


Abstract
Arylamine N-acetyltransferase 1 (NAT1) is a phase II xenobiotic-metabolizing enzyme that plays an important role in the biotransformation of aromatic drugs and carcinogens. NAT1 activity has long been associated with susceptibility to various cancers. Evidence for a role of NAT1 in malignant progression has also been obtained, particularly for breast and prostate cancer. Cisplatin is widely used in chemotherapy against human cancers, and it is thought to act principally by forming DNA adducts. However, recent studies have suggested that some of the pharmacological and/or toxicological effects of cisplatin may be due to the direct targeting and inhibition of certain cellular enzymes. We show here that the exposure of breast cancer cells, known to express functional NAT1 enzyme, to therapeutically relevant concentrations of cisplatin impairs the catalytic activity of endogenous NAT1. Endogenous NAT1 was also found to be inactivated, in vivo, in the tissues of mice treated with cisplatin. Mechanistic studies with purified human NAT1 indicated that this inhibition resulted from the irreversible formation of a cisplatin adduct with the active-site cysteine residue of the enzyme. Kinetic studies suggested that NAT1 interacts rapidly with cisplatin, with a second-order rate inhibition constant of 700 M-1 min-1. This rate constant is one the highest ever reported for the reaction of cisplatin with a biological macromolecule. Few enzymes have been clearly shown to be inactivated by cisplatin. We provide here molecular and cellular evidence suggesting that NAT1 is one of the targets of cisplatin in cells.
Acetylation is a key biotransformation pathway for various aromatic amines, including drugs and carcinogens. Several arylamines, such as 4-aminobiphenyl and benzidine, are important environmental carcinogens (Badawi et al., 1995; Hein, 2002), and changes in the N- and/or O-acetylation of these aromatic compounds have been linked to carcinogenesis (Hein, 2002, 2006). In humans, these reactions are catalyzed by two phase II xenobiotic-metabolizing enzymes (XMEs), arylamine N-acetyltransferase (NAT)1 and NAT2 (Hein, 2002). NAT enzymes thus play a key role in the detoxification and/or activation of several therapeutic drugs and carcinogens. NAT1 and NAT2 have very similar sequences, but they differ markedly in terms of aromatic substrate preference (Grant et al., 1991; Sim et al., 2007). Their tissue distribution also differs. NAT1 is ubiquitous (Rodrigues-Lima et al., 2003; Dairou et al., 2005b), whereas NAT2 is located principally in the liver and colon epithelium (Minchin et al., 2007). However, NAT2 mRNA has been detected in a wider range of tissues, suggesting that the NAT2 enzyme may also be present in other tissues (Husain et al., 2007). Polymorphisms affecting enzyme activity have been found in both genes (Hein, 2002). These interindividual variations have been implicated in differential susceptibility to adverse drug reactions and to various diseases, including cancers (Hein, 2002). In addition, human NATs—particularly NAT1—are influenced by environmental factors, such as certain arylamine substrates, oxidative stress, or androgens, which may either induce or inhibit cellular activity (Atmane et al., 2003; Dairou et al., 2003,2004; Butcher et al., 2007).
Many studies have reported a possible role for NAT1 and NAT2 in carcinogenesis and/or cancer progression (Badawi et al., 1995; Hein, 2002, 2006; Adam et al., 2003). In particular, NAT1 levels have been shown to be high in breast cancers (Adam et al., 2003; Bièche et al., 2004; Wakefield et al., 2008). The overproduction of NAT1 in normal luminal epithelial breast cells induces two of the hallmark traits of cancer: enhanced growth and resistance to toxic compounds, such as etoposide, which is used for chemotherapy (Adam et al., 2003). Recent studies have shown that human NAT1 is induced by androgens in human prostate cancer cells, with possible implications for cancer risk (Butcher et al., 2007). The association of NAT1 with carcinogenesis, particularly for breast cancer, suggests that this XME could be targeted in cancer treatment (Adam et al., 2003; Wakefield et al., 2008). Other phase II XMEs, such as glutathione transferases (GSTs), have been shown to be involved in malignant processes, leading to recent efforts to develop drugs targeting GST (McIlwain et al., 2006).
