


Abstract
Expression of Urate transporter 1 (URAT1/SLC22A12) is restricted to the proximal tubules in the kidney, where it is responsible for the tubular reabsorption of urate. To elucidate the mechanism underlying its tissue-specific expression, the transcriptional regulation of the hURAT1 and mUrat1 genes was investigated. Hepatocyte nuclear factor 1 α (HNF1α) and HNF1β positively regulate minimal promoter activity of the URAT1 gene as shown by reporter gene assays. Electrophoretic mobility shift assays revealed binding of HNF1α and/or HNF1β to the HNF1 motif in the hURAT1 promoter. Furthermore, the mRNA expression of Urat1 is reduced in the kidneys of Hnf1α-null mice compared with wild-type mice, confirming the indispensable role of HNF1α in the constitutive expression of URAT1 genes. It was also shown that the proximal promoter region of mUrat1 was hypermethylated in the liver and kidney medulla, whereas this region was relatively hypomethylated in the kidney cortex. These methylation profiles are in a good agreement with the proximal tubule-restricted expression of mUrat1 in the kidney cortex. Taken together, these results strongly suggest that tissue-specific expression of the URAT1 genes is coordinately regulated by the transcriptional activation by HNF1α/HNF1β heterodimer and repression by DNA methylation.
Urate is an end product of purine metabolism in humans and other higher primates. It is generally recognized that urate works as a scavenger of potentially harmful radicals in the human body. However, hyperuricemia can be associated with health problems such as gout, nephrolithiasis, hypertension, and vascular disease, whereas hypouricemia is primarily characterized by exercise-induced acute renal failure. Thus, the serum urate level must be tightly regulated through complex renal handling processes, which is historically explained by a four-component model (i.e., glomerular filtration, tubular secretion, and pre- and postsecretory reabsorption). After these processes, approximately 90% of the urate filtered through the glomerulus is reabsorbed in humans (Rafey et al., 2003; Enomoto and Endou, 2005; Hediger et al., 2005).
Urate transporter 1 (URAT1/SLC22A12) is a membrane transporter responsible for the reabsorption of urate in the apical membrane of the renal proximal tubules (Enomoto et al., 2002). Mutations in the URAT1 gene, causing functional impairment, are associated with idiopathic renal hypouricemia (Enomoto et al., 2002; Iwai et al., 2004). In addition, fractional excretion of urate in patients with homozygous or compound heterozygous SLC22A12 mutations is not sensitive to uricosuric (probenecid and benzbromarone) and anti-uricosuric (pyrazinamide) drugs, suggesting that these drugs target URAT1 to exert their effects in vivo (Ichida et al., 2004). The mouse homolog of hURAT1, mUrat1, shows the same tissue distribution as that of hURAT1 and is probably involved in the reabsorption of urate in the kidney (Hosoyamada et al., 2004). It can also be an exit pathway for several anionic drugs in the proximal tubules together with the basolateral uptake transporters [organic anion transporter (Oat) 1 and Oat3] (Imaoka et al., 2004).
A previous report suggested the presence of a hepatocyte nuclear factor 1 (HNF1) binding motif in the minimal promoter region of the hURAT1 gene, which is conserved in the proximal promoters of rodent Urat1 genes (Li et al., 2004). HNF1 is known to regulate transcription of many hepatic genes by forming homodimers or heterodimers between two isoforms, HNF1α and HNF1β (Mendel and Crabtree, 1991; Tronche and Yaniv, 1992). Their expression in the kidney and a renal Fanconi syndrome or polycystic kidney disease in Hnf1α- or kidney-specific Hnf1β-null mice, respectively, also imply a role for these transcription factors in the kidney (Pontoglio et al., 1996; Gresh et al., 2004). We have found recently that HNF1α/β is a potent transactivator of the human and mouse OAT3 promoter (Kikuchi et al., 2006). Then it was reported that the mRNA levels of several drug transporters, including Oat1 and Oat3, are reduced in the kidney of Hnf1α-null mice, further confirming the involvement of HNF1α in the transcriptional regulation of drug transporters in the kidney (Maher et al., 2006). The functional importance of HNF1 in the regulation of the URAT1 gene has yet to be investigated. However, considering that the serum levels of urate are reduced and the renal fractional excretion is increased in Hnf1α-null mice (Pontoglio et al., 1996), it is possible that HNF1α directly regulates URAT1 expression.
