonstrated in the baboon brain, which is likely to reflect the differential recovery of 5-HT axon projections.
2. Long-Term Dopamine Depletion and Neuronal Damage.
All the data presented in the previous section have indicated that MDMA produces selective neurotoxic damage to 5-HT neurons in the brains of primates. However, a recent study has challenged this view and demonstrated that MDMA will produce damage to dopamine neurons if given in a specific way. Ricaurte et al. (2002) tried to mimic the way MDMA is sometimes taken recreationally by giving several (normally 3) doses (2 mg/kg) of the drug at 3-h intervals to squirrel monkeys and baboons. In addition to the serotonergic loss, the authors also observed significant loss of dopamine, DOPAC, and dopamine transporter sites in the striatum. Since brain dopamine levels generally decline with age in humans the authors speculated on the possibility that human recreational users might be at greater risk of acquiring Parkinson's disease in later life if they have accelerated the process of dopamine loss.
In some ways these data are analogous to the study on dopamine loss in selenium-deficient mice. In that investigation (Sanchez et al., 2003) showed that selenium deficiency resulted in 5-HT loss occurring in the brain in addition to the expected dopamine loss. That is, in conditions where endogenous free radical trapping activity was exhausted, damage to additional monoamine neurotransmitter systems might occur. In that regard the rat may differ from the mouse and primate since even selenium deficiency did not result in damage to dopaminergic systems in the rat (Sanchez et al., 2003).
3. Complex Brain Function.
Frederick and Paule (1997) examined in squirrel monkeys the effects of repeated escalating doses of MDMA. Chronic treatment with the highest dose resulted in disruption of different aspects of all the operant tasks performed, while performance in the motivation task was more sensitive to disruption than in the other tasks. Operant performance tended to return to pretreatment levels within 2 or 3 weeks after cessation of the chronic treatment regimen, and was stable for the remaining experimental period (up to 20 months). When monkeys that had been exposed to chronic treatment were challenged with an acute dose of MDMA, tolerance was apparent.
However, Winsauer et al. (2002) found that a neurotoxic dose of MDMA failed to disrupt learning in squirrel monkeys even though disruption could be demonstrated when other drugs such as fenfluramine and triazolam were given. Previous studies using rhesus monkeys have also failed to observe a change in operant tasks (Frederick et al., 1995, 1998). Overall, the data indicate that behavioral disruption following an MDMA-induced neurotoxic lesion may only be seen following certain tests, and that 5-HT loss is not invariably associated with a functional change.
A study recently performed in a small cohort of squirrel monkeys demonstrated the re-enforcing properties of MDMA and its stereoisomers in self-administration studies. The attenuation of the behavior following administration of MDL 100,907 indicated the involvement of 5-HT2A receptors in the behavior (Fantegrossi et al., 2002).
V. Effects of MDMA in Humans
A. Problems of Relating Animal and Human Data
1. Doses Used.
It has often been asserted by young recreational ecstasy users that data on adverse effects of MDMA obtained in experimental animals are not relevant to human use, as the doses administered have been much higher than those used by humans. While it is undoubtedly true that many experimental studies have used high doses, it now appears that different strains have different susceptibilities to the drug, as exemplified by the rat (see above). Furthermore, recent studies have demonstrated neurotoxicity in rats following doses that are a fraction of those used in the earlier studies (O'Shea et al., 1998).
It is always difficult to make direct comparisons between results obtained in animal and human studies, since small mammals tend to eliminate drugs at a faster rate than large mammals. However, it has been suggested that the technique of interspecies scaling (see Mordenti and Chappell, 1989) enables prediction of drug elimination in different species based upon the underlying anatomical, physiological, and biochemical similarities among most land mammals. Thus, to achieve a similar effect to that seen in humans, smaller animals require higher doses of drug, estimated according to the relationship Dhuman = Danimal (Whuman/Wanimal)0.7, where D = dose of drug in milligrams and W = body weight in kilograms. So, for example, a single neurotoxic dose of MDMA (5 mg/kg) administered to a 1-kg monkey can be calculated to equate to a dose in a 70-kg human of 98 mg or 1.4 mg/kg (McCann and Ricaurte, 2001). A dose of MDMA of 10 to 15 mg/kg that produces substantial damage in the brain of Dark Agouti rats is thus equivalent to a human dose of 140 to 190 mg in a 70-kg human. However, the validity of interspecies dose scaling in relation to MDMA has been questioned by Vollenweider et al. (2001), who point out the problems of taking into account metabolism, active metabolites, and the paucity of pharmacokinetic data on MDMA in rodents (see Fitzgerald et al., 1990).
Ecstasy tablets have been reported to generally contain between 80 and 150 mg of MDMA (Schifano, 1991; Henry, 1992). However, some recent analyses have reported doses of up to 250 mg/tablet (www.dancesafe.org/labtesting). Therefore, a possible neurotoxic dose may be being ingested when 2 to 3 tablets are taken (extrapolating from rat data). Peroutka (1987) reported that the amount of drug taken in a single dose ranged from 60 to 250 mg (1-4 mg per kilogram body weight), while Bolla et al. (1998) stated that, in a sample of 30 persons who had used MDMA on at least 25 separate occasions (and thus perhaps not a typical cohort), an average monthly dose was 441 mg (range: 55-4000). The weakness of this estimation, however, is that the authors had to estimate the average dose contained in each tablet ingested. It is, however, worth noting that a 10 mg/kg dose of MDMA administered to Dark Agouti rats produces a plasma MDMA concentration of 6.3 nmol/ml 45 min post-injection (Colado et al., 1995). This value is within the range occurring in humans following MDMA ingestion; for example, ingestion of a 150 mg tablet resulted in a plasma concentration of 5.2 nmol/ml (Dowling et al., 1987).
A further complication in extrapolating from experimental animal to human is the lack of clinical pharmacokinetic information. However, a recent study has provided clinical data indicating that MDMA has saturable kinetics (de la Torre et al., 2000a,b), meaning that metabolism of the drug is relatively slower at higher doses. If we assume that many of the acute adverse effects of the compound (particularly hyperthermia) are linearly related to the concentration of the parent compound (MDMA), then high doses may result in a disproportionately high risk compared to lower doses. The final, possibly self-evident problem with ingesting an illegally synthesized and marketed tablet is that the dose and purity are unknown, leading to further complication when trying to assess what might be a “safe” dose.
2. Interpreting Clinical Data.
The sections that follow review the clinical problems that have been reported to occur in recreational users of MDMA. However, it is important to realize that there are problems with all such reports. The existing preclinical data strongly suggest that MDMA can produce neurotoxic damage in the brain. Therefore, any prospective clinical study involving MDMA administration is constrained by ethical considerations. Even administration of low doses of MDMA (1.7 mg/kg) has been publicly questioned and discussed (McCann and Ricaurte, 2001; Vollenweider et al., 2001). We have to, therefore, generally rely on retrospective studies, and such data must be interpreted cautiously. All studies can be criticized for the fact that accurate information on both the doses taken and the duration of drug use is not available. This is because the drug has been synthesized and supplied illicitly and most subjects are unreliable sources of information on use. In addition, many subjects are poly-drug users so that it cannot be stated unequivocally that any event seen results solely from MDMA use. Alternatively, the problem may relate to the combination of MDMA with another recreational compound. This problem is compounded by the fact that a significant percentage of ecstasy tablets contain psychoactive compounds other than MDMA. Subjects are, therefore, sometimes taking drug combinations without realizing it. However, if we are considering long-term neurotoxicity data it is worth reiterating that, of the major recreational drugs, only MDMA and other amphetamine analogs have been clearly demonstrated to produce neurotoxicity. It is, therefore, difficult to attribute the neurotoxicity to “other drugs,” although one cannot rule out the possibility that the neurotoxicity is due to a combination of the MDMA and other compounds ingested.
In addition, many reports on psychiatric abnormalities have been in the form of case studies. Given that the psychiatric disorders described are relatively common, it has been difficult to offer the reports as conclusive proof of a causal link. Obviously an acute toxic psychosis may occur, and since amphetamine derivatives are well documented in acutely producing a true schizophrenia-like psychosis, such effects are likely to occur following MDMA use. However, this form of psychosis is “drug induced” and will be a time-limited adverse event. Such effects appear to be rare. More commonly, the drug has been suggested to be associated with a range of psychiatric illnesses. The question then arises as to whether it is producing new cases of illness. As pointed out elsewhere (Green et al., 1995) there are three different interpretations of these data.
-
If high-risk individuals are more likely to misuse the drug, then the association is largely statistical and drug misuse will not increase the total number of psychiatric problems in the community. Given the demographics of use, this seems an unlikely scenario;
-
If the drug increases the risk of chronic psychiatric problems in a vulnerable pool of individuals with a high predisposition to develop the illness (see McGuire et al., 1993), then again the effect of the drug in producing new cases is rather unimportant;
-
If the drug increases morbid risk in subjects with only a modest risk of illness, then there must be concern when the drug is used widely within a youth culture. There may, with time, be an increase in admissions with psychiatric problems. Since neurotoxic damage may well not be apparent for some time and there is evidence that this type of damage can be occult and not appear for some years (see Vingerboets et al., 1994), the possibility must be considered that a public health problem may occur in the future (see Green and Goodwin, 1996).
