


Abstract
Although repression of inflammatory gene expression makes glucocorticoids powerful anti-inflammatory agents, side effects limit usage and drive the search for improved glucocorticoid receptor (GR) ligands. In A549 pulmonary cells, dexamethasone and the prototypical dissociated ligand RU24858 (Mol Endocrinol11:1245-1255, 1997) repress interleukin (IL)-1β-induced expression of cyclooxygenase (COX)-2 and IL-8. Although RU24858 is a weaker GR ligand, both glucocorticoids showed similar efficacies on transrepression of nuclear factor κB (NF-κB)-dependent transcription, whereas RU24858 yielded less than 12% of the response to dexamethasone on a classic glucocorticoid response element (GRE) reporter (transactivation). Modest NF-κB-dependent transrepression (∼40%), along with analysis of IL-8 transcription rate and the accumulation of unspliced nuclear RNA, indicates that transrepression does not fully account for the repression of genes such as IL-8. This was confirmed by the finding that mRNA degradation is increased by both dexamethasone and RU24858. Analysis of IL-1β-induced steady-state mRNA levels for IL-8 and COX-2 show that dexamethasone- and RU24858-dependent repression of these genes is attenuated by inhibitors of transcription and protein synthesis. Because similar effects were observed with respect to COX-2 and IL-8 protein expression, we conclude that glucocorticoid-dependent gene expression is necessary for repression by both glucocorticoids. Despite RU24858 being defective at classic GRE-dependent transactivation, both dexamethasone and RU24858 induced the expression of potentially anti-inflammatory genes and metabolic genes, suggesting the importance of nontraditional glucocorticoid-dependent gene expression. Thus, classic transactivation- and transrepressionbased screens for anti-inflammatory “dissociated” GR ligands may be flawed because they may not reflect the effects on real glucocorticoid-inducible genes.
Glucocorticoids suppress the expression and/or release of multiple inflammatory mediators, cytokines, chemokines, adhesion molecules, and other inflammatory proteins, making these agents first-line therapy in the treatment of inflammatory diseases such as asthma (Barnes, 2001; Rhen and Cidlowski, 2005). However, the clinical use of anti-inflammatory glucocorticoids is limited by numerous side effects, including osteoporosis, arterial hypertension, obesity, diabetes, cataracts, skin thinning, and muscle weakness (Barnes, 2001; Rhen and Cidlowski, 2005).
Glucocorticoids are believed to act via the glucocorticoid receptor (GR), which in its inactive form is retained in the cytoplasm in a multiprotein complex containing heat shock proteins and immunophilins (Rhen and Cidlowski, 2005). After binding of steroid ligand to GR, a conformational change results in the dissociation of the complex and translocation of GR to the nucleus. Nuclear GR may then dimerize and bind specific DNA sequences, known as glucocorticoid response elements (GREs), to promote transcription (transactivation) of responsive genes (Rhen and Cidlowski, 2005). However, ligand-activated GR may also inhibit, or transrepress, transcription factors such as nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) via processes that are believed to be independent of GR dimerization and GR DNA binding (Barnes, 2001; Rhen and Cidlowski, 2005). Thus, transgenic (GRdim/dim) mice, which harbor an A458T mutation within the GR dimerization loop that renders GR defective in both DNA binding and classic GRE-dependent transcriptional responses, are still capable of repressing inflammatory gene expression (Reichardt et al., 1998, 2001). Furthermore, such dimerization loop mutations do not prevent glucocorticoid-mediated repression of AP-1 and NF-κB-dependent transcription (Heck et al., 1994; Reichardt et al., 1998, 2001). Because NF-κB and AP-1 are key regulators of many inflammatory genes, these data support the widely held belief that transrepressive mechanisms account for the dominant anti-inflammatory benefits of glucocorticoids (Barnes, 2001). Conversely, the undesirable metabolic effects of GR are often attributed to transactivation, and this concept has led to the search for novel, or disassociated, GR ligands that selectively activate the transrepressive but not the transactivation functions of GR (Heck et al., 1994; Vayssiere et al., 1997; Barnes, 2001; Uings and Farrow, 2005).