Cisplatin (cis-diaminedichloroplatinum II) is an important anticancer drug used in chemotherapy (Cullen et al., 2007). It is thought to act by forming intrastrand and interstrand cross-links with DNA, leading to cell death (Hasinoff et al., 2005), but the precise mechanism by which cisplatin affects tumor cells remains unclear, and many cellular targets may be involved (Cullen et al., 2007). Indeed, only approximately 1% of intracellular cisplatin is bound to DNA, most of the drug being available for interactions with nucleophilic sites on other molecules, such as proteins (Cullen et al., 2007). This suggests that some of the pharmacological and toxicological effects of cisplatin may be due to the inhibition of certain cellular enzymes. A limited number of enzymes have been identified as possible cellular targets of cisplatin. These enzymes include DNA polymerase (Duman et al., 1999), caspases 3 and 8 (Shin et al., 2005), and topoisomerase II (Hasinoff et al., 2005).
We showed, in cultured human breast cancer cells and in mouse tissues, that cisplatin inhibits the enzymatic functions of human NAT1 in vivo. Further analysis showed that NAT1 was rapidly (k = 700 M-1 min-1inact and irreversibly inactivated by cisplatin, through the formation of a cisplatin-NAT complex at the active site of the enzyme. Thus, our results identify NAT1 as a new enzymatic target of cisplatin, suggesting that the inhibition of this XME by cisplatin may contribute to the pharmacological and/or toxicological effects of this drug in cells.
Materials and Methods
Materials. Cisplatin, p-aminosalicylic acid, acetyl (Ac)CoA, CoA, 1,4-dithiothreitol (DTT), reduced glutathione (GSH), protease inhibitor cocktail, and nickel-agarose resin were obtained from Sigma (St.-Quentin Fallavier, France). Anti-fluorescein Fab' fragments conjugated to peroxidase and fluorescein-conjugated iodoacetamide were obtained from Roche Applied Science (Paris, France). The Bradford protein assay kit was supplied by Bio-Rad (Marne la Coquette, France). All other reagents were purchased from Sigma or Euromedex (Souffelweyersheim, France).
Cisplatin Treatment and Total Cell Extracts. MCF-7 and MDA-MB-231 cells (all human breast cancer cells expressing functional NAT1 enzyme; Wakefield et al., 2008) were cultured as monolayers in 100-mm Petri dishes at 37°C in Dulbecco's modified Eagle's medium (Invitrogen, Paris, France) supplemented with 20% fetal bovine serum and penicillin/streptomycin.
At ∼90% confluence, cell monolayers were washed with PBS and exposed to various concentrations of cisplatin (obtained by dilution of a 100 mM stock solution in 75% dimethyl sulfoxide/25% H2O) in 10 ml of PBS for 30 min at 37°C. Control cells were treated with PBS only. The monolayers were then washed with PBS and scraped into 1 ml of lysis buffer (20 mM Tris-HCl, pH 7.5, 0.2% Triton X-100, and protease inhibitors). Extracts were briefly sonicated and centrifuged for 15 min at 13,000g. Supernatants (total cell extracts) were removed, and their protein concentration determined using the Bradford reagent following the manufacturer's instructions with bovine serum albumin as a standard. All cell extracts were adjusted to the same protein concentration, by adding lysis buffer, and they were used for enzyme assays and Western blotting.
We investigated whether cisplatin inhibited NAT1 function in vivo, by treating C57BL/6J mice with 0.6 mg of cisplatin/mouse, as described by Shin et al. (2005). One hour after cisplatin injection (corresponding to peak cisplatin concentration in most tissues; Korst et al., 1998), mice were killed by cervical dislocation, and endogenous murine NAT2 activity (this enzyme being the murine counterpart of the human NAT1 isoform) was assessed in tissues producing murine Nat2 and preferentially targeted by cisplatin, such as the liver, kidney, and blood (Korst et al., 1998).
Production and Purification of Recombinant Human NAT1.Escherichia coli BL21 (DE3) cells containing a pET28a-based plasmid were used to produce 6x-His-tagged NAT1, as described previously (Dairou et al., 2004). Purified NAT1 was reduced by incubation with 10 mM DTT for 10 min at 4°C and dialyzed against 25 mM Tris-HCl, pH 7.5. Purity was assessed by SDS-PAGE, and protein concentrations were determined using the Bradford reagent following the manufacturer's instructions with bovine serum albumin as a standard.