HNF1α and HNF1β are expressed in the polarized epithelia of a variety of tissues, including liver, kidney, intestine, stomach, and pancreas (Blumenfeld et al., 1991; De Simone et al., 1991). Thus, positive regulation by HNF1 alone cannot account for the predominant expression of URAT1 in the kidney. Our previous study suggests that the expression of hOAT3 is negatively regulated by DNA methylation in addition to the positive regulation by HNF1α and HNF1β (Kikuchi et al., 2006). DNA methylation is one of the most well-characterized mechanisms underlying the epigenetic regulation of gene expression (Bird, 2002; Shiota, 2004). Methylation of the cytosine residue in the CpG dinucleotide negatively regulates gene transcription through the recruitment of chromatin remodeling factors, and many reports highlight the key role of DNA methylation in tissue-specific gene transcription. Because several CpG dinucleotides were found in the 5′-flanking region of hURAT1 and mUrat1 (Fig. 1), it is of great interest to determine whether the concerted effect of DNA methylation and HNF1α/β regulates tissue-specific expression of these genes. The present study was aimed to clarify the involvement of HNF1α and HNF1β as genetic factors and DNA methylation as an epigenetic factor in the tissue-specific expression of human and mouse URAT1 genes.
Materials and Methods
Isolation of the 5′-Flanking Sequence of hURAT1 and mUrat1 Gene. The minimal promoter region of hURAT1 gene (-253/+83) (Li et al., 2004) was amplified by PCR using human genomic DNA as a template and the forward (h - 253) and reverse (h + 83) primers with an artificial KpnI and HindIII site, respectively (Table 1). The PCR product was subcloned into the KpnI and HindIII restriction sites of pGL3-Basic (Promega, Madison, WI), yielding the hURAT1 minimal promoter construct, hURAT1_-253/+83-HNF1wt. The transcriptional start site of the mUrat1 gene was determined using the public database Database of Transcriptional Start Sites (available at http://dbtss.hgc.jp/) with the ref sequence identification for mUrat1 (NM_009203), and based on high homology with the 5′-flanking region of hURAT1 (Fig. 1). Because the position of transcriptional start site in the clone MS2926204 is nearest to that of hURAT1 transcriptional start site, which was experimentally determined in the previous report, we chose this site as the transcriptional start site of mUrat1 gene. The mUrat1 putative promoter region was isolated from mouse genomic DNA by a PCR-based approach using the forward (m - 261) and reverse (m + 80) primers (Table 1). The PCR product was subcloned into pGL3-Basic as described above, yielding mUrat1_-260/+80. The sequences of all constructs were verified by DNA sequencing.
Oligonucleotides used for the production of promoter fragments, mobility shift assays, site-directed mutagenesis, quantitative PCR, and bisulfite PCR Regarding the oligonucleotides used for the mobility shift assays and site-directed mutagenesis, the HNF1-motif in the hURAT1 promoter region is underlined. Boldface type indicates the difference in the sequence of the per and mut compared with the wild-type sequence found in the hURAT1 promoter.

Alignment of the proximal promoter of human and mouse URAT1. Nucleotide sequences of the proximal promoter region of human and mouse URAT1 genes were aligned using Genetyx version 8 to show the high homology of the 5′-flanking region between species. Nucleotide numberings are relative to the transcriptional start sites indicated by an arrow (+1), and homologous sequences between species are boxed. Canonical HNF1 binding motif in the promoter is shaded, and CpG dinucleotides in each sequence are reverse-colored.
Site-Directed Mutagenesis. Mutations in the HNF1 motif located within the hURAT1 minimal promoter were introduced using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with internal mutated oligonucleotides (Table 1). The introduction of mutations was verified by DNA sequencing.