B. Pharmacokinetics of MDMA
de la Torre et al. (2000a) investigated the pharmacokinetics of a single dose of MDMA (50, 75, 100, 125, or 150 mg) in recreational MDMA users. An initial assessment of plasma levels of MDMA and MDA indicated a nonproportional dose-dependent kinetics of MDMA and its metabolites. While there was no significant difference between doses with regard to urinary clearance of MDMA, nonrenal clearance was dose-dependent. Non-renal clearance was reduced by 50% following administration of 125 mg MDMA, indicating an impairment of hepatic clearance of the drug. Analysis of the urinary recovery (excretion) of MDMA and its main metabolites [MDA, HMMA, and 4-hydroxy-3-methoxyamphetamine (HMA)] demonstrated that approximately 50% of MDMA was recovered within 24 h, regardless of dose. While HMMA recovery was almost constant for all doses studied, MDMA recovery increased in a nonproportional dose-response pattern. Analysis of plasma samples demonstrated that HMMA was the major product in plasma following administration of 50, 75, and 100 mg MDMA, while MDMA was the main product following 125- and 150-mg doses. Thus, these data demonstrated that with increasing MDMA dose, the rise in MDMA concentrations does not follow the same proportionality, which might indicate nonlinearity. One explanation of this finding is that MDMA metabolism becomes saturated at higher doses. Alternatively, there may be some interaction between MDMA metabolites within its metabolic pathways. For example, the maintenance of methoxy groups in positions 3 and 4 of the methylenedioxyamphetamine benzene ring provides such molecules with an increased affinity for CYP2D6 compared to their respective O-demethylated products (i.e., HMMA versus N-Me-α-MeDA). Thus the nonlinear pharmacokinetics of MDMA could be associated with inhibition of its O-demethylenation, with MDMA and HMMA playing important roles.
Fallon et al. (1999) administered racemic MDMA (40 mg) to eight nondrug-using subjects and measured blood and urine concentrations of (+)-S-MDMA, (-)-R-MDMA, (+)-S-MDA, and (-)-R-MDA at regular intervals thereafter, using gas chromatography. The maximum observed plasma concentrations (Cmax) of both MDMA enantiomers were attained within 4 h after drug administration. Both the mean Cmax value and overall plasma concentrations (at all time points) of (-)-MDMA were significantly greater than those of (+)-MDMA. In addition, the mean elimination half-life (t1/2) of (+)MDMA was significantly shorter than that of (-)MDMA. There was no difference in the renal clearance of the two MDMA enantiomers, indicating that nonrenal (metabolic) clearance is the primary stereoselective process. The majority of urinary excretion occurred within the first 24 h, approximately 2% of the dose was recovered during the 24 to 72 h period. The mean urinary recovery of the enantiomers of MDMA and MDA during the 0 to 24 h period was (-)-MDMA, 21.4%; (+)-MDMA, 9.3%; (-)-MDA, 1.0%; (+)-MDA, 1.4%. These data indicate that MDMA undergoes substantial enantioselective disposition in humans, the (+)-enantiomer being more pharmacologically active with a shorter half-life, lower peak plasma concentrations, and increased clearance.
An investigation of MDMA metabolism in human and rat liver microsomes provided information regarding the different CYP450 isozymes involved in the different metabolic pathways. In particular, demethylenation of MDMA to N-Me-α-MeDA in humans is catalyzed by CYP1A2, CYP2D6, and CYP3A4, and in rats by CYP2D1 and CYP3A2. N-demethylation of MDMA to MDA in humans and rats is primarily catalyzed by CYP1A2, and to a minor extent by CYP2D6 and CYP2D1 in humans and rats, respectively (Maurer et al., 2000). The elimination half-life of MDMA is about 8 to 9 h (Mas et al., 1999; de la Torre et al., 2000b).
C. Acute Effects
1. Physiological Effects.
The acute adverse physiological effects that occur during the peak period after MDMA ingestion by humans include elevated blood pressure and heart rate, nausea, chills, sweating, tremor, jaw clenching, bruxism, hyperreflexia, urinary urgency, muscle aches or tension, hot and cold flushes, nystagmus, and insomnia (see McCann et al., 1996).
After administering an oral dose of MDMA (125 mg), Mas et al. (1999) noted acute, significant increases in systolic and diastolic blood pressure, heart rate, and plasma concentrations of prolactin and cortisol. Oral temperature did not increase significantly. Harris et al. (2002) in a study in a small cohort (n = 8) of MDMA-experienced users also noted increases in plasma cortisol, prolactin, and dehydroepiandrosterone (DHEA) levels and reported an increased heart rate. Five volunteers reported jaw clenching. In a study investigating the subjective effects of MDMA in 100 recreational users on a university campus, the major acute effects reported were tachycardia, dry mouth, bruxism, and/or trismus (Peroutka et al., 1988).
Liechti and Vollenweider (2000a) examined the effect of the serotonin uptake inhibitor citalopram on MDMA-induced physiological responses to determine whether carrier-mediated release of presynaptic serotonin was responsible for various effects. Orally administered MDMA (1.5 mg/kg) produced a significant increase in both systolic and diastolic blood pressure and heart rate. These symptoms were attenuated by citalopram pre-treatment. MDMA also modestly increased body temperature (contrasting with the study of Mas et al., 1999 above), a response that citalopram did not modify. A similar study was also performed that examined the effect of the dopamine D2 receptor antagonist haloperidol on the physiological and psychological responses to MDMA. Results indicated that D2 receptor blockade did not alter any of the physiological effects listed above (Liechti and Vollenweider, 2000b). Both studies are necessarily limited by the fact that only a single dose of citalopram and haloperidol could be administered rather than dose-response studies being undertaken.
Hyperthermia is one of the major symptoms of acute MDMA-induced toxicity, and body temperatures of over 43°C have been reported. This can lead to other often fatal toxicological problems including rhabdomyolysis, disseminated intravascular coagulation (which results in widespread bleeding and tissue necrosis), and acute renal failure. Other physiological symptoms that have been reported during the first few hours following ingestion of MDMA include tachycardia, coagulopathy, thrombocytopenia, delayed leukocytosis, acidosis, hypoglycemia, pulmonary congestion, edema, and hepatitis (Simpson and Rumack, 1981; Brown and Osterloh, 1987; Dowling et al., 1987; Chadwick et al., 1991; Henry et al., 1992; Screaton et al., 1992; Barrett and Taylor, 1993; Green et al., 1995; McCann et al., 1996; Milroy et al., 1996).
In addition, potentially fatal neurological effects can occur following MDMA ingestion, including subarachnoid hemorrhage, intracranial hemorrhage or cerebral infarction, and cerebral venous sinus thrombosis. These complications may arise from short-term hypertension, cerebral angiitis, or dehydration (McCann et al., 1996; Milroy et al., 1996; Rutty and Milroy, 1997). Necrosis of liver and heart tissue has also been reported following post-mortem examination of individuals where death was associated with the use of amphetamine derivatives (Milroy et al., 1996; Rutty and Milroy, 1997).
2. Cerebral Blood Flow and Brain Activity.
[H2-15O]PET has been used to measure regional cerebral blood flow (rCBF), 75 min after administration of a single dose of MDMA (1.7 mg/kg) to MDMA-naïve subjects. MDMA produced a significant bilateral increase in rCBF in the ventromedial frontal-, the inferior temporal- and the medial occipital-cortex, and the cerebellum. It also produced a bilateral decrease in rCBF in the superior temporal cortex, the thalamus, the preparacentral cortex, in addition to significant decreases in the left amygdala. These regional changes in rCBF paralleled the psychological effects of MDMA, such as mood enhancement and increased sensory perception (Gamma et al., 2000).
By using quantitative electroencephalography (EEG), it has been shown that the extent of MDMA use was positively correlated with a global increase in alpha rhythm power (8-12 Hz) across the brain and in beta rhythm power (12-20 Hz) in the left posterior quadrant. In addition, EEG coherence, which provides a measure of the synchronization of neuronal firing between two cortical locations, was negatively correlated with MDMA use (Dafters et al., 1999).
Although quantitative EEG measures can be used to identify the extent and severity of cerebral pathology (see Salansky et al., 1998), its use of scalp surface electrodes does not provide information regarding intracerebral distribution of neuronal signals. Low-resolution electromagnetic tomography (LORETA), however, enables three-dimensional functional imaging of brain electrical activity. Frei et al. (2001) administered MDMA (1.7 mg/kg) to 16 MDMA-naive subjects before measurement of neuronal electrical activity using a combination of EEG and LORETA. Significant differences were observed in the spatial distribution of brain electrical activity between MDMA-treated and control subjects in all seven frequency bands and under both “eyes open” and “eyes closed” test conditions. In general, MDMA treatment led to a decrease in activity in the slow and medium EEG frequency bands, while activity within the high-frequency bands was increased. In particular, β-band activity was increased in the limbic and paralimbic brain regions, which might contribute to MDMA-induced positive mood enhancement.