Enzyme Assay. NAT1 activity was detected as described previously in cells or tissue extracts (Dairou et al., 2004), in a total volume of 100 μl. Samples (50 μl) were first incubated with 200 μM p-aminobenzoic acid in assay buffer (25 mM Tris-HCl, pH 7.5) at 37°C for 5 min. AcCoA (400 μM) was added to start the reaction, and the samples were incubated at 37°C for various periods (up to 30 min). The reaction was quenched by adding 100 μl of ice-cold aqueous trichloroacetic acid [20% (w/v)], and proteins were recovered by centrifugation for 5 min at 12,000g. 4-Dimethylaminobenzaldehyde [DMAB; 800 μl, 5% (w/v) in 9:1 acetonitrile/water] was added, and absorbance was measured at 450 nm. The amount of residual arylamine was determined from a standard curve. All assays were performed in triplicate, such that a straight line was obtained for the initial reaction rate. Enzyme activities were normalized according to the protein concentration of extracts, and they are expressed as a percentage of control NAT1 activity.
Recombinant NAT1 activity was determined with the 5,5′-dithio-bis-(2-nitrobenzoic acid) assay, as described previously (Dairou et al., 2004).
Reaction of Recombinant NAT1 with Cisplatin. Stock solutions of cisplatin (100 mM) were prepared in 75% dimethyl sulfoxide/25% H2O (v/v). In all subsequent experiments, the final concentration of purified recombinant NAT1 during the preincubation steps with cisplatin was 0.4 μM, giving a final concentration in the enzyme assay of 4 nM (total volume of the assay, 1 ml). The effect of cisplatin on NAT1 activity was assessed by incubating the purified enzyme (0.4 μM final concentration) with various concentrations of cisplatin in 25 mM Tris-HCl, pH 7.5 (total volume of 10 μl), for 1 h at 37°C. Samples were then assayed, and NAT1 activity in absence of cisplatin was taken as 100%.
We tested the ability of reducing agents to protect NAT1 from the effects of cisplatin by treating the purified protein with cisplatin in the presence or absence of various concentrations of GSH or DTT and then determining residual NAT1 activity. Control assays were carried out in the presence of GSH or DTT only and gave 100% NAT1 activity. We assessed the reactivation of the cisplatin-treated enzyme by reducing agents, by treating the enzyme with cisplatin (100 μM final concentration) for 1 h at 37°C in a total volume of 10 μl, and then with GSH or DTT (final volume 50 μl) for 15 min at 37°C. Residual NAT1 activity was determined as described above. Controls including GSH or DTT only were carried out and gave 100% NAT1 activity.

Impairment of human NAT1 and murine Nat2 enzymatic functions by cisplatin in cultured human breast cancer cells and mouse tissues, respectively. A, human breast cancer cells (MCF-7, dark gray bars; MDA-MB-231, light gray bars) cultured in Petri dishes were exposed to cisplatin in 10 ml of PBS for 1 h at 37°C. Cells were washed with PBS and total extracts were prepared. Residual human NAT1 activity in cell extracts, adjusted to the same protein concentration, was assessed using DMAB, as described previously (Sinclair et al., 1998). Results are means of treatments performed in triplicate. Activity was measured in triplicate. Results are presented as a percentage of NAT1 activity present in the control extract. Error bars indicate the S.D. value. *, p < 0.05 versus NAT1 activity in control. Cisplatin dose-dependent differences were all statistically significant at p < 0.05, except for 250 and 500 μM ciplatin concentrations (MCF-7 cells). B, PBS or cisplatin (0.6 mg/mouse) was injected intraperitoneally into adult C57BL/6J mice (n = 6), as described by Shin et al. (2005). One hour after treatment, mice were killed, and mouse NAT2 activity in lysates from the liver, kidney, and blood tissues was measured with the DMAB assay. Results are presented as a percentage of the Nat2 activity present in control extracts. Error bars indicate S.D. values. The p values for comparisons with Nat2 activity in control extracts are indicated.
Substrate protection experiments were carried out by incubating NAT1 with various concentrations of AcCoA or CoA (final concentrations 0.5-3 mM) in 25 mM Tris-HCl, pH 7.5, for 10 min at 37°C. Samples were then incubated with 100 μM cisplatin for 1 h at 37°C and assayed. Assays carried out in these conditions with AcCoA or CoA alone gave 100% NAT1 activity.
Fluorescein-Conjugated Iodoacetamide Labeling of Human NAT1. Purified NAT1 (0.4 μM final concentration) was incubated with or without (control) various concentrations of cisplatin (12.5-100 μM final concentration) in 25 mM Tris-HCl, pH 7.5, for 30 min at 37°C. Samples were then incubated with fluorescein-conjugated iodoacetamide (20 μM final concentration) for 10 min at 37°C. Samples were then analyzed by SDS-PAGE under reducing conditions followed by Western blotting with peroxidase-conjugated antifluorescein Fab' fragments. Quantitation of intensities in Westernblots was carried out using the MultiGauge 3.0 program (FujiFilm, St. Quentin-En-Yvelines Cedex, France).