The continued analysis of transrepression now suggests that glucocorticoids may differentially affect the interaction with the basal transcriptional machinery, the phosphorylation of RNA polymerase II, the recruitment of coactivators/corepressors, and may involve chromatin remodelling via histone deacetylases to repress inflammatory gene transcription (De Bosscher et al., 2000; Nissen and Yamamoto, 2000; Adcock et al., 2004; Garside et al., 2004). However, there is also considerable evidence that glucocorticoid-mediated repression of inflammatory genes involves significant post-transcriptional and/or translational mechanisms (Newton, 2000; Stellato, 2004). Thus, the repression of inflammatory genes, including cyclooxygenase (COX)-2, interleukin (IL)-8, and inducible nitric-oxide synthase, involves mechanisms that regulate mRNA stability (Ristimaki et al., 1996; Newton et al., 1998; Chang et al., 2001; Lasa et al., 2001; Korhonen et al., 2002). Furthermore, a requirement for de novo protein synthesis in glucocorticoid-dependent repression has been highlighted in several cases (Ristimaki et al., 1996; Newton et al., 1998; Newton, 2000; Chang et al., 2001; Lasa et al., 2001; Korhonen et al., 2002). In this context, glucocorticoids are known to target p38 mitogen activated protein kinase (MAPK)-dependent mRNA stabilization of inflammatory genes (Lasa et al., 2001). This occurs via the glucocorticoid-dependent induction of mitogen activated protein kinase phosphatase (MKP)-1, which in turn inhibits p38 MAPK and causes mRNA destabilization (Clark and Lasa, 2003).
In addition, a number of other genes that show anti-inflammatory properties are also known to be glucocorticoid-inducible and could contribute to the anti-inflammatory effects of these drugs (Newton, 2000; Clark and Lasa, 2003; Rogatsky et al., 2003). Therefore, the rationale for the use of novel dissociated GR ligands as improved anti-inflammatory compounds with reduced side effect profiles remains equivocal (Newton, 2000; Belvisi et al., 2001; Schacke et al., 2004). Therefore, we have chosen COX-2 and IL-8 as model inflammatory genes to examine the effect of RU24858, a prototypical dissociated glucocorticoid (Vayssiere et al., 1997). In this study, we specifically address the requirement for steroid-inducible gene expression in the repression by both dexamethasone and the dissociated ligand RU24858.
Materials and Methods
Cell Culture. A549 cells and stable reporter lines were grown as described previously (Chivers et al., 2004), and used at confluence after overnight incubation with serum-free media (SFM). Dexamethasone (Sigma, Poole, Dorset, UK) was dissolved in Hanks' balanced salt solution (Sigma), RU486 (Sigma) was dissolved in ethanol, and RU24858 (a gift from Sanofi-Aventis Pharmaceuticals, Bridgewater, NJ) was dissolved in dimethyl sulfoxide. Drugs were added 1 h before stimulation with IL-1β (R&D Systems, Oxon, UK) unless otherwise stated. Final concentrations of dimethyl sulfoxide and ethanol were never in excess of 0.2% (v/v). This had no effect on GRE-dependent transcription, NF-κB-dependent transcription, or the expression of COX-2 or prostaglandin (PG) E2 release (data not shown).
Analysis of IL-8 and PGE2 Release, COX/Prostaglandin E Synthase Activity, and COX-2 Protein Expression. Culture medium was collected for measurement of IL-8 and PGE2 release by ELISA (R&D Systems, Minneapolis, MN) and radioimmunoassay (Sigma) using commercially available antibodies. For the measurement of combined COX/prostaglandin E synthase (PGES) activity, cells were rinsed with SFM, before incubation at 37°C for 10 min with SFM containing 30 μM arachidonic acid (Sigma). PGE2 release was measured as above and taken as an index of COX/PGES activity. For Western blot analysis of COX-2, total proteins were harvested in reporter lysis buffer (Promega, Madison, WI) supplemented with complete protease inhibitor cocktail (Roche, Mannheim, Germany), before analysis using Santa Cruz Biotechnology antibodies (Santa Cruz, CA) as described previously (Chivers et al., 2004).