RNA Isolation and Quantitative PCR. Total RNA was isolated from the kidneys of 7- to 14-week-old male (n = 3) and female (n = 4) wild-type or Hnf1α-null adult mice (Lee et al., 1998) and treated with DNase I to remove the contaminated genomic DNA, followed by reverse-transcription using a random-nonamer primer (Takara, Shiga, Japan). To quantify the mRNA expression of mUrat1 in wild-type or Hnf1α-null mice, real-time quantitative PCR was performed as described previously using the primers shown in Table 1 (Kikuchi et al., 2006). The mRNA expression of mUrat1 was normalized by the mRNA expression of Gapdh and statistically analyzed by Student's t test. Asterisks (* and **) represent significant differences (P < 0.05 and P < 0.01, respectively) between wild-type and Hnf1α-null mice of each gender.
Cell Culture and Transfections. Cell culture, transfections, and luciferase assays were performed as described previously (Kikuchi et al., 2006). For transactivation assays in HEK293 cells, 5 ng of empty pcDNA3.1(+) vector, 5 ng of HNF1α expression vector, 2.5 ng of HNF1α and HNF1β expression vectors, or 5 ng of HNF1β expression vector was cotransfected with 0.5 μg of the corresponding promoter construct and 0.05 μg of internal standard pRL-SV40. The promoter activity was measured as relative light units of firefly luciferase per unit of Renilla reniformis luciferase.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay. Nuclear extracts were prepared from HepG2, Caco-2, and HEK293 parent cells, or HEK293 cells transiently transfected with pcDNA3.1(+), either HNF1α or HNF1β expression vector, or both expression vectors as described previously (Kikuchi et al., 2006). Transient transfection of the expression vectors into HEK293 cells was performed using FuGENE6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions. Three kinds of double-stranded oligonucleotides (wt, per, and mut) were obtained by hybridizing the single-stranded complementary oligonucleotide with sense sequences shown in Table 1. Sequence “wt” corresponds to the wild-type HNF1 motif in the hURAT1 promoter, and “per” corresponds to the perfect consensus sequence for the HNF1 motif, whereas “mut” denotes the wild-type sequence mutated within the motif. Five micrograms of nuclear extracts from HepG2, Caco-2, or HEK293 parent cells or 3 μg of nuclear extracts form HEK293 cells transfected with several expression vectors was used in the electrophoretic mobility shift assays. Competition and supershift assays were performed as described previously with Dig Gel Shift Kit, Second Generation (Roche Diagnostics).
Sodium Bisulfite Genomic Sequencing. Genomic DNA from liver, kidney cortex, or kidney medulla of ddY male mice at 8 weeks of age was extracted using a Get Pure DNA Kit (Dojindo Molecular Technologies, Gaithersburg, MD). One microgram of genomic DNA digested with EcoRI was denatured by adding NaOH to give a final concentration of 0.3 M and was incubated for 15 min at 37°C. After the incubation, sodium metabisulfite, pH 5.0, and hydroquinone were added to give final concentrations of 2.0 M and 0.5 mM, respectively, and the mixture was incubated for 16 h at 55°C in the dark. The modified DNA was purified using the Wizard DNA Clean-Up System (Promega), and the bisulfite reaction was terminated by adding NaOH to give a final concentration of 0.3 M and incubating for 15 min at 37°C. The solution was neutralized by adding NH4OAc, pH 7.0, and the DNA was ethanol-precipitated, dried, and resuspended in 10 mM Tris-HCl and 1 mM EDTA, pH 8.0. The DNA fragment covering the proximal promoter region of the mUrat1 gene was amplified by PCR using the following primers: -419-B-F, and +134-B-R (Table 1). The PCR products were cloned into pGEM-T Easy vector (Promega), and 10 clones randomly picked from each of two independent PCRs were sequenced to determine the presence of methylated cytosines.
Results
Involvement of HNF1α/β in the Promoter Activity of URAT1 Genes. A previous report suggested the presence of cis-elements required for the basal promoter activity of the hURAT1 gene in the region from -253 to -39 base pairs relative to the transcriptional start site (Li et al., 2004). The minimal promoter construct of hURAT1 was transfected into three human-derived cell lines, and luciferase activities were measured. The hURAT1 promoter showed a significant increase in luciferase activity compared with the promoterless pGL3-Basic plasmid in HepG2 and Caco-2 cells, whereas the promoter activity was negligible in HEK293 cells (Fig. 2A).