Kinetic Analysis of Cisplatin-Dependent NAT1 Inactivation. NAT1 (0.4 μM final concentration) was incubated with cisplatin (final concentrations of 0-125 μM) at 37°C in 25 mM Tris-HCl, pH 7.5. At various time intervals, aliquots were removed and assayed for residual activity. The equation for the rate of inactivation of purified NAT1 by cisplatin can be written -d[NAT1]/dt = kinact · [NAT1] · [cisplatin], where [NAT1] is the concentration of active enzyme, and kinact is the second-order rate constant. As cisplatin is present in substantial excess over NAT1, the apparent first-order inactivation rate constants (kobs = kinact · [cisplatin]) can be calculated for each cisplatin concentration, from the slope of the natural log (ln) of percentage of residual activity plotted against time. The second-order rate constant was determined from the slope of kobs against cisplatin concentration. All kinetic data were obtained with KaleidaGraph version 3.5 (Abelbeck/Synergy, Reading, PA).
SDS-PAGE and Western Blotting. Samples were mixed with reducing 4× SDS sample buffer, boiled for 3 min at 100°C, and separated by SDS-PAGE. Gels were stained with Coomassie Brilliant Blue R-250. For Western blotting, proteins were electrotransferred onto nitrocellulose membrane. The membrane was blocked by incubation with Tris-buffered saline/Tween 20 (TBS) supplemented with 5% nonfat milk powder for 1 h in TBS. Antibodies were added (1/20,000), and the membrane was incubated for 1 h in TBS. SuperSignal reagent (Pierce Chemical, Rockford, IL) was used for detection.

Inactivation of purified recombinant NAT1 by cisplatin and labeling of NAT1 cysteine residues by fluorescein-conjugated iodoacetamide after cisplatin treatment. A, recombinant NAT1 (0.4 μM) was incubated with cisplatin at the indicated final concentrations in 25 mM Tris-HCl, pH 7.5, for 1 h at 37°C. Residual NAT1 activity was then determined, using the 5,5′-dithio-bis-(2-nitrobenzoic acid) assay, as described previously (Dairou et al., 2004). These results are the means of treatments carried out in triplicate. NAT1 activity was determined in triplicate. Results are presented as a percentage of the NAT1 activity of the control. Error bars indicate S.D. values. *, p < 0.05 versus NAT1 activity in control. Cisplatin dose-dependent differences were all statistically significant at p < 0.05. B, recombinant NAT1 (0.4 μM) was incubated for 1 h at 37°C with the indicated concentrations of cisplatin, as described in Fig. 2. The reaction mixture was further incubated with fluorescein-conjugated iodoacetamide (20 μM final concentration) for 10 min at 37°C. Samples were then subjected to SDS-PAGE under reducing conditions, followed by Western blotting with an anti-fluorescein antibody. Cisplatin was omitted from the control. C, quantitation of signals in Western blots (relative to control) are shown. Error bars indicate S.D. values. *, p < 0.05 versus control. Cisplatin dose-dependent differences were all statistically significant at p < 0.05.
Statistical Analysis. Data are means ± S.D. of three independent experiments performed in triplicate, unless otherwise stated. One-way analysis of variance was performed and followed by Student's t test (unpaired and paired), between two groups using Stat-View 5.0 (SAS Institute, Cary, NC).
Results
Cisplatin Inhibits the Endogenous NAT1 Enzyme.Cisplatin is known to interact with DNA, but it has also been shown to target certain key cellular enzymes, inhibiting their catalytic functions (Shin et al., 2005).
We assessed the effect of cisplatin on cellular NAT1 by exposing cultured human breast cancer cells (MCF-7 and MDA-MB-231 cells), which are known to express this XME (Wakefield et al., 2008), to various concentrations of this chemotherapeutic drug. The exposure of MCF-7 cells to cisplatin at clinically relevant concentrations (<400 μM) (Sfikakis et al., 1996; Tegeder et al., 2003) caused significant dose-dependent inhibition of the endogenous NAT1 enzyme (Fig. 1A). The IC50 value for the inactivation of cellular NAT1 in MCF-7 was close to 100 μM. Similar results were obtained with MDA-MB-231 breast cancer cells (Fig. 1A). We investigated whether cisplatin treatment inhibited NAT1 in vivo, by treating C57BL/6J mice (n = 6) with cisplatin, as described by Shin et al. (2005). As shown in Fig. 1B, a single cisplatin treatment significantly decreased murine Nat2 (murine counterpart of human NAT1) enzymatic function in the liver (∼25% inhibition), kidney (∼40% inhibition), and blood cells (∼50% inhibition). These data support the hypothesis that NAT1 is an enzymatic target of cisplatin.