Reporter Cell Lines and Luciferase Assay. A549 cells containing the NF-κB-dependent reporter 6κBtkluc, which contains six copies of the consensus NF-κB binding site (GGG ACT TTC C) derived from the human immunodeficiency virus enhancer have been described previously (Chivers et al., 2004). The GRE-dependent reporter, pGL3.neo.TATA.2GRE, contains two copies of a consensus GRE site (TGT ACA GGA TGT TCT) positioned upstream of a minimal β-globin promoter driving a luciferase gene and a separate neomycin gene to confer resistance to G418. A549 cells harboring this reporter have also been described previously (Chivers et al., 2004). Reporter cells were harvested in reporter lysis buffer (Promega) 6 h after treatment, and luciferase activity was determined using a commercial kit (Promega).
RNA Isolation, Northern Blot Analysis, and Nuclear Run on Transcription Assay. Total RNA was isolated using the RNeasy mini kit (QIAGEN Ltd., Crawley, UK). Northern blot analysis and nuclear run-on assays were performed according to standard procedures as described previously (Newton et al., 1998).
Semiquantitative Reverse Transcriptase-Polymerase Chain Reaction. Total RNA (1 μg) was incubated at 70°C for 5 min before reverse transcription in a total reaction volume of 20 μl including 1 mM concentration of each dNTP, 10 ng/μl random hexamers, 0.4 U/μl avian myeloblastosis virus reverse transcriptase, 1× avian myeloblastosis virus buffer, and 1.6 U/μl RNasin (all from Promega). The reaction was incubated at 37°C for 60 min. Reverse transcriptions were terminated by heating to 90°C for 4 min, and the resulting cDNA was then diluted 1:5 with nuclease-free water.
PCR was carried out by adding 5 μl of diluted cDNA to 20 μl of reaction mixture giving final concentrations of 0.2 mM dNTPs, 1× NH4 buffer [16 mM (NH4)2SO4, 67 mM Tris-HCl, pH 8.8, and 0.1% (v/v) Tween 20], 1.5 mM MgCl2, 0.1 U/μl Taq polymerase, and 5 ng/μl of both the upstream and downstream PCR primers (Sigma-Genosys, Cambridge, UK). The primer sequences are shown in Table 1. After an initial denaturation at 94°C for 2 min, RT products were amplified for 22 to 30 cycles (dependent on expression level of each gene) of PCR using 30-s denaturation at 94°C, 30-s primer annealing at 58 to 62°C (dependent on primer), 30-s primer extension at 72°C, and a final extension of 72°C for 10 min. Annealing temperatures were the following: COX-2, GAPDH, RGS-2, and GILZ, 58°C; IL-8, 60°C; MKP-1, 59°C; AMP N, 59°C; and MET 1X, 62°C. PCR products were then subjected to electrophoresis on 2% agarose gels containing 0.5 μg/ml ethidium bromide and visualized using ultraviolet illumination and the GelWorks documentation and analysis system (GelWorks ID Intermediate, Cambridgeshire, UK). In each case, the number of amplification cycles used was carefully optimized to allow the detection of cDNA within the exponential phase of the reaction (i.e., where starting cDNA concentrations are proportional to the product generated) and thereby enable relative quantification (see Supplemental Data for further details). Representative examples are shown and these provide validation of this approach (Supplemental Fig. S1, A and B).
Primers for semiquantitative RT-PCR
Real-Time TaqMan PCR Analysis. After cDNA synthesis, above, TaqMan PCR was performed using 2.5 μl of cDNA in a reaction volume of 20 μl essentially according to the manufacturer's specification (Applied Biosystems Inc., Foster City, CA) using a premade master mix and an ABI 7900HT instrument (Applied Biosystems). Analysis of COX-2 and GAPDH was carried out using the validated off-the-shelf assays, hs00153133_m1 and 432631E, respectively (Applied Biosystems). IL-8 was amplified using the primers 5′-CTG GCC GTG GCT CTC TTG-3′ (forward) and 5′-TTA GCA CTC CTT GGC AAA ACT G-3′ (reverse) with the 5-carboxyfluorescein/5-carboxytetramethylrhodamine-linked probe 5′-CCT TCC TGA TTT CTG CAG CTC TGT GTG AA-3′, which had been designed using Primer Express version 2 software (Applied Biosystems). All samples were analyzed in duplicate. Relative cDNA concentrations were determined from a cDNA standard curve that was analyzed simultaneously with the test samples (see Supplemental Fig. S1, C and D, for an example).