3. Psychological Effects.
While the acute psychological effects that occur during the peak period following MDMA ingestion generally include euphoria and reduction of negative thoughts, adverse effects that follow subacute MDMA ingestion include depression, irritability, panic attacks, visual hallucinations, and paranoid delusions (Brown and Osterloh, 1987; Whitaker-Azmitia and Aronson, 1989; Creighton et al., 1991; McCann et al., 1996; Davison and Parrott, 1997).
Davison and Parrott (1997) studied 20 recreational drug users, aged 18 to 31 years, each of whom had used MDMA at least once. The subjects reported feelings of elation, increased energy, happiness, exhilaration, warmth, friendliness, calmness, relaxation, and confusion in addition to heightened perception of sound, color, and touch while “on MDMA.” When “coming off” the drug, the subjects reported feelings of lethargy, moodiness, irritability, insomnia, depression, and paranoia. Individuals who had used MDMA more than once stated that their first experience (“trip”) had been “the most intense,” and that subsequent trips were not weaker, but that the nature of drug-induced sensations was “known and expected” (Davison and Parrott, 1997). Recreational poly-drug users reported significantly higher feelings of elation, agreeability, and emotional composure when under the influence of MDMA compared to when taking amphetamine or LSD, while feelings of increased energy and confidence were reportedly similar under the influence of MDMA or amphetamine (Parrott and Stuart, 1997). Since the acute release of 5-HT is presumably followed by a period where central neurotransmitter stores must be replenished (McKenna and Peroutka, 1990), there have been several studies examining the mood of subjects that had taken MDMA several days earlier. Two studies have reported recreational users of MDMA have “low mood” several days after the acute dose (Curran and Travill, 1997; Parrott and Lasky, 1998). Furthermore, female users showed higher depression scores than male users or male or female control subjects (Verheyden et al., 2002). This mood change was not related to measures of long-term use of MDMA. Users were also more susceptible than controls to aggression.
Whitaker-Azmitia and Aronson (1989) reported three cases where acute anxiety episodes resulted from MDMA ingestion. None of the individuals had a personal or family history of panic disorder, and one of the patients had never taken MDMA before. These panic attacks were short-lived and did not recur when MDMA was taken subsequently. Visual hallucinations and paranoid delusions that can persist for days or weeks have also been reported by some users of MDMA (Brown and Osterloh, 1987; Creighton et al., 1991; Davison and Parrott, 1997).
The effects of pretreatment with a series of 5-HT and dopamine antagonists and uptake inhibitors (ketanserin, haloperidol, and citalopram) on MDMA-induced psychological responses were investigated in an attempt to determine the role of different neurotransmitters in such responses (Liechti and Vollenweider, 2000b; Liechti et al., 2000a,b; 2001). Approximately 60 min after oral MDMA administration (1.5 mg/kg), the predominant effects noted were a state of well being, extroversion, and sociability, in addition to moderate depersonalization and feelings of “unreality”; an altered perception of time; altered sensory perception; and moderate psychomotor activation; these effects lasted for 3.5 to 4 h (Liechti and Vollenweider, 2000b; Liechti et al., 2000a,b; 2001).
Pretreatment with a single high dose of citalopram inhibited most of the psychological effects of MDMA. However, MDMA-induced increases in emotional excitability and sensitivity were unaffected by citalopram, indicating that these psychological effects might not involve an action at the 5-HT uptake site (Liechti et al., 2000a). Such data are consistent with the observation that chronic treatment with serotonin reuptake inhibitors, such as citalopram and paroxetine, prevent the occurrence of MDMA-induced euphoria (Stein and Rink, 1999), but not with the preliminary study of McCann and Ricaurte (1993) that fluoxetine did not block the reinforcing subjective effect of MDMA in four subjects. Anecdotal evidence (www.ecstasy.org) suggests that the reinforcing effects of MDMA are prevented in some subjects taking MDMA but not others, which indicates that effects may relate to doses ingested of either of the drugs. It does seem reasonable to propose that many of the reinforcing properties of MDMA are associated with dopamine release that should not be attenuated by selective serotonin uptake inhibitors. This view is supported by the observation that pretreatment with the 5-HT2 receptor antagonist, ketanserin, resulted in a significant reduction in MDMA-induced perceptual changes and emotional excitation while having little effect on positive mood and well being (Liechti et al., 2000b). Haloperidol pretreatment also tended to reduce the euphoria and positive mood-state. The authors concluded that, although some nonspecific dysphoric effects of haloperidol could account for these results, they indicated an involvement of dopamine in MDMA-induced psychological responses (Liechti and Vollenweider, 2000b). These authors have recently reviewed all this work in detail (Liechti and Vollenweider, 2001).
Prepulse inhibition (PPI) of the startle response is used as an operational measure of sensorimotor gating whereby excess stimuli are filtered out, enabling an individual to focus on relevant environmental stimuli. The mechanisms underlying this phenomenon have been shown to be deficient in patients suffering from schizophrenia, obsessive-compulsive disorder, and Huntington's disease (Braff et al., 2001). MDMA has been previously shown to impair PPI in animal models (Kehne et al., 1992, 1996b; Dulawa and Geyer, 2000), and such effects of MDMA-like drugs have been demonstrated to be reduced by pretreatment with selective serotonin reuptake inhibitors (Kehne et al., 1992; Martinez and Geyer, 1997). In a study of the effect of MDMA on PPI in human subjects, the startle reflex was measured as an eye blink response to an acoustic stimulus and the percentage PPI was calculated as the reduction in startle magnitude in the presence of a prepulse compared to the response to the main stimulus alone. MDMA treatment resulted in an increase in startle magnitude and percentage PPI compared to control subjects. Citalopram pretreatment reduced the MDMA-induced increase in percentage PPI, haloperidol pretreatment had no effect on either startle magnitude or percentage PPI, and ketanserin pretreatment further increased percentage PPI in MDMA-treated subjects. These results indicate that MDMA enhances PPI in humans via a mechanism involving serotonin (Liechti et al., 2001).
D. Long-Term Effects
1. Cerebral Serotonin.
a. Biochemical Studies.
Kish et al. (2000) reported severe depletion (50-80%) of striatal 5-HT and 5-HIAA in the brain (measured 21 h post mortem) of a 26-year-old male who had taken MDMA regularly for 9 years. Post-mortem examination revealed a blood MDMA concentration of 4.4 μg/ml and a concentration in the occipital cortex of approximately 1 μg per gram of tissue. The subject had also taken cocaine and heroin during the months before his death, but since neither of these drugs has previously been demonstrated to alter striatal serotonin concentration, the authors suggested that the results seen were most likely to be due to chronic use of MDMA. However, since the death probably resulted from an acute overdose of MDMA, a plausible explanation for the data is that the monoamine loss resulted from the acute effect of the drug.
More reliable evidence for long-term changes have come from McCann et al. (1998) and Ricaurte et al. (2000), who used PET with the 5-HT transporter ligand [11C]McN-5652 to examine 5-HT transporter binding in recreational users of MDMA. Experimental subjects had used MDMA on at least 25 occasions but had been abstinent for 3 weeks or more, while control subjects had no previous MDMA exposure. There was a lower density of brain 5-HT transporter sites in MDMA users, which positively correlated with the extent of previous MDMA use. However, there was no correlation between the duration of abstinence from MDMA use and the decrease in [11C]McN-5652 binding. Although none of the subjects had neuropsychiatric conditions or used other illicit drugs known to cause serotonergic neurotoxicity, the findings do not exclude the possibility that decreased density of 5-HT transporter sites are secondary to preexisting differences in serotonergic function. It is also impossible to rule out the possibility that subjects with a reduced density of 5-HT transporter binding sites are predisposed to misusing recreational drugs such as MDMA. Semple et al. (1999), using single photon emission computed tomography (SPECT) with the serotonin transporter ligand [123I]β-CIT, demonstrated a reduction in radioligand binding to the 5-HT uptake site in the MDMA user group but also found a correlation between the regional uptake of the radioligand and duration of abstinence. Reneman et al. (2001), also using [123I]β-CIT, divided subjects into groups of moderate or heavy MDMA users based on consumption of fewer or more than 50 tablets, respectively, and compared these to ex-MDMA users (who had taken more than 50 tablets but none within 1 year of the study) and drug-free controls. They reported significantly lower binding in female, but not male, heavy MDMA users compared to controls, but no differences between moderate or ex-MDMA users and controls. The effect seen in female heavy MDMA users was not related to greater MDMA use reported by the subjects since this was actually larger for the male heavy MDMA users when measured both in absolute terms and per kilogram of body weight. This may suggest a difference in the susceptibility of men and women to the neurotoxic effects of ecstasy.