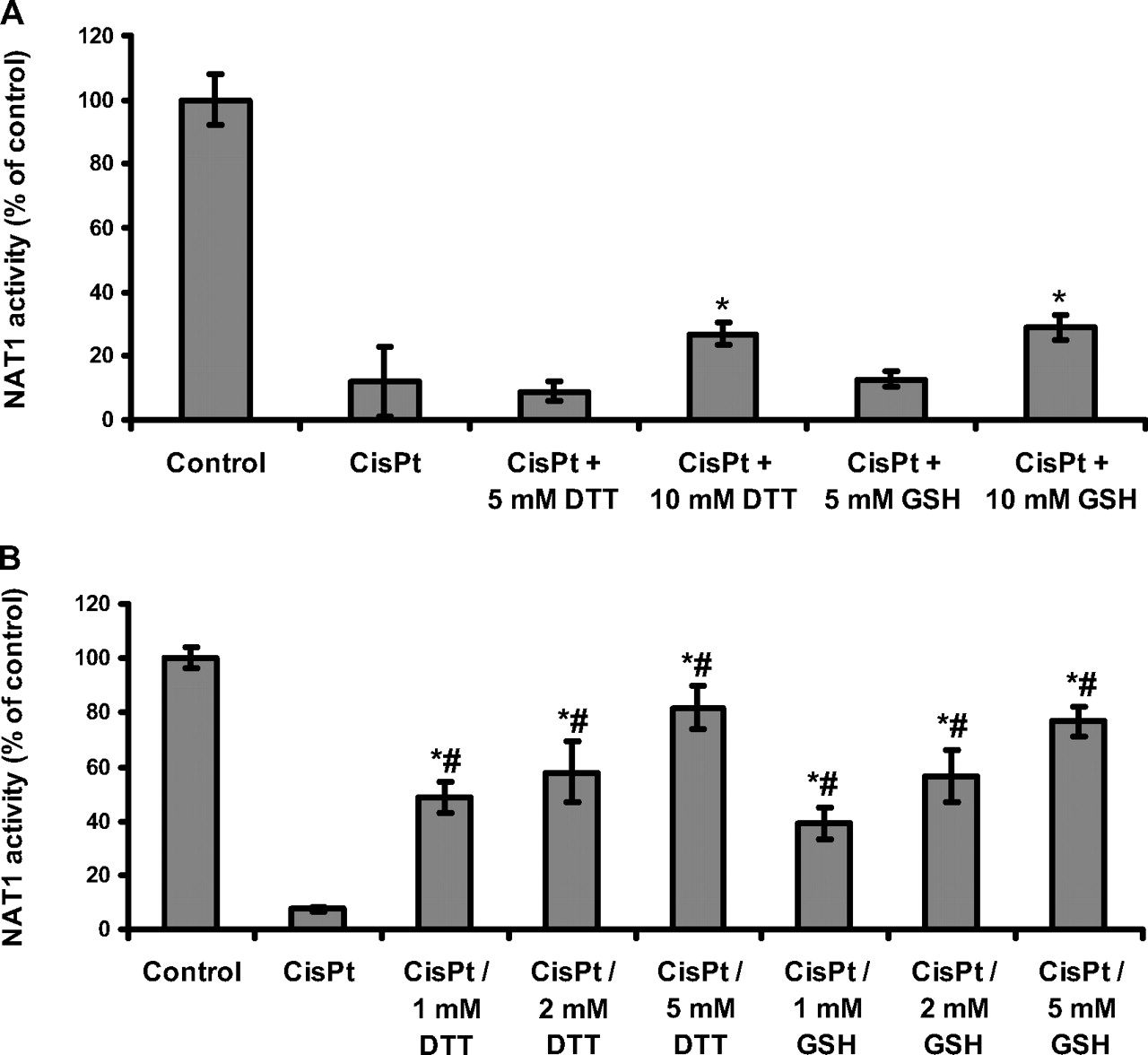
Effect of reducing agents on cisplatin-dependent NAT1 inactivation. A, recombinant NAT1 (0.4 μM) was incubated with cisplatin (100 μM final concentration) in 25 mM Tris-HCl, pH 7.5, for 1 h at 37°C. Inactivated NAT1 was then incubated with various concentrations of reducing agent (GSH and DTT) for 1 h at 37°C, and enzyme activity was measured. NAT1 activity was determined in triplicate. Results are presented as a percentage of the NAT1 activity of the control. Error bars indicate S.D. values. *, p < 0.05 versus cisplatin-inactivated enzyme. Dose-dependent differences were statistically significant (p < 0.05) for the concentrations of DTT and for the concentrations of GSH. B, NAT1 (0.4 μM) was incubated with 100 μM cisplatin in the presence of GSH or DTT in 25 mM Tris-HCl, pH 7.5, for 1 h at 37°C. Cisplatin was omitted for the control. These results are the means of treatments in triplicate. NAT1 activity was determined in triplicate. Results are presented as a percentage of the NAT1 activity present in the control. Error bars indicate S.D. values. *, p < 0.05 versus cisplatin-inactivated enzyme; #, p < 0.05 versus control NAT1 (100% active enzyme). Dose-dependent differences were all statistically significant (p < 0.05) for DTT concentrations and for GSH concentrations.