Analysis of Unspliced Nuclear RNA for IL-8 and GAPDH. Cells were harvested by scraping on ice before a brief centrifugation. The cell pellet was resuspended in 150 μl of ice-cold 10 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 1.5 mM MgCl2, and 0.65% Nonidet P-40 supplemented with 20 U RNasin (Promega). After 10 min on ice and a 10-s vortex, the intact nuclei were pelleted at 13,000 rpm in a bench-top centrifuge. The cytoplasmic lysate was removed, and RNA was prepared using the RNeasy kit (QIAGEN). The nuclear pellet was also processed according to the RNeasy method, except that the on-column DNase I digestion was extended to 30 min. Preparation of cDNA was as above.
To detect the presence of unspliced RNA intermediates, amplification primers were designed using Primer Express software (Applied Biosystems) such that that one member of each pair was positioned within an exon and the other primer was positioned in the adjoining intron. This ensures that mRNA, which has undergone splicing to remove introns, cannot give rise to an amplification product. Primers spanning the IL-8 (accession number NM_000584.2) exon 1-intron A junction were 5′-CTC TTG GCA GCC TTC CTG AT-3′ (forward) and 5′-CTG TTT CTG AAT AAA AAG GAT GTT TGT TAC-3′ (reverse). Primers spanning the GAPDH (accession number NM_002046) exon 2-intron B junction were 5′-CCA CAT CGC TCA GAC ACC AT-3′ (forward) and 5′-CGC TGA CCT TGA GCT CTC CTT-3′ (reverse). Amplicon sizes were 151 and 184 base pairs, respectively. After PCR optimization and validation, 5-carboxyfluorescein/minor groove binder TaqMan probes (Applied Biosystems) for IL-8, 5′-TGA AGG TAA GCA CAT CTT TCT GAC CTA CAG CG-3′, and GAPDH, 5′-TCA ACG GGT GAG TTC G-3′, were designed that spanned the junction site and therefore could only bind products from unspliced RNA or genomic DNA, and these were used for real-time analysis using standard conditions (above). In all cases, relative quantification was performed using a serial cDNA dilution. The presence of copurified genomic DNA was assessed by analysis of cDNA that had been prepared in the absence of reverse transcriptase. Any sample in which the genomic DNA contributed to more than 5% of the total unspliced GAPDH signal was excluded from the analysis (see Supplemental Data for details).
Microarray Analysis. Total RNA (5 μg) was prepared as above and assayed for quality using RNA LabChips (Agilent Technologies, Palo Alto, CA). RNA was reverse-transcribed to generate cDNA and was subsequently transcribed in vitro to generate biotin-labeled cRNA before fragmentation and hybridization with the GeneChip expression arrays (Human genome U95Av2 and B arrays) as specified by the manufacturer (Affymetrix Inc., Santa Clara, CA). The array was subsequently washed and stained with a streptavidin/phycoerythrin-conjugated antibiotin to visualize hybridized cRNA, and the GeneChip was scanned to quantify gene expression. After global normalization, analysis was performed using the P-FOLD algorithm for Bayesian estimation of -fold changes (Theilhaber et al., 2001).
Data Presentation and Statistical Analysis. Densitometry was performed using TotalLab software (Nonlinear Dynamics, Newcastle, UK). All graphical data are presented as means ± S.E. Multiple comparisons were analyzed by analysis of variance with a Bonferroni post test, and paired analyses were by Mann-Whitney U test as appropriate.