Reneman et al. (2000a) used SPECT with the 5-HT2A receptor ligand [123I]R91150 in addition to measuring relative cerebral blood volume (rCBV) via dynamic MR imaging. Mean cortical 5-HT2A receptor binding ratios were significantly lower in current MDMA users compared to abstinent users (average abstinent period, 18 weeks) and control subjects. These data indicated a down-regulation of 5-HT2A receptors in MDMA users, possibly due to MDMA-induced 5-HT release. The authors suggested that the higher binding of [123I]R91150 in the abstinent MDMA user group indicated an up-regulation of postsynaptic 5-HT2A receptors due to MDMA-induced 5-HT depletion. There was no significant difference in mean rCBV values between MDMA users and control subjects. However, in MDMA users a positive correlation was observed between cortical 5-HT2A receptor binding ratios and rCBV values in the globus pallidus and occipital cortex. Recently this group, again using SPECT, reported that MDMA users do not appear to suffer any reduction in nigrostriatal dopamine neurons. However, subjects regularly using amphetamine in addition to MDMA did display a 20% loss in binding (Reneman et al., 2002b).
The evidence for brain serotonin neuron damage in MDMA users as obtained by neuroimaging has recently been critically evaluated by Kish (2002), who concluded that the methodological flaws in the studies (use of poly-drug users and reliability and validity of the SPECT measurements) meant that none of the current data provided definitive answers as to whether MDMA use did or did not produce a long-term neurotoxic lesion of 5-HT nerve endings in the brain. Such strong criticism appears unwarranted. First, the PET data has been validated and replicated in baboons (Szabo et al., 1995). Second, recent studies on both SPECT and serotonin transporter drugs have demonstrated the validity of such techniques in measuring MDMA-induced neurotoxicity (Reneman et al., 2002d; Szabo et al., 2002). Finally, no other recreational drugs, other than amphetamines, have been demonstrated to produce serotonergic loss in the central nervous system.
b. Serotonin Function.
Central 5-HT function has also been assessed by neuroendocrine challenge tests: intravenous infusion of the 5-HT precursor L-tryptophan leads to an increase in serum prolactin concentration, which is likely to occur via enhanced synthesis and release of 5-HT. This response is blunted in depressed patients compared with healthy controls, and is enhanced by antidepressant drugs with effects on 5-HT function (see Charney et al., 1982; Heninger et al., 1984; Price et al., 1989). A group of nine regular users of MDMA did not differ in their baseline prolactin concentration compared to control subjects. Following intravenous infusion of L-tryptophan a significant increase in prolactin concentration was observed in control subjects, but not in the MDMA user group. However, this apparent difference in response failed to reach statistical significance, possibly because of the small sample size (Price et al., 1989). A similar, but statistically significant, alteration in the prolactin response was observed following a D-fenfluramine challenge. Gerra et al. (1998) demonstrated that D-fenfluramine-induced increases in both prolactin and cortisol were significantly lower in an MDMA user group compared to control subjects. Furthermore, prolactin secretion is believed to be controlled by activation of 5-HT2A and 5-HT2C receptors, while cortisol secretion might occur following 5-HT2C receptor stimulation in the presence of a 5-HT1A antagonist (see Meltzer and Maes, 1995a,b; Palazidou et al., 1995). Thus the altered prolactin and cortisol responses in MDMA users might indicate a reduced sensitivity of postsynaptic 5-HT1A and 5-HT2A/5-HT2C receptors (Gerra et al., 1998). MDMA users administered the mixed 5-HT agonist and releaser meta-chlorophenylpiperazine (m-CPP), a compound that increases plasma cortisol and prolactin, probably by an action at postsynaptic 5-HT2C receptors (Mazzola-Pomietto et al., 1996), demonstrated blunted cortisol and prolactin responses compared to control subjects (McCann et al., 1999a).
A subsequent study (Gerra et al., 2000) aimed to determine whether the alterations in serotonergic function, as indicated by blunted D-fenfluramine-induced prolactin and cortisol responses, were reversible. MDMA users were tested 3 weeks and 1 year after abstaining from taking the drug. At both time points there was no difference in the basal concentrations of prolactin and cortisol between MDMA users and control subjects. Following 3 weeks of abstinence, both prolactin and cortisol responses were significantly lower in MDMA users compared to control subjects. After 1 year of abstinence the prolactin concentration response was still significantly lower in MDMA users and similar to that recorded after 3 weeks of abstinence. However, there was no significant difference in the cortisol responses between the two experimental groups after 1 year of abstinence from MDMA. These results suggest the presence of a long-lasting impairment of brain serotonergic function in recreational users of MDMA. The recovery of the cortisol response, while the prolactin response remained lowered, might indicate that MDMA affects different brain regions and 5-HT receptors to different extents; 5-HT1A receptors are believed to be involved in prolactin secretion (see Palazidou et al., 1995) and, therefore, serotonergic functions mediated by this receptor subtype might be subject to more persistent dysfunction.
Recreational users of MDMA (>25 occasions) and control subjects have been tested for CSF monoamine metabolite (5-HIAA, HVA, and MHPG) concentrations. The MDMA users had significantly lower levels of CSF 5-HIAA than control subjects, the reduction being greater in females (46%) than males (20%). An apparent negative correlation between CSF 5-HIAA and the number of MDMA exposures was not statistically significant, and there was no correlation between CSF 5-HIAA and the duration or frequency of MDMA use. There was no overall difference in CSF HVA or MHPG concentrations between the two groups (McCann et al., 1994, 1999b). The apparent negative correlation between CSF 5-HIAA and MDMA consumption was, however, statistically significant in the study of Bolla et al. (1998), who noted that in their experimental group the mean concentration of 5-HIAA in the CSF of MDMA users was lower than the control group, and that the CSF 5-HIAA levels decreased with increasing MDMA dose.
In an attempt to determine whether MDMA use was associated with changes in neurons and glial cells Chang et al. (1999) used proton magnetic resonance spectroscopy (1H MRS) to measure brain concentrations of N-acetylaspartate (NA), a neuronal marker, and myoinositol (MI), a tentative glial marker. Concentrations of creatine (CR), choline compounds (CHO), and glutamate/glutamine were also assessed, in addition to metabolite ratios, using CR as internal standard. Initial MRI scans demonstrated no significant brain atrophy or white matter lesions in either MDMA users or control subjects. 1H MRS demonstrated that, in MDMA users, both MI and MI/CR were elevated in the parietal white matter, and CHO/CR was elevated in the occipital gray matter. The duration of MDMA use was correlated with the concentration of MI in parietal white matter and frontal cortex. NA, CR, and CHO concentrations were similar in MDMA users and control subjects in all brain regions examined. The elevation of MI concentration indicated increased glial content in the brains of recreational users of MDMA, while the normal NA and glutamate/glutamine concentrations indicated a lack of persistent neuronal damage or ischemic lesions. The latter could be due to minimal 5-HT neurotoxicity following recreational doses of MDMA (1.5-3 mg/kg) or the occurrence of neuronal recovery. In contrast to these data, Reneman et al. (2002c) reported marked reductions in NA/CR and NA/CHO ratios in frontal gray matter, but not in occipital gray matter or right parietal white matter. No changes in MI/CR ratio were observed. Discrepancies between both studies could be due to the lifetime exposure to MDMA and to age-associated differences.
2. Physiological Effects.
Longer-term physiological effects that can result from chronic use of MDMA include the development of temporomandibular joint (TMJ) syndrome (affecting the joint of the lower jaw), dental erosion, and myofacial pain, which are secondary to the acute effects of trismus and bruxism (McCann et al., 1996). In addition, aplastic anemia has been reported following recreational use of MDMA, and in both cases the condition spontaneously recovered 7 to 9 weeks after onset (Marsh et al., 1994).
While hepatotoxicity has also been reported in recreational users of MDMA (Brown and Osterloh, 1987; Henry, 1992; Henry et al., 1992; McCann et al., 1996; Milroy et al., 1996), it is probable that contaminants in ecstasy or other tablets may have played a major role. Varela-Rey et al. (1999) investigated the effects of MDMA on type I collagen production in cultured hepatic stellate cells, a cell type primarily responsible for collagen synthesis in the liver. α1(I) procollagen mRNA levels increased concentration-dependently, being over twofold greater than levels in control cells after 24 h incubation. The induction of α1(I) procollagen 1 mRNA expression was seen to correlate with a depletion of intracellular glutathione levels and a transient increase in hydrogen peroxide levels, while lipid peroxidation was unaltered. Thus MDMA exerts a profibrogenic effect on hepatic stellate cells, which is mediated by oxidative stress but does not appear to involve lipid peroxidation (Varela-Rey et al., 1999).