To investigate the involvement of HNF1α or HNF1β in the hURAT1 minimal promoter, the HNF1 motif found in the hURAT1 promoter was disrupted by site-directed mutagenesis, and the promoter activity was measured in HepG2 and Caco-2 cells in which endogenous HNF1α and/or HNF1β is expressed (Kikuchi et al., 2006). Mutation in the HNF1 motif completely abolished the promoter activity in both cell lines (Fig. 2B). Direct confirmation of the importance of HNF1α or HNF1β for the promoter activity of URAT1 genes was obtained by cotransfection assays in HEK293 cells in which neither HNF1α nor HNF1β is endogenously expressed (Fig. 2C). Exogenous expression of HNF1α and/or HNF1β markedly increased luciferase activity of the hURAT1 wild-type reporter (-253/+83-HNF1wt) compared with the pcDNA3.1(+)-transfected control, whereas the luciferase activity of pGL3-Basic was unaffected. The luciferase activity driven by hURAT1 HNF1-mutated reporter (-253/+83-HNF1mut) was not transactivated by exogenously expressed HNF1α and/or HNF1β to the same degree as that of the wild-type construct as expected. The activity of mUrat1 promoter (mUrat1_-261/+80) was also stimulated by forced expression of HNF1α and/or HNF1β. These results strongly suggest that the minimal promoter activity of human and mouse URAT1 genes predominantly depends on the function of HNF1α or HNF1β.

HNF1α and HNF1β predominantly regulate URAT1 promoters. A, analysis of hURAT1 minimal promoter. HepG2 (□), Caco-2 ( ), and HEK293 cells (▪) were transiently transfected with promoterless pGL3-Basic plasmid or minimal promoter construct of hURAT1 (hURAT1_-253/+83-HNF1wt) together with the internal standard pRL-SV40 to normalize the transfection efficiency. B, mutational analysis of the HNF1 motif. HepG2 (□) and Caco-2 cells (
), and HEK293 cells (▪) were transiently transfected with promoterless pGL3-Basic plasmid or minimal promoter construct of hURAT1 (hURAT1_-253/+83-HNF1wt) together with the internal standard pRL-SV40 to normalize the transfection efficiency. B, mutational analysis of the HNF1 motif. HepG2 (□) and Caco-2 cells ( ) were transfected with pGL3-Basic, wild-type (hURAT1_-253/+83-HNF1wt), or HNF1-mutated (hURAT1_-253/+83-HNF1mut) promoter construct. C, exogenous expression of HNF1α and HNF1β. HEK293 cells were transfected with pGL3-Basic, wild-type, or the HNF1-mutated promoter construct of hURAT1 (hURAT1_-253/+83-HNF1wt or hURAT1_-253/+83-HNF1mut, respectively), or the mUrat1 promoter construct (mUrat1_-261/+80), together with empty pcDNA3.1(+) vector (white bars), HNF1α expression vector (black bars), HNF1α and HNF1β expression vectors (gray bars), or HNF1β expression vector (light gray bars). The promoter activity was measured as described under Materials and Methods and was shown as the induction factor over the background activity measured in cells transfected with pGL3-Basic in each cell line (A and B) or cells transfected with pGL3-Basic together with pcDNA3.1(+) (C). All results are presented as the mean ± S.E. of triplicate samples.