We investigated the molecular mechanisms by which cisplatin inactivates endogenous and recombinant NAT1 further, by carrying out biochemical and kinetic analyses on the recombinant NAT1 enzyme, as reported below.
In Vitro Dose-Dependent Inactivation of Purified Human NAT1 by Cisplatin. As observed with cellular NAT1, the exposure of purified NAT1 to cisplatin (up to 100 μM) for 1 h led to the dose-dependent inactivation of the enzyme (Fig. 2A). Fifty percent inhibition of the enzyme was obtained with concentrations of cisplatin as low as 20 μM. At a concentration of 100 μM cisplatin, full inhibition (>97% inhibition) of the enzyme was obtained.
Modification of the Cysteine Residues of NAT1 by Cisplatin. Cisplatin has been shown to form adducts with certain reactive protein thiol groups (Hasinoff et al., 2005). We therefore assessed the reaction of cisplatin with NAT1 cysteine residues, using a method based on the sulfhydryl-reactive reagent iodoacetamide-fluorescein, as described previously (Dairou et al., 2004). The incubation of NAT1 with various concentrations of cisplatin resulted in the dose-dependent modification of cysteine residues, as indicated by the disappearance of fluorescein-conjugated iodoacetamide labeling (Fig. 2B). The dose-dependent inactivation of purified NAT1 by cisplatin (Fig. 2A) was strongly correlated with dose-dependent modification of the cysteine residues of the enzyme (Fig. 2, B and C). Thus, cisplatin clearly forms adducts with the cysteine residues of NAT1, and the timing of this process coincides with functional impairment.
Effect of Reducing Agents on the Cisplatin-Dependent Inactivation of NAT1. Cisplatin reacts with certain reactive thiol groups to form quasi-irreversible cisplatin-thiol adducts (Hagrman et al., 2003, 2004; Hasinoff et al., 2005). We investigated whether the cisplatin-dependent inhibition of NAT1 could be reversed by adding excess of reducing agent. No significant reactivation of the enzyme was observed after the addition of a high concentration (5 mM) of GSH (Fig. 3A). Similar data were obtained with the non-physiological reducing agent DTT. Up to 30% enzyme reactivation was observed at the very high concentrations (10 mM) of GSH or DTT.

Kinetic analysis of cisplatin-dependent NAT1 inactivation. A, recombinant NAT1 enzyme (0.4 μM) was treated with various concentrations of cisplatin at 37°C in 25 mM Tris-HCl, pH 7.5. At various time intervals, aliquots were removed and assayed for residual NAT1 activity. Plots of ln percentage of residual activity against time are shown. The apparent first-order inactivation constant (kobs) are calculated from the linear regressions. No cisplatin (▪); 25 μM (•); 50 μM(▴); and 125 μM(♦). B, determination of the second-order rate inactivation constant (kinact) by plotting kobs values against cisplatin concentrations. C, determination of the order of the reaction by replotting ln kobs against ln cisplatin concentration.
We investigated whether the cellular reducing agent GSH could prevent the cisplatin-dependent inactivation of NAT1. At a GSH concentration of 1 mM, corresponding to 10-fold excess of GSH over cisplatin and a 2500-fold excess of GSH over NAT1 enzyme), GSH protected NAT1 only partially (∼50% protection) against cisplatin-dependent inactivation (Fig. 3B). A concentration of 2 mM GSH gave ∼58% protection. At much higher concentrations of GSH (5 mM final concentration), ∼75% residual NAT1 activity was obtained. Similar results were obtained with DTT. Full protection against cisplatin-induced NAT1 inactivation was obtained only at very high concentrations of GSH or DTT (10 mM final concentration; data not shown). These data indicate that the physiological reducing agent GSH provides strong protection against cisplatin-dependent NAT1 inactivation only at high concentrations (5-10 mM), which suggests that cisplatin reacts more rapidly with NAT1 than with GSH. We addressed this issue by carrying out kinetic analyses of the ciplatin-NAT1 reaction.