3. Psychological Effects.
Longer-term psychological effects resulting from recreational use of MDMA have been reported to persist long after cessation of drug use (Creighton et al., 1991; McCann and Ricaurte, 1991, 1992; McCann et al., 1994, 1996, 1999a; Bolla et al., 1998; McGuire, 2000). Visual hallucinations and paranoid delusions can form part of the peak effects of the drug but may sometimes persist for days or weeks together with anxiety, depression and panic disorder, cognitive impairment, and other alterations in behavior (Creighton et al., 1991; McCann and Ricaurte, 1991, 1992; McGuire and Fahy, 1991; Schifano, 1991; McCann et al., 1994, 1996, 1999a; McGuire et al., 1994; Bolla et al., 1998; Parrott and Lasky, 1998; Morgan, 1999; 2000; McGuire, 2000; Parrott et al., 2000; Bhattachary and Powell, 2001).
Regular use of MDMA has been reported to result in chronic psychosis (Creighton et al., 1991; McGuire and Fahy, 1991) but subject numbers have been small. Two of the case studies reported by Creighton et al. (1991) also involved persistent visual hallucinations, where two patients had suffered from frequent flashbacks over a period of several weeks following MDMA ingestion, although one patient also misused many other psychoactive compounds.
A recent study examining recreational drug users in the United Kingdom and Italy reported that heavy ecstasy poly-drug users scored more highly on phobic anxiety, obsessive-compulsive behavior, anxiety, psychoticism, and somatization than control subjects. However, problems did not appear to be specific to MDMA use as they were also seen in other recreational poly-drug users (Parrott et al., 2001).
McCann et al. (1999a) compared m-CPP-induced changes in behavior between MDMA users and control subjects by using a series of behavioral assessment scales. In each case, MDMA users demonstrated higher positive scores (“happy,” “energetic,” “content,” and “elated”) and lower negative scores (“sad,” “tired,” and “worried”). MDMA users also demonstrated greater positive- and fewer negative-mood responses to m-CPP treatment compared to control subjects. In addition, 32% of control subjects experienced an m-CPP-induced panic attack compared to 4% of the MDMA users. This could indicate that long-term MDMA use results in decreased anxiety or alternatively may indicate that underlying personality differences in individuals who take MDMA, such as sensation-seeking, could be involved. The authors suggested that the lowered sensitivity of MDMA users to m-CPP-induced anxiety indicated down-regulation of postsynaptic 5-HT2C receptors, which are thought to mediate these effects of m-CPP (McCann et al., 1999a).
4. Cognitive Impairment.
There is substantial evidence that some recreational MDMA users display selective cognitive deficits (Krystal et al., 1992; Parrott and Lasky, 1998; Parrott et al., 1998; Fox et al., 2001) and studies suggest that problems continue in the drug-free state (Bolla et al., 1998; Morgan, 1999; Gouzoulis-Mayfrank et al., 2000; Rodgers, 2000; Verkes et al., 2001) and may be more pronounced in heavy drug users (Morgan, 2000; Wareing et al., 2000). The cognitive deficits appear to be more apparent in tasks known to be sensitive to temporal functioning (Fox et al., 2002). Bolla et al. (1998) compared the verbal and visual memory performance of a group who had used MDMA on at least 25 occasions (and had abstained from use for >2 weeks) with a control group with no prior exposure. The MDMA user group displayed impaired immediate verbal memory and delayed visual memory. The mean concentration of 5-HIAA in the CSF was lower in MDMA users compared to control subjects and CSF 5-HIAA levels decreased with increasing MDMA dose. Furthermore, the lower the 5-HIAA concentration, the worse the memory performance. The data thus indicate that MDMA-induced brain 5-HT neurotoxicity might account for such deficits.
McCann et al. (1999b) assessed cognitive performance in MDMA users with a computerized psychological test battery. Abstinent MDMA users demonstrated performance deficits in several cognitive tests and, in particular, deficits in the working memory task were seen to directly correlate with the extent of MDMA use. However, the cognitive deficits did not correlate with a reduction in CSF 5-HIAA, in contrast to the findings of Bolla et al. (1998). Bhattachary and Powell (2001) performed a similar investigation of cognitive functioning and found that regular users of MDMA demonstrated poorer immediate and delayed verbal recall than nonusers, the degree of impairment correlating with lifetime drug ingestion. While Bolla et al. (1998) and McCann et al. (1999b) found visual memory impairments in MDMA users, Bhattachary and Powell (2001) found no difference. In a study by Heffernan et al. (2001) global impairments in prospective memory were detected.
The evidence that impaired serotonergic function may be associated with memory deficits in MDMA users is further extended by correlations between alterations in cortical 5-HT2A receptor binding (Reneman et al., 2000b), or altered D-fenfluramine-induced cortisol responses (Verkes et al., 2001), and memory deficits. Reneman et al. (2000b) demonstrated higher overall 5-HT2A receptor binding ratios (using SPECT with [123I]R91150) in the brains of an MDMA user group compared to control subjects. These differences reached statistical significance in the occipital cortex, and the authors suggested that the increased binding was due to MDMA-induced 5-HT depletion resulting in up-regulation of 5-HT2 receptors. The MDMA users also demonstrated significant deficits in delayed memory tasks, which directly correlated with the increase in 5-HT2A receptor binding ratios (Reneman et al., 2000b). Verkes et al. (2001) demonstrated a significantly reduced cortisol response to D-fenfluramine in MDMA users compared to control subjects. MDMA users also had significantly longer reaction times to visual and auditory stimuli, lower visual recall, and lower working memory scores. The reduced cortisol response was demonstrated to correlate significantly with visual recall scores, indicating a significant association between chronic MDMA use, diminished memory performance, and serotonergic neuroendocrine functional deficits (Verkes et al., 2001).
A potential confound in cognitive testing in MDMA users is the additional use of other illicit drugs, and could aid explanation of the variation in results reported by different authors. MDMA users often take cannabis to alleviate the negative effects of an ecstasy “comedown,” making it difficult to recruit subjects for studies who have not also used cannabis (Croft et al., 2001). Recent animal data even suggest the possibility of synergistic effects between cannabinoids and MDMA (Braida and Sala, 2002). Morgan (1999) used the Rivermead Behavioral Memory Test to compare “everyday memory” in MDMA users with that of subjects taking other drugs (alcohol, cigarettes, cannabis, amphetamine, LSD, and cocaine). The MDMA users demonstrated significantly poorer immediate and delayed recall compared to both other drug users and control subjects, indicating that the deficits in recall performance were primarily associated with chronic use of MDMA (Morgan, 1999). In a study of 11 MDMA/cannabis users, 18 cannabis users, and 31 matched controls, cognitive deficits (including learning, memory, verbal word fluency, speed of processing, and manual dexterity) in MDMA/cannabis users were no worse than those of the cannabis group. It appeared, therefore, that the poorer performance of the drug user groups compared to controls was not caused by MDMA ingestion. However, a possible explanation for this result is that MDMA did cause cognitive impairment, but the lack of difference between MDMA/cannabis and cannabis groups was due to some interaction between the drugs. The authors suggested that cannabis might attenuate the effects of MDMA alone, perhaps through cannabis-related dopamine down-regulation, protecting against MDMA-induced serotonergic deficits. The results indicate the need for caution in interpretation of MDMA-induced cognitive deficits and the requirement to account for cannabis use in human MDMA research (Croft et al., 2001).
Finally, it appears that many of the neuropsychological performance problems reported to occur in MDMA users, including impaired working memory and verbal recall, are not reversed by prolonged abstinence, suggesting the existence of a selective neurotoxic lesion (Morgan et al., 2002).
5. Cerebral Blood Flow.
Since 5-HT is involved in modulation of cerebral blood flow (Cohen et al., 1996; Nobler et al., 1999), studies have been conducted to determine whether MDMA use results in altered rCBF in recreational users. For example, Chang et al. (2000) administered MDMA to abstinent users and measured rCBF using MRI and SPECT. Little difference in baseline rCBF was observed between abstinent MDMA users and control subjects. In the subjects who were administered MDMA and subjected to a second scan, global and regional CBF were decreased in most regions compared to baseline values and compared to the matched control subjects. The lack of pronounced effect recorded in MDMA users suggests that long-term use of MDMA does not significantly affect 5-HT-mediated regulation of CBF. However, regional reductions in CBF can be observed for 2 to 3 weeks following administration of MDMA (Chang et al., 2000).
VI. Metabolism of MDMA
A. Pathways of Metabolism
Several pathways are probably involved in MDMA metabolism in the rat: N-demethylation, O-dealkylation, deamination, and conjugation (O-methylation, O-glucoronidation, and/or O-sulfation), resulting in the formation of 14 in vivo metabolites (Lim and Foltz, 1988, 1991a,b). Initially, MDMA is metabolized to MDA by N-demethylation; 3,4-dihydroxymethamphetamine (DHMA; N-methyl-α-methyldopamine, N-Me-α-MeDA) by demethylenation; and 2-hydroxy-4,5(methylenedioxy)methamphetamine (6-HO-MDMA) by ring hydroxylation (Lim and Foltz, 1988, 1991a,b; Tucker et al., 1994; Fig. 3).