) were transfected with pGL3-Basic, wild-type (hURAT1_-253/+83-HNF1wt), or HNF1-mutated (hURAT1_-253/+83-HNF1mut) promoter construct. C, exogenous expression of HNF1α and HNF1β. HEK293 cells were transfected with pGL3-Basic, wild-type, or the HNF1-mutated promoter construct of hURAT1 (hURAT1_-253/+83-HNF1wt or hURAT1_-253/+83-HNF1mut, respectively), or the mUrat1 promoter construct (mUrat1_-261/+80), together with empty pcDNA3.1(+) vector (white bars), HNF1α expression vector (black bars), HNF1α and HNF1β expression vectors (gray bars), or HNF1β expression vector (light gray bars). The promoter activity was measured as described under Materials and Methods and was shown as the induction factor over the background activity measured in cells transfected with pGL3-Basic in each cell line (A and B) or cells transfected with pGL3-Basic together with pcDNA3.1(+) (C). All results are presented as the mean ± S.E. of triplicate samples.
Interaction of HNF1α/β with the hURAT1 Promoter. Interaction of digoxigenin-labeled oligonucleotide probes (wt, per, and mut) with nuclear proteins of HepG2, Caco-2, or HEK293 cells was assessed by electrophoretic mobility shift assays. The wt probe corresponds to the wild-type HNF1 motif in the hURAT1 promoter, and the per probe corresponds to the consensus sequence for the HNF1 motif, whereas the mut probe has the same mutation in the HNF1 motif as that used in the luciferase assays. A nonspecific band, which was formed with every probe and abolished by 25-fold molar excess of both per and mut competitor, was observed in all cell lines. In the competition assays (Fig. 3A), one (band a) or two shifted bands (bands a and b) were formed with nuclear extracts derived from HepG2 or Caco-2 cells, respectively, when the wt and per probe were used (lanes 1, 4, 6, and 9). The bands a and b were abolished by an excess of unlabeled per but not by mut oligonucleotide (lanes 2, 3, 7, and 8), and these bands were not formed when the mut probe was used (lanes 5 and 10). These results suggest that bands a and b represent the binding of HNF1α or HNF1β to the HNF1 motif in the hURAT1 promoter. In contrast, there were no specific bands in HEK293 cells (lanes 11 and 12), consistent with the lack of HNF1α and HNF1β in this cell line.
Supershift analysis with antibodies against HNF1α or HNF1β revealed specific interaction of HNF1α and/or HNF1β with the HNF1 motif in the hURAT1 promoter (Fig. 3B). Addition of an anti-HNF1α antibody supershifted band a in HepG2 cells (lanes 3 and 4) and bands a and b in Caco-2 cells (lanes 8 and 9). Addition of an anti-HNF1β antibody resulted in the supershift of band b in Caco-2 cells (lanes 10 and 11), whereas band a was not supershifted (lanes 5, 6, 10, and 11). These results suggest that the bands a and b reflect the binding of HNF1α/HNF1α homodimer and HNF1α/HNF1β heterodimer, respectively. Binding of exogenously expressed HNF1α or HNF1β to the HNF1 motif was also demonstrated. Another shifted band showing faster mobility than HNF1α/HNF1β heterodimer was detected when the nuclear extracts of HEK293 cells transfected with HNF1β alone were incubated with the labeled probe, and this band was supershifted by anti-HNF1β antibody but not by anti-HNF1α antibody (data not shown). This result suggests that HNF1β/HNF1β homodimer can also interact with the HNF1 motif in the hURAT1 promoter.
Impaired Expression of mUrat1 in Hnf1α-Null Mice. Real-time PCR analysis revealed that the expression of mUrat1 mRNA in the kidney is much lower in Hnf1α-null mice compared with wild-type mice of both genders (Fig. 4). In contrast to a previous report (Hosoyamada et al., 2004), there was no significant difference in the expression level of mUrat1 between male and female wild-type mice. On the other hand, the expression of mUrat1 mRNA in male mice was significantly higher than that in female in Hnf1α-null mice. The difference in Hnf1α-dependent expression (wild-type minus Hnf1α-null mice) between male and female mice did not reach statistical significance.