Results
Dexamethasone and RU24858 Repress PGE2 Release and Inflammatory Gene Expression. Previous studies have documented the repression of IL-6 and inducible nitricoxide synthase by RU24858 and have suggested a lower potency, and possibly efficacy, relative to dexamethasone (Vanden Berghe et al., 1999; Korhonen et al., 2002). A549 cells were therefore stimulated with IL-1β (1 ng/ml) to induce the expression of the inflammatory genes, COX-2, and IL-8 after treatment with various concentrations of either dexamethasone or RU24858. Initially, PGE2 release was measured, and in both cases, this was reduced to near basal levels (95 and 90% repression, respectively), suggesting similar levels of efficacy (data not shown). In contrast, EC50 values for inhibition of PGE2 by dexamethasone and RU24858 were 2.8 ± 1.8 and 53.9 ± 12.8 nM (P =< 0.05), respectively, indicating a reduced potency for RU24858 relative to dexamethasone. Parallel analysis of combined COX/PGES also revealed significant repression by both steroids (Fig. 1A). Again, a lower potency was noted for RU24858 (EC50 = 783 nM) compared with dexamethasone (EC50 = 3.76 nM) (P =< 0.001). In addition, maximal concentrations of dexamethasone reduced COX/PGES activity to basal levels (94.7% inhibition), whereas the repression by RU24858 was significantly different from this and only reached 71.2% (P < 0.01). Although COX/PGES activity in this system is believed to predominantly reflect COX-2 activity, we also performed Western blot analysis to examine COX-2 protein expression (Fig. 1B). Densitometric analysis of these experiments revealed essentially similar inhibition characteristics to that observed for COX/PGES activity.

Inhibition of IL-8, COX-2, and COX/PGES activity by glucocorticoids. A549 cells were treated with concentrations of dexamethasone (▪) or RU24858 (•) for 1 h before stimulation with IL-1β (1 ng/ml) or not stimulated (NS). After 24 h, COX/PGES activity (A), COX-2 and GAPDH protein (B), and IL-8 release (C) were assayed by radioimmunoassay, Western blot, and ELISA, respectively. D, after 6 h, cells were harvested for Northern blot analysis of COX-2, IL-8, and GAPDH. All data are n = 5to8. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Analysis of IL-8 release showed a substantial increase after IL-1β treatment, and this was repressed by both dexamethasone and RU24858 (Fig. 1C). Both dexamethasone and RU24858 repressed IL-8 release to near basal levels (92.2 and 75.5%, respectively). This difference between dexamethasone (1 μM) and RU24858 (10 μM) was significant (P < 0.01). Again, RU24858 (EC50 = 128 nM) was considerably less potent than dexamethasone (EC50 = 2.55 nM). In parallel experiments, Northern blot analysis showed a concentration-dependent inhibition of both COX-2 and IL-8 mRNA by both dexamethasone and RU24858 (Fig. 1D). In each case, dexamethasone was more potent than RU24858, and this was consistent with the protein data above.
The observed differences in potency between dexamethasone and RU24858 prompted us to confirm that RU24858 was able to activate GR. Immunohistological analysis revealed that after administration of either dexamethasone (1 μM) or RU24858 (10 μM), GR translocated to the nucleus (Supplemental Fig. S2A). In addition, competitive ligandbinding studies were performed to analyze the binding affinity of RU24858 for GR. This revealed a Ki of 110.0 ± 24.0 nM for RU24858 compared with a Ki of 4.9 ± 1.3 nM for dexamethasone (Supplemental Fig. S2B). These data therefore document the activation of GR by both dexamethasone and RU24858 and suggest that the differences in potency with respect to the above outputs are primarily accounted for by the lower binding affinity of RU24858 to GR.
The role of GR in the repression of COX-2 and IL-8 was further confirmed by analysis of the receptor antagonist, RU486. At concentrations of 1 μM and above, RU486 by itself may elicit repression of COX-2, NF-κB-dependent transcription, and IL-8 release (Chivers et al., 2004) (Fig. 2) (and data not shown). Therefore, the lower concentrations of 0.1 μM dexamethasone and 1 μM RU24858 were selected for analysis of antagonism by RU486. In each case, the repression of IL-1β-induced COX-2 and IL-8 mRNA by dexamethasone and RU24858 was progressively reversed by increasing concentrations of RU486 (Supplemental Fig. S3). Similar antagonism was revealed at the level of COX-2 protein expression, COX/PGES activity, and IL-8 release (data not shown).