Postulated pathways of MDMA metabolism (after Lim and Foltz, 1988, 1991; Hiramatsu et al., 1990; Zhao et al., 1992; Tucker et al., 1994; Colado et al., 1995; de la Torre et al., 2000). Structures in brackets are postulated intermediate compounds in the formation of 4-hydroxy-3-methoxyamphetamine and 5,6-dihydroxy-1,2-dimethylindole.
Lim and Foltz (1988) identified and determined the distribution of MDMA metabolites in the rat via ion trap mass spectrometry based on their electron ionization and chemical ionization mass spectra. In vitro metabolism was measured by incubating brain and liver samples in an MDMA-containing mixture for 2 h (Lim and Foltz, 1988). The distribution of the metabolites is shown in Table 1 (Lim and Foltz, 1988, 1991a,b).
Distribution of MDMA and its metabolites in the rat (after Lim and Foltz, 1988, 1991a, b). Distribution determined via gas chromatography with an ion trap detector, in samples obtained following in vivo and in vitro metabolism of MDMA in Sprague-Dawley rats. (Note that only liver samples were examined for the presence of 2,4,5-trihydroxymethamphetamine, 2,4,5-trihydroxyamphetamine, and 5,6-dihydroxy-1,2-dimethylindole.)
Hiramatsu et al. (1990) investigated the metabolism of MDMA to N-Me-α-MeDA in rat liver microsomes. The demethylenation reaction only occurred in intact microsomes, in the presence of an NADPH generating system, and was inhibited by a carbon monoxide/oxygen (9:1) atmosphere. The reaction was proportional to the concentration of cytochrome P450 (CYP450) in the incubation mixture, and was inhibited by SKF-525A, indicating that conversion of MDMA to N-Me-α-MeDA is mediated by the CYP450 monooxygenase system. The subsequent metabolism of N-Me-α-MeDA was also demonstrated to require intact microsomes and an NADPH generating system, and was inhibited in a CO/O2 atmosphere. However, this reaction was insensitive to SKF-525A (Hiramatsu et al., 1990). In addition, it was found that ascorbate blocked the initial oxidation. Since these reactions appeared to be oxygen-dependent, the authors investigated the effects of compounds that blocked the production of different oxygen species. Addition of SOD to the MDMA incubation mixture resulted in an approximately fourfold increase in the levels of N-Me-α-MeDA, indicating involvement of the superoxide anion in the further metabolism of N-Me-α-MeDA. In contrast, hydrogen peroxide, singlet oxygen, or hydroxyl radicals (·OH) were not involved. Incubation of N-Me-α-MeDA itself with liver microsomes resulted in its rapid metabolism, which was prevented by addition of SOD. Addition of glutathione (GSH) to the MDMA incubation mixture resulted in the formation of an electrochemically active product that was dependent upon the presence of GSH, intact microsomes, and an NADPH generating system. Similar results were obtained when GSH was added to an N-Me-α-MeDA incubation mixture. These results thus indicated that the in vitro metabolism of MDMA resulted in production of a compound capable of forming adducts with GSH via a SOD-sensitive pathway (Hiramatsu et al., 1990).
Lin et al. (1992) performed a similar investigation using rat brain microsomes. The demethylenation of MDMA and MDA to N-Me-α-MeDA and α-MeDA, respectively, was again shown to be dependent upon the presence of intact microsomes and NADPH, and the involvement of a CYP450 monooxygenase was demonstrated. However, the addition of SKF-525A or α-naphthoflavone, a selective CYP450 1A inhibitor, had no effect, indicating that a different CYP450 isozyme is involved in the brain compared to the liver. Addition of ·OH scavengers (thiourea and benzoate) suppressed demethylenation activity, indicating the participation of ·OH in the reaction (Lin et al., 1992). These data indicate that brain microsomes have the potential to oxidize MDMA via a CYP450-dependent system and an ·OH-dependent system.
Kumagai et al. (1994) investigated the properties of the enzymes responsible for MDMA demethylenation in rat liver microsomes. At low MDMA concentrations, liver microsomes prepared from female Dark Agouti rats, which are deficient in the CYP2D1 isozyme, demonstrated approximately 9% of the demethylenation activity seen in microsomes prepared from male SD rats, indicating the involvement of CYP2D1 in MDMA demethylenation in the rat. The authors suggested that demethylenation activity involved CYP2D isozymes at low MDMA concentrations, and that phenobarbital-inducible isozymes enhanced this activity at higher concentrations (Kumagai et al., 1994).
Tucker et al. (1994) demonstrated the demethylenation of MDMA to N-Me-α-MeDA in Saccharomyces cerevisiae yeast microsomes expressing human CYP2D6. Microsomes prepared from control yeast that did not contain CYP2D6 did not demethylenate MDMA, while yeast heterologously expressing CYP2D6 demonstrated linear demethylenation of MDMA to N-Me-α-MeDA. Microsomes prepared from the livers of human extensive metabolizers produced significantly more N-Me-α-MeDA than microsomes prepared from the liver of a poor metabolizer (CYP2D6 mutation). These results indicated that CYP2D6 is involved in the hepatic demethylenation of MDMA in humans (Tucker et al., 1994).
B. Pharmacology of Metabolites
1. 3,4-Methylenedioxyamphetamine.
MDA is a major metabolite of MDMA and plasma levels in rats rise rapidly following MDMA administration (for example see Colado et al., 1995). Levels in the brain rise in a parallel manner and plateau between 1 and 3 h after administration (Chu et al., 1996). Acutely, MDA increases locomotor activity from 15 to 180 min following its administration (Yeh and Hsu, 1991). The (+)-stereoisomer of MDA is more potent than the (-)-enantiomer at producing 5-HT-mediated behavior (Hiramatsu et al., 1989) and also induces hyperthermia in mice and rats (Miller and O'Callaghan, 1994; Colado et al., 1995).
MDA produces an acute release of 5-HT, this being reflected in a loss in cerebral 5-HT content, and (+)MDA is more potent than (-)-MDA in producing 5-HT depletion (Schmidt, 1987b; Johnson et al., 1988). MDA, like MDMA, increases 5-HT release in the n. accumbens, although MDA is reported to be less potent (Kankaanpaa et al., 1998). MDA, MDMA, and MDEA in decreasing order of potency increase striatal dopamine release in vivo and in vitro (Nash and Nichols, 1991; O'Loinsigh et al., 2001).
Multiple doses of MDA cause a marked reduction in TPH activity and concentrations of 5-HT and 5-HIAA in several serotonergic nerve terminal regions. However, no alteration in striatal tyrosine hydroxylase activity was seen 3 days after its last administration (Stone et al., 1986, 1987b). Fine 5-HT axon terminals are extremely vulnerable to MDA, whereas beaded axons with large varicosities and raphe cell bodies survive (O'Hearn et al., 1988; Mamounas et al., 1991). MDA was noted to be more potent than MDMA as a serotonergic neurotoxin in Dark Agouti rats (Colado et al., 1995). The neurotoxic effects of MDA on serotonergic projections to the forebrain and brain stem are completely blocked by prior administration of the 5-HT reuptake inhibitor fluoxetine (Harvey et al., 1993). No change has been found in striatal dopamine concentrations 3 days after the last of a series of doses of MDA (Stone et al., 1986, 1987b), or in the number of [3H]mazindol-labeled dopamine uptake sites (Battaglia et al., 1987).
Recently it has been reported that 4 weeks following repeated administration of MDA to rats there is a reduction in exploratory behaviors (distance moved, mean velocity, and wall rearing) compared with saline-treated animals. However, no change was observed in the performance on the elevated plus maze between MDA and saline-treated groups (Harkin et al., 2001).
Overall, therefore, it is generally difficult to separate the pharmacological actions of MDA from those of MDMA, and it is reasonable to suppose that most of the acute and long-term behavioral and biochemical changes appearing to occur in vivo after MDMA administration result from the action not only of MDMA, but also of its major metabolite, MDA.
2. Neurotoxicity of Other Metabolites.
Although systemic administration of MDMA results in long-term damage to serotonergic nerve terminals in rats, direct intracerebral injection of MDMA does not produce serotonergic neurotoxicity. It has therefore been suggested that although the parent compound is probably responsible for the acute 5-HT and dopamine-releasing properties of MDMA it is unlikely to be responsible for the neurotoxic effects, and that systemic metabolism is required for the development of toxicity (Schmidt and Taylor, 1988; Hiramatsu et al., 1990; Lim and Foltz, 1991b; Miller et al., 1995, 1996, 1997; Bai et al., 1999, 2001; Zhao et al., 1992). This hypothesis is supported by the fact that MDMA-induced 5-HT depletion is attenuated by pretreatment with the CYP450 inhibitor SKF-525A and potentiated by pretreatment with phenobarbital, which induces CYP450 isozymes and enhances N-demethylenation of MDMA in vitro (Gollamudi et al., 1989).