Epigenetic Regulation of the mUrat1 Gene. Eight CpG dinucleotides, primary targets of DNA methylation in the vertebrate genome, are located in the 5′-flanking sequence up to -500 base pairs of mUrat1 gene; seven of them are within the minimal promoter region (Fig. 1). To elucidate the role of DNA methylation in the tissue-specific expression of the mUrat1 gene, the methylation status of each CpG site was analyzed by sodium bisulfite genomic sequencing in the liver, kidney cortex, and kidney medulla, and the total methylation profiles in these tissues were compared (Fig. 5). These CpGs were heavily methylated in the liver and kidney medulla, in which there is no expression of mUrat1. In contrast, the region was relatively hypomethylated in the kidney cortex, in which mUrat1 is predominantly expressed. These results suggest that tissue-specific expression of mUrat1 gene is regulated through DNA methylation-mediated gene silencing, and the proximal promoter region of the mUrat1 gene can be regarded as a tissue-dependent differentially methylated region (T-DMR).
Discussion
In the present study, the involvement of both genetic and epigenetic mechanisms in the transcriptional regulation of human and mouse URAT1 genes was demonstrated. A pivotal role for HNF1α and HNF1β in the minimal promoter activity of URAT1 genes was confirmed by reporter gene assays (Fig. 2), and interaction of HNF1α/HNF1α homodimer, HNF1α/HNF1β heterodimer, and HNF1β/HNF1β homodimer with the HNF1 motif in the hURAT1 promoter was shown by electrophoretic mobility shift assays (Fig. 3).
In the kidney, HNF1α normally exists as a heterodimer with HNF1β, although the HNF1α/HNF1α homodimer is a more potent transactivator than the heterodimer in vitro. HNF1β/HNF1β homodimer is also detectable in nuclear extracts from kidney, but the transactivation potency is the lowest among the three species (Rey-Campos et al., 1991; Pontoglio et al., 1996). Because expression of HNF1α is confined to the proximal tubules whereas that of HNF1β is observed along the entire nephron (Lazzaro et al., 1992; Pontoglio et al., 1996), formation of HNF1α/HNF1β heterodimer will be restricted to the proximal tubules in the kidney, and that of HNF1β/HNF1β homodimer is possible in the entire nephron. The expression of human and mouse URAT1 genes is predominantly in the proximal tubules (Enomoto et al., 2002; Hosoyamada et al., 2004), which is consistent with the distribution of the HNF1α/HNF1β heterodimer. This suggests that the HNF1α/HNF1β heterodimer is required for constitutive expression of human and mouse URAT1. In contrast, the contribution of HNF1β/HNF1β homodimer is debatable. Although the HNF1β/HNF1β homodimer can also transactivate the mUrat1 promoter, the expression of mUrat1 was markedly reduced in the kidney of Hnf1α-null mice (Fig. 4). This supports an essential role for the HNF1α/HNF1β heterodimer, which could not be compensated for by the HNF1β/HNF1β homodimer.
Eight CpG dinucleotides in the 5′-flanking region of the mUrat1 gene appeared hypermethylated in the liver and kidney medulla, whereas they were relatively hypomethylated in the kidney cortex (Fig. 5). Two mechanisms are proposed for DNA methylation-dependent gene silencing: 1) DNA methylation directly interrupts the binding of transcription factors to their recognition sequences, including the CpG dinucleotide; and 2) methyl CpG binding proteins bound to the methylated cytosine recruit chromatin remodeling factors, such as histone deacetylases, and cause the neighboring chromatin configuration to condense, thereby preventing many transcription factors from accessing their recognition sequences within that region (Shiota, 2004). The HNF1 motif in the mUrat1 promoter is located within the T-DMR, but the motif itself does not contain the CpG dinucleotide. It is possible that methylation of DNA in the T-DMR indirectly inhibits the binding of HNF1α or HNF1β to the promoter through chromatin remodeling events. Hypermethylation of the T-DMR may explain the absence of mUrat1 expression regardless of the formation of HNF1α/HNF1α homodimers in the liver. In the kidney, the promoter region is hypomethylated in the cortex but hypermethylated in the medulla. Together with the proximal tubule-restricted distribution of the HNF1α/HNF1β heterodimer, the regional difference in the methylation status seems to lead to the constitutive expression of mUrat1 in the proximal tubules in the cortex. In humans, the methylation profiles of hURAT1 promoter remain unknown. Considering the similar frequency of CpG dinucleotides in the minimal promoter region between human and mouse (Fig. 1), DNA methylation will be also involved in the tissue/region-specific expression of hURAT1.