Kinetics and Stoichiometry of Inactivation by Cisplatin. We further analyzed the inhibition of NAT1 by cisplatin, by carrying out a time course study of inactivation of the enzyme. Semilogarithmic plots of percentage of residual activity against time for various concentrations of cisplatin gave straight lines, indicating that the inactivation followed apparent first-order kinetics (Fig. 4A). Replotting the observed pseudo-first order rate constants (kobs) against cisplatin concentrations gave a straight line passing through the origin (Fig. 4B), consistent with a single-step bimolecular process. The second-order rate constant (kinact) for the inactivation of NAT1 by cisplatin was estimated at 700 M-1 min-1. These data are consistent with the results reported above, and they support that, even in the presence of high concentrations of GSH, cisplatin significantly inactivates NAT1 (Fig. 4). The stoichiometry of NAT1 inactivation by cisplatin was determined by replotting ln kobs against ln cisplatin concentration (Fig. 4C), as the first-order rate constant for each cisplatin concentration could be expressed as kobs = kinact. [cisplatin]n, where n is the order of cisplatin in its reaction with NAT1. We obtained a value for n of ∼1, indicating that NAT1 inactivation by cisplatin is due to the reaction of one molecule of cisplatin with a single site of NAT1.
Identification of the Site Involved in Cisplatin-Dependent Inactivation. Cysteine labeling (Fig. 2, B and C) and kinetics data suggested that the cisplatin-dependent inactivation of NAT1 was probably due to the modification of a single cysteine residue of the enzyme. We therefore used the catalytic cysteine protection assay (Dairou et al., 2004, 2005a) to investigate whether a cisplatin adduct at the active site cysteine residue was responsible for NAT1 inactivation. This assay is based on the specific acetylation, by AcCoA, of the catalytic cysteine residue of NAT enzymes (resulting in the formation of a covalent acetyl-enzyme complex), protecting this residue against further chemical modification (Minchin et al., 2007). CoA cannot acetylate the catalytic cysteine residue, so this residue remains susceptible to chemical modification after incubation with CoA. AcCoA protected NAT1 activity in a dose-dependent manner, with a residual NAT1 activity of 66 ± 2% obtained when the enzyme was incubated with cisplatin in the presence of 2 mM AcCoA (Table 1). Conversely, a residual activity of 7 ± 2% was obtained with 2 mM CoA. Our data suggest that cisplatin inactivates the NAT1 enzyme by forming an adduct with the catalytic cysteine residue of the enzyme.
Protection of NAT1 against cisplatin-dependent inactivation by AcCoA and CoA
NAT1 was incubated with cisplatin (100 μM final concentration) in the presence of AcCoA or CoA at the indicated final concentrations, as described under Materials and Methods.
Discussion
Many studies have associated NAT1 and NAT2 activities with an increased risk of cancer (Hein, 2002, 2006). In particular, there is increasing evidence to suggest that NAT1 plays a biological role in breast cancer progression (Adam et al., 2003; Bièche et al., 2004; Wakefield et al., 2008) and that this XME could be targeted in treatment (Wakefield et al., 2008).