A series of studies have been performed to identify MDMA metabolites and to investigate their potential neurotoxic effects. Zhao et al. (1992) compared the neurotoxicological properties of MDMA with those of two of its metabolites, 6-HO-MDMA and 2,4,5-trihydroxymethamphetamine (Tri-HO-MA), 1 week after systemic or central administration to Sprague-Dawley rats. Although systemic administration of MDMA produced a large depletion of regional brain 5-HT, systemic, i.c.v., and intrastriatal administration of 6-HO-MDMA had no effect on brain levels of 5-HT or dopamine, indicating that this metabolite does not play a direct role in the neurotoxic actions of MDMA. Administration (i.c.v.) of Tri-HO-MA resulted in a moderate depletion of striatal dopamine but had no effect on 5-HT levels. Similar results were obtained by Johnson et al. (1992). Intrastriatal administration of Tri-HO-MA, however, resulted in significant depletion of both dopamine and 5-HT, and intracortical administration resulted in a significant depletion of 5-HT.
A potential pathway of metabolism of MDMA and MDA in the rat results in the formation of ortho-quinones, quinone-thioethers, and the GSH conjugates 5-(glutathion-S-yl)-α-methyldopamine (5-GSyl-α-MeDA) and 2,5-bis-(glutathion-S-yl)-α-methyldopamine (2,5-bis-(glutathion-S-yl)-α-MeDA) (see Bai et al., 2001). These metabolites have been investigated for their involvement in MDMA- or MDA-induced neurotoxicity. For example, Miller et al. (1995) investigated the further metabolism of the α-MeDA metabolite 5-GSyl-α-MeDA. Following a single (i.c.v.) administration, 5-GSyl-α-MeDA was rapidly metabolized to form 5-(cystein-S-yl)-α-methyldopamine (5-(CYS)-α-MeDA), the metabolite reaching maximal concentrations within 30 to 60 min after administration of 5-GSyl-α-MeDA. 5-(CYS)-α-MeDA was also rapidly metabolized, and the resulting compound, 5-(N-acetylcystein-S-yl)-α-methyldopamine (5-(NAC)-α-MeDA), reached maximal concentrations within 2 h after 5-GSyl-α-MeDA administration. 5-(NAC)-α-MeDA was eliminated relatively slowly from the brain, concentrations in the pons/medulla, cortex, striatum, and hippocampus being virtually unchanged 2 to 6 h after administration of 5-GSyl-α-MeDA. The enzyme γ-glutamyl transpeptidase (γ-GT) is present in high concentrations in the endothelial cells of the blood-brain barrier and cleaves the γ-glutamyl bond of GSH; therefore, metabolism of GSH and its S-conjugates should reflect regional distribution of this enzyme. This was, in fact, demonstrated to occur as regional differences in brain γ-GT activity positively correlated with the total formation of 5-(CYS)-α-MeDA and 5-(NAC)-α-MeDA. However, whether or not 5-GSyl-α-MeDA and its metabolites are involved in MDMA- and MDA-mediated neurotoxicity would depend upon their ability to cross the blood-brain barrier (Miller et al., 1995).
Miller et al. (1996) demonstrated that a single administration of both MDA (s.c.) and 5-GSyl-α-MeDA (i.c.v.) caused an increased turnover of brain dopamine, shown by initial increases (1 h post-administration) and subsequent decreases (up to 7 days post-administration) in the concentration of dopamine and its metabolites. Acute increases in 5-HT turnover were also observed following i.c.v. administration of 5-GSyl-α-MeDA, but long-term serotonergic toxicity did not occur. Brain uptake of 5-GSyl-α-MeDA was shown to decrease following pretreatment with GSH and to increase following systemic pretreatment with acivicin, an inhibitor of γ-GT. The authors stated that these results might indicate competition between 5-GSyl-α-MeDA and GSH for the putative GSH transporter (Miller et al., 1996).
Miller et al. (1997) subsequently investigated the effects of multiple-dose administration of MDA (s.c.) and the α-MeDA metabolites 5-GSyl-α-MeDA, 5-(NAC)-α-MeDA, and 2,5-bis-(glutathion-S-yl)-α-MeDA (i.c.v.). Rats were administered four consecutive 12 hourly doses of each compound and were sacrificed 1 week later. MDA treatment resulted in significant depletion of 5-HT in the striatum, hippocampus, and cortex, while no such decreases were observed following administration of either 5-GSyl-α-MeDA or 5-(NAC)-α-MeDA. However, 2,5-bis-(glutathion-S-yl)-α-MeDA treatment resulted in significant reductions in overall cortical 5-HT levels and in ipsilateral hippocampal 5-HT and 5-HIAA concentrations. In addition, 2,5-bis-(glutathion-S-yl)-α-MeDA treatment resulted in modest reductions in striatal 5-HT, while striatal dopamine, DOPAC, and HVA were unaffected. Administration of each of the α-MeDA metabolites produced the same behavioral responses as MDA (hyperactivity, fore-paw treading, Straub tail, and low posture), but only 2,5-bis-(glutathion-S-yl)-α-MeDA produced serotonergic neurotoxicity that was restricted to the terminal areas. These results imply a dissociation between acute behavioral changes and long-term neurotoxicity, and subtle differences between the effects of 2,5-bis-(glutathion-S-yl)-α-MeDA and MDA suggests that other metabolites are likely to be involved in MDA-induced neurotoxicity (Miller et al., 1997).
Bai et al. (1999) extended the findings of Miller et al. (1997) by administering multiple doses of 2,5-bis-(glutathion-S-yl)-α-MeDA, 5-GSyl-α-MeDA, or 5-(NAC)-α-MeDA via direct injections into the striatum, cortex, and hippocampus, and neurotransmitter levels were analyzed 1 week after the last injection. Intrastriatal administration of 5-GSyl-α-MeDA resulted in significant depletion of 5-HT in the striatum and cortex, while intracortical administration resulted in significant 5-HT depletion in the cortex (and the striatum, following the higher dose only). 5-HT depletion was not observed in the hippocampus following either intrastriatal or intracortical administration, and 5-HT concentrations in the contralateral striatum and cortex, respectively, were also unchanged. Intrastriatal and intracortical administration of 2,5-bis-(glutathion-S-yl)-α-MeDA also resulted in 5-HT depletion in the striatum and cortex, while the hippocampus was again unaffected. Intrastriatal, intracortical, and intrahippocampal administration of 5-(NAC)-α-MeDA resulted in significant depletions of 5-HT in the striatum, cortex, and hippocampus, respectively. In addition, dopaminergic and noradrenergic systems were unaffected, indicating that the thio-ether metabolites of α-MeDA exhibit selectivity for serotonergic systems (Bai et al., 1999).
In a recent study investigating the involvement of glutathione in the production of neurotoxic metabolites O'Shea et al. (2002) depleted cerebral and peripheral glutathione stores with inhibitors of glutathione synthesis. Although these compounds produced a neuroprotective effect, evidence suggested that this action was due to a body temperature-lowering effect rather than true neuroprotection. Further studies are clearly required to clarify the involvement of glutathione in the neurotoxic process.
VII. Conclusions
There has been a steady increase in both interest in and knowledge of MDMA over the last 20 years, the number of yearly publications jumping from none in 1984 and 2 in 1985 to 25 in 1986 and over 100 in 2000. We now know much about the pharmacology of this compound in experimental animals, both in terms of its acute actions and its longer-term neurotoxic effects. In general, its effects are consistent across species, with the notable exception of the mouse. Importantly, its acute effects in humans are also very similar to those seen in experimental animals. What is uncertain is whether the clear and consistent long-term neurotoxic effects seen in animals can and do occur in humans. There are data suggesting that damage may occur in the human brain, and this should be a cause for concern. Nevertheless, it should be remembered that in common with every other drug, be it therapeutic or recreational, MDMA obeys common pharmacological principles, and adverse effects (both acute and long-term) are related to both dose and frequency of administration. Just because MDMA is a recreational drug does not make it inherently dangerous. The major problems in investigating the clinical effects of MDMA are the facts that prospective studies are generally unethical (so retrospective studies must be performed), the purity of the ingested drug, the doses taken, and frequency of administration are unknown, and many of the subjects are poly-drug users either by choice or unknowingly because of the impure nature of the tablets ingested.
Finally, it should be emphasized that despite several years of intensive effort by various laboratories, we still do not know the mechanisms by which MDMA produces long-term damage to serotonin nerve endings. We do have data strongly implicating some metabolic and other steps, including free radical formation. However, the full sequence of events and the identity of a specific chemical neurotoxic entity (if there is only one) have yet to be determined. Nevertheless, data obtained to date have proven to be valuable in enhancing our knowledge of the neuropharmacology of monoamines and neurotoxic degeneration.
Acknowledgments
We thank all the colleagues with whom we have had the pleasure of working on MDMA over the years. M.I.C. thanks Plan Nacional sobre Drogas (Ministerio del Interior), Ministerio de Ciencia y Tecnologia (Grant SAF2001-1437), Ministerio de Sanidad (Grant FIS01/0844), and Fundacion MapfreMedicina for financial support.