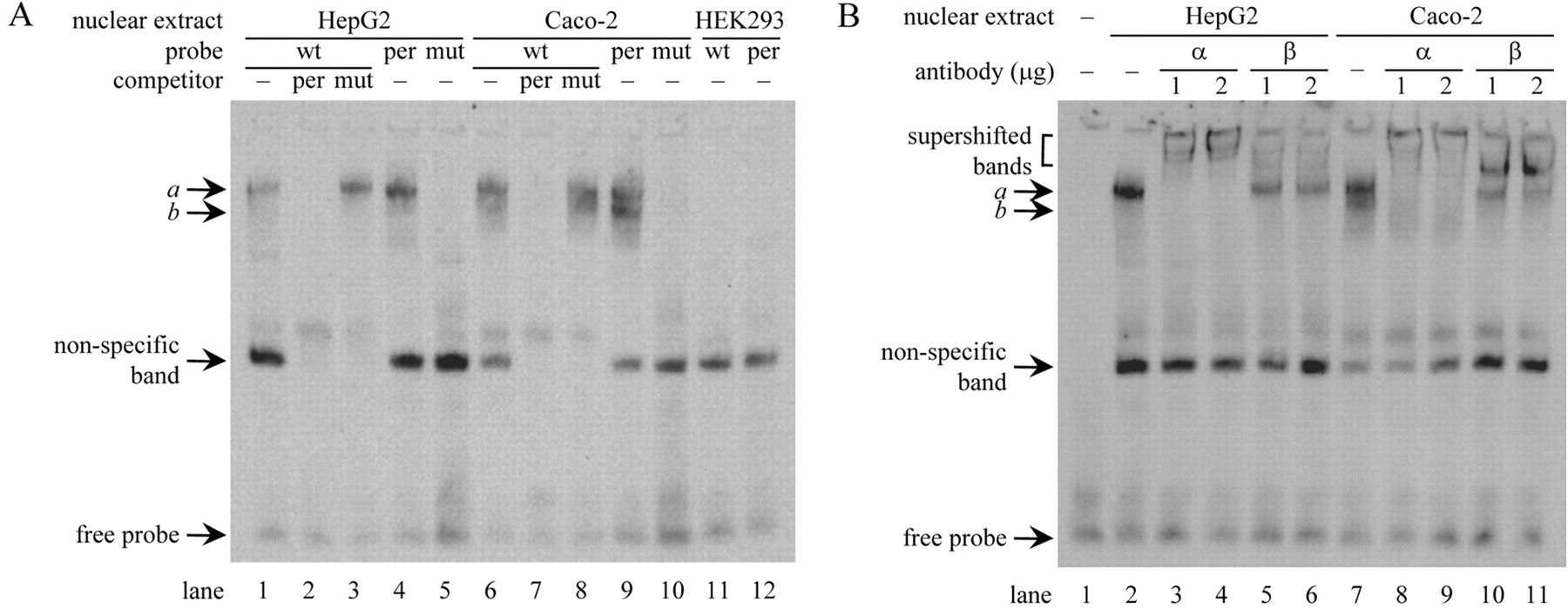
HNF1α and HNF1β interact with the hURAT1 promoter. A, competition assays. Three kinds of digoxigenin-labeled probe (Table 1) were incubated with nuclear extracts of HepG2, Caco-2, or HEK293 cells, with or without a 25-fold excess of unlabeled competitor (per or mut) as indicated. B, supershift assays. The wt probe was incubated with nuclear extracts of HepG2 or Caco-2 cells with or without an increasing amount of specific antibody against HNF1α (α) or HNF1β (β) as indicated. The DNA-protein complex was detected as described under Materials and Methods.

Relative mRNA expression of mUrat1 in wild-type and Hnf1α-null mice. mRNA expression of mUrat1 in the kidneys of male or female wild-type (WT, □) and Hnf1α-null mice (null, ▪) was measured by real-time quantitative PCR using specific primers (Table 1), and the data were normalized by the mRNA expression of Gapdh. The relative mRNA expression was given as a ratio with respect to the mRNA expression of mUrat1 in male wild-type mice that was taken as 100%. Results are presented as the mean ± S.E. of three (male) or four (female) mice. *, P < 0.05, and **, P < 0.01, significantly different between wild-type and Hnf1α-null mice.