RU24858 Is a Dissociated Steroid in A549 Cells. To examine the ability of RU24858 to activate classic GRE-dependent transcription, A549 cells harboring a previously characterized 2× GRE luciferase reporter were treated with increasing concentrations of dexamethasone or RU24858 (Chivers et al., 2004) (Fig. 2A). Dexamethasone resulted in greater than 25-fold activation of the reporter (EC50 = 93 nM). By contrast, RU24858 was essentially silent, giving rise to less than 12% of the response to dexamethasone at the maximum test concentrations. The steroid antagonist RU486 produced a concentration-dependent loss of GRE-dependent transcription produced by dexamethasone and failed to alter the already low RU24858 response (Fig. 2B).
Because IL-1β-dependent induction of COX-2 and IL-8 is highly NF-κB-dependent in A549 cells (Catley et al., 2005), we made use of a previously characterized NF-κB-dependent reporter to examine transrepression by RU24858 (Chivers et al., 2004). This luciferase reporter is driven by six classic NF-κB sites and is proven to be NF-κB-dependent (Catley et al., 2005). Reporter activity was partially repressed by dexamethasone to a level that was approximately 60% of the response to IL-1β alone (Fig. 2C). RU24858 also achieved this level of transrepression indicating that both steroids are equally transrepression-competent in this system. Although the relative potency of dexamethasone (EC50 = 2.2 nM) to RU24858 (EC50 = 1090 nM) was maintained, it was noteworthy that the response to dexamethasone showed more than a 40-fold enhanced potency relative to that observed on the GRE reporter (i.e., on transactivation). This distinction between the potency for transactivation verses transrepression has been reported previously, but the mechanistic basis for this effect remains unclear (Jonat et al., 1990). Prior addition of RU486 resulted in a loss of transrepression by both dexamethasone and RU24858 at concentrations of up to 0.1 μM RU486 (Fig. 2D). Above this level, RU486 alone showed considerable ability to repress NF-κB-dependent transcription (Fig. 2D). Given that RU486 binds GR with a lower Ki than dexamethasone (Chivers et al., 2004), these effects are likely to be nonspecific.
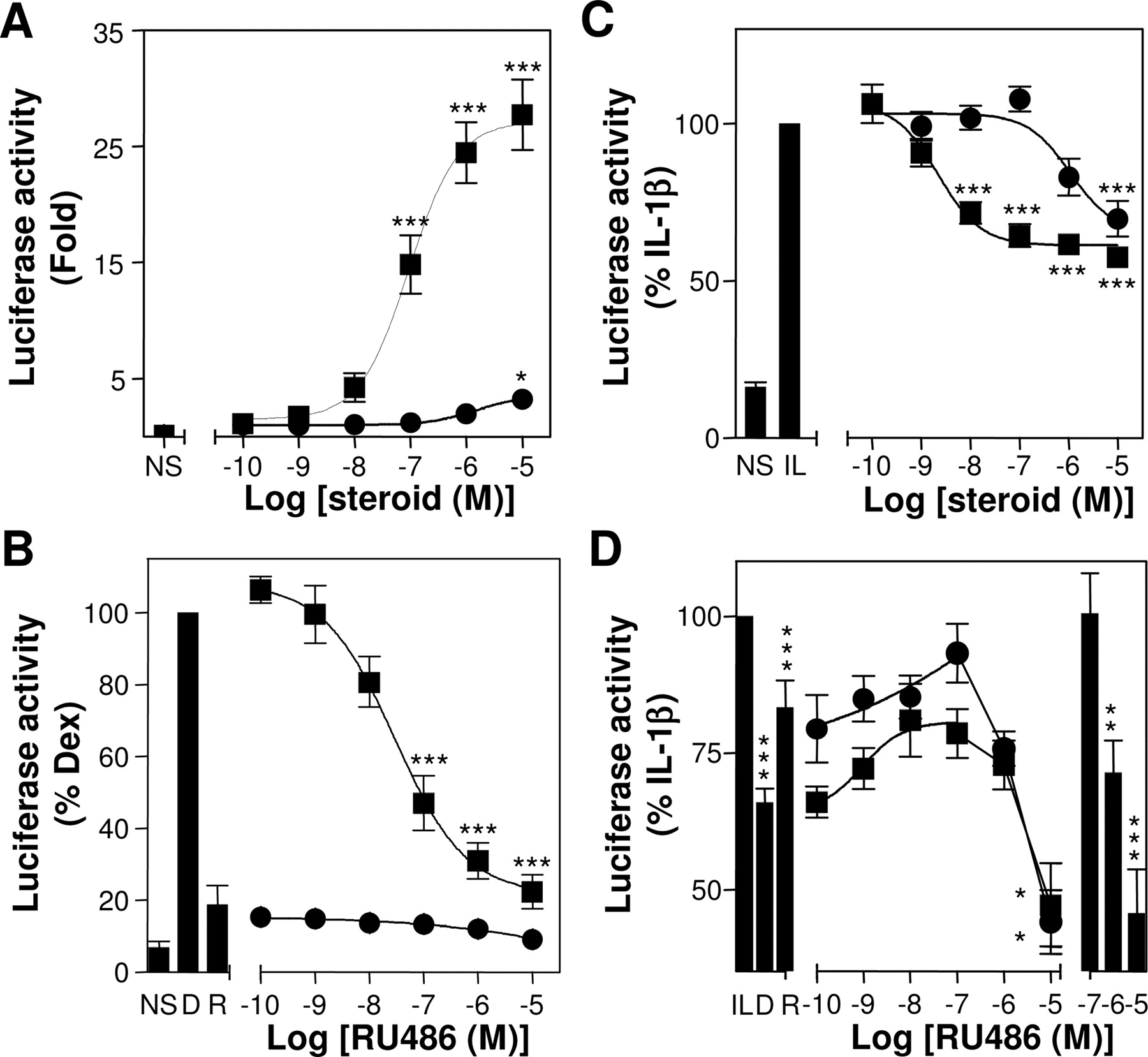
Effect of dexamethasone and RU24858 on GRE-dependent transactivation and NF-κB-dependent transrepression. A, 2× GRE A549 reporter cells were cultured with various concentrations of dexamethasone (▪) or RU24858 (•). After 6 h, cells were harvested for luciferase assay. Data are expressed as -fold induction and are plotted as means ± S.E.M. B, 2× GRE A549 reporter cells were treated with dexamethasone (0.1 μM) (▪) or RU24858 (1 μM) (•) in the presence of