Footnotes
-
↵1 Present address: Department of Neurology, Johns Hopkins Bayview Medical Center, 5501 Hopkins Bayview Circle, Baltimore, MD 21224.
-
↵2 Abbreviations: MDMA, 3,4-methylenedioxymethamphetamine (“ecstasy”); MDA, 3,4-methylenedioxyamphetamine; LSD, d-lysergic acid diethylamide; 5-HT, 5-hydroxytryptamine (serotonin); MDEA, N-ethyl-3,4-methylenedioxyamphetamine (“Eve”); PCA, p-chloroamphetamine; MDBA, 3,4-methylenedioxybutylamphetamine; TPH, tryptophan hydroxylase; Ta, ambient temperature; MAO, monoamine oxidase; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, 4-hydroxy-3-methoxyphenylacetic acid (homovanillic acid); GBR 12909, 1-[2-bis(4-fluorophenyl)methoxy]ethyl]-4-3-phenylpropyl]piperazine; PKC, protein kinase C; DOI, 2,5-dimethoxy-4-iodoamphetamine; 5-MeODMT, 5-methoxy-N,N-dimethyltryptamine; NE, norepinephrine; DA, dopamine; IEG, immediate early gene; MK-801, (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (dizocilpine); SCH 23390, R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzapine; PBN, α-phenyl-N-tert-butyl nitrone; 2,3-DHBA, 2,3-dihydroxybenzoic acid; SD, Sprague-Dawley; PND, postnatal day; MR, metabolic rate; EWL, evaporative water loss; MDL 11,939, α-phenyl-1-(2-phenylethyl)-4-piperidinemethanol; 8-OH-DPAT, 8-hydroxy-2-(di-n-propylamino)tetralin; MDL 100,907, R-(+)-a-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol; GR 127935, 2′-methyl-4′-(5-methyl-[1,2,4]oxadiazol-3-yl)-biphenyl-4-carboxylic acid [4-methoxy-3-(4-methyl-piperazin-1-yl)-phenyl] amide; DNMTP, delayed nonmatch to place; 5,7-DHT, 5,7-dihydroxytryptamine; PPI, prepulse inhibition; 5-HIAA, 5-hydroxyindoleacetic acid; SOD, superoxide dismutase; NOS, nitric oxide synthase; AR-R17477AR, N-(4-(2-((3-chlorophenylmethyl) amino)-ethyl)phenyl) 2-thiophene carboxamidine; GFAP, glial fibrillary acidic protein; [125I]RTI-55, [125I]3β-(4-iodophenyl)tropane-2β-carboxylic acid methyl ester tartrate; [125I]MIL, N-1-methyl-2-[125I]lysergic acid diethylamide; [123I]R91150, [123I]4-amino-N-[1-[3-(4-fluorophenoxy) propyl]-4-methyl-4-piperidinyl]-5-iodo-2-methoxy-benzamide; SPECT, single photon emission computed tomography; CSF, cerebrospinal fluid; PET, positron emission tomography; [11C]McN-5652, 1,2,3,5,6,10b-hexahydro-6-[4-([11C]methylthio)-phenyl]pyrrolo-[2,1-α]-isoquinoline; HMMA, 4-hydroxy-3-methoxymethamphetamine; α-MeDA, α-methyldopamine (3,4-dihydroxyamphetamine); N-Me-α-MeDA, N-methyl-α-methyldopamine (3,4-dihydroxymethamphetamine); CYP450, cytochrome P450; rCBF, regional cerebral blood flow; EEG, electroencephalography; LORETA, low-resolution electromagnetic tomography; [123I]β-CIT, 2β-carbomethoxy-3β-(4-iodophenyl)tropane; rCBV, regional cerebral blood volume; MRI, magnetic resonance imaging; m-CPP, 1-(3-chlorophenyl)piperazine; MHPG, 3-methoxy-4-hydroxyphenyl glycol; 1H MRS, proton magnetic resonance spectroscopy; NA, N-acetylaspartate; MI, myoinositol; CR, creatine; CHO, choline compounds; DHMA, 3,4-dihydroxymethamphetamine (N-methyl-α-methyldopamine); 6-HO-MDMA, 2-hydroxy-4,5(methylenedioxy)methamphetamine; GSH, glutathione; Tri-HO-MA, 2,4,5-trihydroxymethamphetamine; 5-GSyl-α-MeDA, 5-(glutathion-S-yl)-α-methyldopamine; 2,5-bis-(glutathion-S-yl)-α-MeDA, 2,5-bis-(glutathion-S-yl)-α-methyldopamine; 5-(CYS)-α-MeDA, 5-(cystein-S-yl)-α-methyldopamine; 5-(NAC)-α-MeDA, 5-(N-acetylcystein-S-yl)-α-methyldopamine; 6-HO-MDA, 2-hydroxy-4,5-(methylenedioxy)amphetamine; Tri-HO-A, 2,4,5-trihydroxyamphetamine; γ-GT, γ-glutamyl transpeptidase; NMDA, N-methyl-d-aspartate
-
Article, publication date, and citation information can be found at http://pharmrev.aspetjournals.org.
-
DOI: 10.1124/pr.55.3.3.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
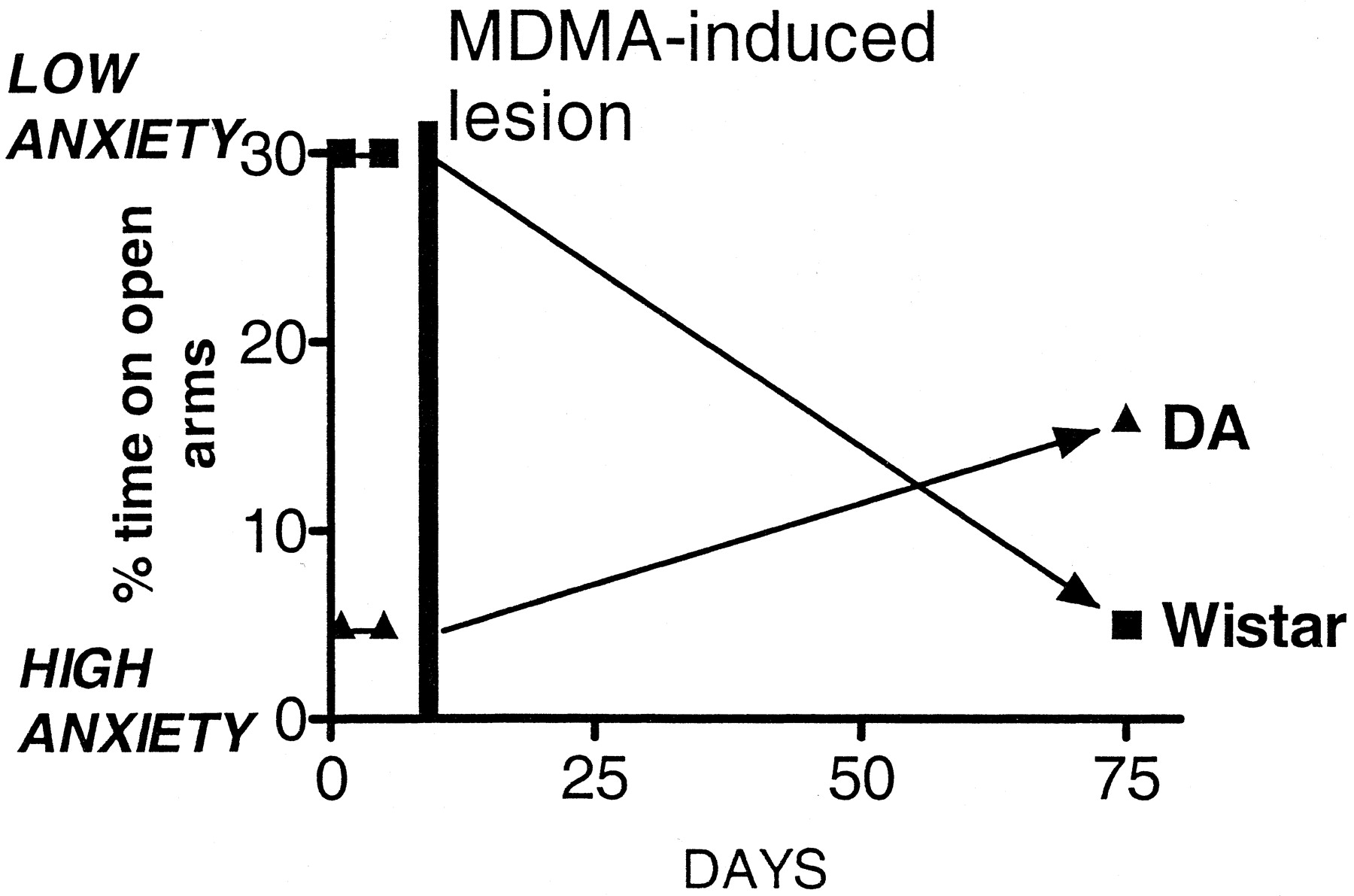{kind=link}
{kind=link}