DNA methylation profile of the mUrat1 promoter. Top, a schematic diagram of the mUrat1 5′-flanking region. The vertical lines and numbers indicate the positions of cytosine residues of CpGs relative to the transcriptional start site (TSS, +1). The position of the HNF1 motif is shown by a rectangle. Bottom, DNA methylation status of individual CpGs. Bisulfite sequencing analysis was performed with genomic DNAs extracted from mouse liver, kidney cortex, and kidney medulla. ○ and • represent unmethylated and methylated cytosines, respectively.
Mice lacking HNF1α suffer from severe renal Fanconi syndrome, a dysfunction in renal proximal tubular reabsorption (Pontoglio et al., 1996). The renal fractional excretion of urate is higher in mutant animals compared with wild-type controls, resulting in a lower serum urate level. In the present study, in vitro and in vivo evidence of the importance of HNF1α in the expression of URAT1 genes was provided; accurate tissue-specific expression of this gene is essential for the reabsorption of filtered urate in the kidney. The altered renal excretion of urate in mutant mice is thus accounted for by impaired expression of Urat1. It was reported that expression of transporters responsible for reabsorption of filtered glucose, phosphate, and bile acids is also absent or lower in Hnf1α-null mice (Pontoglio et al., 2000; Shih et al., 2001; Cheret et al., 2002). Taken together, the expression of transporters required for the reabsorption of filtered solutes in the kidney proximal tubules is largely impaired in mutant mice, leading to renal Fanconi syndrome.
In addition to renal Fanconi syndrome, Hnf1α-null mice exhibit a phenotype similar to non–insulin-dependent diabetes mellitus (Lee et al., 1998; Pontoglio et al., 1998). In humans, mutations in HNF1α and HNF1β cause maturity-onset diabetes of the young (MODY) types 3 and 5, respectively (Yamagata et al., 1996; Horikawa et al., 1997). MODY is an autosomal dominant inherited disease that is responsible for 2 to 5% of non–insulin-dependent diabetes mellitus (Ledermann, 1995), and MODY3 accounts for 20 to 75% of patients with MODY, whereas MODY5 is a rare condition (Froguel and Velho, 1999; Winter and Silverstein, 2000; Ryffel, 2001). It was reported that the serum urate levels are low in patients with both type I and type II diabetes mellitus because of the elevated renal clearance (Shichiri et al., 1987; Magoula et al., 1991; Golik et al., 1993). The increase in the urate clearance has been ascribed to an elevated glomerular filtration rate and/or a defect in tubular urate reabsorption. It is thus possible that the expression of URAT1 mRNA is reduced in patients with MODY3 and MODY5, resulting in hypouricemia.
In conclusion, clear evidence is provided for the involvement of both genetic (HNF1α and HNF1β) and epigenetic (DNA methylation) mechanisms in establishing the tissue-specific expression of mouse and probably human URAT1 genes. This is the first demonstration of the presence of T-DMR in the promoter region as far as transporters in the kidney are concerned.
Acknowledgments
We thank Dr. Shun Sato for his technical assistance and helpful discussions.
Footnotes
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.039701.
-
ABBREVIATIONS: OAT, organic anion transporter; URAT1, urate transporter 1; HNF1, hepatocyte nuclear factor 1; PCR, polymerase chain reaction; Gapdh, glyceraldehyde-3-phosphate dehydrogenase; T-DMR, tissue-dependent differentially methylated region; MODY, maturity-onset diabetes of the young; HEK, human embryonic kidney; wt, wild-type hepatocyte nuclear factor 1 motif in the hURAT1 promoter; per, the consensus sequence for the hepatocyte nuclear factor 1 motif; mut, mutated hepatocyte nuclear factor 1 motif; m, mouse; h, human.
- Received July 6, 2007.
- Accepted September 10, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}