various concentrations of RU486. Cells were harvested after 6 h, and luciferase activity, expressed as a percentage of dexamethasone (0.1 μM), was plotted as a mean ± S.E.M. The effects of dexamethasone (0.1 μM) (D) and RU24858 (1 μM) (R) alone are also shown. C, 6κBtk A549 reporter cells were cultured with various concentrations of dexamethasone (▪) or RU24858 (•) for 1 h before stimulation with IL-1β (1 ng/ml). After 6 h, cells were harvested for luciferase assay. D, 6κBtk reporter cells were treated with dexamethasone (0.1 μM) (▪) or RU24858 (1 μM) (•) in the presence of various concentrations of RU486 before IL-1β stimulation as in B. Data expressed as a percentage of the activation induced by IL-1β are plotted as means ± S.E.M. The effect of dexamethasone (0.1 μM) (D) and RU24858 (1 μM) (R) is shown, as is the effect of RU486 alone (on the right). All data (A-D) are n = 6 to 10. NS, unstimulated. IL, IL-1β. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Although both the 2× GRE and the 6κBtk A549 transcriptional reporters were shown to be dexamethasone-sensitive, the repression of IL-1β-induced PGE2 release was also analyzed to confirm full dexamethasone-responsiveness of each reporter cell line. In each case, the inhibition curves produced by dexamethasone were virtually identical with wild-type A549 cells (Supplemental Fig. S4). Thus, these lines were fully competent for the above analyses.
Transcriptional Repression of Inflammatory Genes. In terms of inflammatory gene expression, the above data suggest that classic GRE-mediated transcriptional responses cannot, in this model, account for the ability of glucocorticoids to repress the expression of COX-2 and IL-8. However, the relatively modest repression of NF-κB-dependent transcription also seems insufficient to explain the full repression that is observed at the RNA and protein levels of these NF-κB-dependent genes. In a previous study, we showed that IL-1β-induced COX-2 transcription rate was also modestly (∼40%) repressed by dexamethasone (Newton et al., 1998). We suggest that this is reasonably consistent with the observed repression of the NF-κB reporter. To provide further insight on the role of transcriptional repression, we performed nuclear run on analysis for IL-8 (Fig. 3A). These data, like those observed previously for COX-2, revealed a robust increase in IL-8 transcription after IL-1β treatment. After a 1-h preincubation with dexamethasone (1 μM), there was no obvious effect on IL-1β-induced IL-8 transcription rate at 2 h (Fig. 3A). By 6 h after stimulation, IL-8 transcription had decreased to ∼50% of the response to IL-1β alone, and this is consistent with effects observed on the NF-κB reporter (above).