Cisplatin is one of the leading chemotherapeutic agents used to treat various human cancers (Ott and Gust, 2007). The efficacy of cisplatin against several breast cancer cell lines in vitro suggests that this compound is potentially useful for the clinical treatment of breast cancer (Ott and Gust, 2007). The cytostatic/cytotoxic effects of cisplatin have largely been attributed to its ability to form adducts with DNA (Hasinoff et al., 2005), but effects of cisplatin on other cellular targets may also contribute to its biological effects (Cullen et al., 2007). We investigated the possibility of NAT1 being a target of cisplatin in vitro and in vivo. We show here that the endogenous NAT1 expressed by MCF-7 and MDA-MB-231 human breast cancer cells is inactivated by short-term exposure (1 h) of these cells to therapeutically relevant cisplatin levels. Cisplatin also significantly inhibited endogenous Nat2 activity (mouse Nat2 is the murine counterpart of human NAT1) in tissues (liver, blood, and kidney) from cisplatin-treated mice, providing further support for the notion that NAT1 is an enzymatic target of cisplatin in cells. Further biochemical and kinetic studies using recombinant purified NAT1 confirmed that cisplatin impairs NAT1 function through the almost irreversible formation of an adduct with the catalytic cysteine of the enzyme. The second-order rate constant for NAT1 inactivation by cisplatin was found to be 700 M-1 min-1, indicating that the catalytic cysteinecisplatin adduct forms rapidly. This rate constant is the highest ever reported for a reaction between a biological macromolecule and cisplatin. Indeed, cisplatin reacts with DNA with a second-order rate constant of 125 M-1 min-1 (Jestin et al., 1998). Metallothioneins and GSH, two of the most important cellular thiol-containing molecules protecting cells against cisplatin through thiol-adduct formation, react with the drug with second-order rate constants lower than those for NAT1 inactivation by factors of 18 (k = 38 M-1 min-1) and 430 (k = 1.6 M-1 min-1), respectively (Hagrman et al., 2003, 2004). Other thiol-containing molecules, such as 2-(3-aminopropylamin-o)ethanethiol (WR 1065) and sodium-2-mercaptoethanesulfonate (mesna), which are often administered to mitigate cisplatin toxicity, react 1000 (k = 0.6 M-1 min-1) and 160 (k = 4.3 M-1 min-1) times more slowly with the drug than NAT1 (Dabrowiak et al., 2002). Our data show that, even in the presence of a high concentration of GSH (5 mM final concentration), cisplatin (at a final concentration one fiftieth that of GSH) was still able to react with and inactivate NAT1, consistent with the kinetic results discussed above. These data may also explain why cellular levels of metallothioneins (∼1 mM; Hagrman et al., 2003) and GSH (∼2 mM; Boubakari et al., 2004) in cells and mouse tissues do not fully protecting endogenous NAT1 against cisplatin-dependent inactivation. The reactive nature of the catalytic cysteine residue of NAT1 (Dupret and Rodrigues-Lima, 2005; Minchin et al., 2007) probably accounts for the strong reactivity of cisplatin with this residue and the subsequent rapid inactivation of the enzyme through cysteine-cisplatin adduct formation. Likewise, caspases 3 and 8 were recently shown to be inactivated by cisplatin, through reaction of the drug with the catalytic cysteine residue of these proteases (Shin et al., 2005).
Phase II XMEs other than NAT1 have also been shown to react with cisplatin. GST-π isozymes have been shown to bind cisplatin at their active site and to catalyze the conjugation of GSH to the drug, thereby possibly contributing to cisplatin resistance (Townsend et al., 2002). Other studies have also suggested that certain GST isoforms may be inhibited by exposure to cisplatin, but the underlying molecular mechanism remains unclear (Sadzuka et al., 1994; Dwivedi et al., 1996; Khynriam and Prasad, 2002).
This study provides molecular and cellular evidence to suggest that NAT1, a carcinogenesis-associated XME, is a primary target of cisplatin in vitro and in vivo. We showed that cisplatin reacts rapidly with the active-site cysteine residue of the enzyme, forming a cisplatin-cysteine adduct, resulting in functional impairment. There is increasing evidence to suggest that NAT1 plays a biological role in cancer progression (Adam et al., 2003; Butcher et al., 2007), particularly in breast cancers, in which it may increase cell growth (Wakefield et al., 2008). Interestingly, tamoxifen, a chemotherapeutic drug active against breast (Veronesi et al., 2005) and prostate (Mimeault et al., 2007) cancer has been shown to impair NAT1 in breast cancer cells (Lee et al., 2004). Overall, our results suggest that the irreversible inactivation of NAT1 by cisplatin may at least partly contribute to the pharmacological and/or toxicological effects of cisplatin.
Footnotes
-
This work was supported by Association pour la Recherche sur le Cancer, Association Française contre les Myopathies, la Chancellerie des Universités de Paris (Leg Poix) and la Caisse d'Assurance Maladies des Professions Libé-rales de Province. J.-M.D. and F.R.-L. contributed equally to this work.
-
ABBREVIATIONS: XME, xenobiotic-metabolizing enzyme; NAT, arylamine N-acetyltransferase; Ac, acetyl; DTT, 1,4-dithiothreitol; GSH, reduced glutathione; PBS, phosphate-buffered saline; PAGE, polyacrylamide gel electrophoresis; DMAB, 4-dimethylaminobenzaldehyde; TBS, Tris-buffered saline/Tween 20.
- Received January 14, 2008.
- Accepted February 29, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}