Analysis of Unspliced Nuclear RNA as a Surrogate of Transcription Rate. Various studies have documented the ability to detect unspliced mRNA precursors in the nucleus and have described the use of this approach as a surrogate for the nuclear run on assay (Lipson and Baserga, 1989; Elferink and Reiners, 1996). In pilot studies, we have been able previously to detect IL-1β-dependent induction of nuclear intermediates for COX-2 (data not shown), and such an approach has also been described for COX-2 in mouse fibroblasts (Gilbert et al., 1994). PCR primers were therefore designed that span the IL-8 exon 1-intron A and the GAPDH exon 2-intron B boundaries. The presence of these intermediates was then assessed in RNA prepared from the nuclei of cells that had been treated with IL-1β for 2 and 6 h or not treated (Fig. 3B). Qualitative PCR, using 32 cycles for unspliced IL-8, revealed low to undetectable levels of IL-8 product in untreated cells, but an intense band in IL-1β-treated samples. By comparison, the product for GAPDH seemed to be uniformly present in unstimulated and stimulated samples when analyzed using 34 cycles (Fig. 3B). In each case, product sizes were of the expected sizes, and amplification from samples prepared without reverse transcriptase resulted in either no or barely detectible product. To provide a more quantitative assessment of the relative levels of these products, TaqMan probes were designed that spanned the exon-intron boundary delimited by each set of amplification primers. TaqMan PCR analysis of the samples shown in Fig. 3B is depicted in the Supplemental Data (Supplemental Fig. S5). These data very clearly confirm the high level of IL-8 inducibility and furthermore show that genomic contamination is not a problem to the analysis.
To examine the effect of dexamethasone on the accumulation of nuclear intermediates, A549 cells were treated, as in Fig. 3A, with dexamethasone and then with IL-1β for either 2 or 6 h. After 2 h, the accumulation of unspliced IL-8 mRNA was hugely up-regulated from essentially undetectable levels in unstimulated cells (Fig. 3C). Prior treatment with dexamethasone produced a statistically significant, but rather modest, 28% drop in unspliced nuclear IL-8. By 6 h after stimulation, the level of unspliced IL-8 intermediates had decreased dramatically, presumably due to either reductions in the transcription rate or an increased efficiency of splicing. However, the repression by dexamethasone was now increased to 60% of the IL-1β-treated value at 6 h (i.e., the level of repression exerted by dexamethasone on events in the nucleus was increased in a time-dependent manner). Taken together with the nuclear run-on data, these data support a modest initial repressive effect of dexamethasone but suggest that by 6 h, this repression may increase to between 50 and 60%. Parallel examination of cytoplasmic IL-8 mRNA revealed a considerably greater level of inhibition at each time point and suggest that events within the nucleus cannot fully account for the inhibition of cytoplasmic IL-8 mRNA by dexamethasone. Finally, analysis of RU24858, not previously possible using nuclear run-on, suggests a lesser inhibitory effect compared with dexamethasone (Fig. 3D).

Effect of dexamethasone on IL-8 transcription rate and accumulation of unspliced RNA intermediates. A, A549 cells were treated with dexamethasone (1 μM) (Dex) for 1 h before stimulation with IL-1β (1 ng/ml). Cells were harvested at the times indicated, and nuclei were prepared for run-off transcription reactions. Radiolabeled transcripts were hybridized to immobilized probes for IL-8 and GAPDH. Representative blots are shown, and after densitometric analysis, data from three such experiments, expressed as a percentage of IL-1β, are plotted as means ± S.E.M. ▪, IL-1β-treated;□, IL-1β + Dex. B, A549 cells were not treated or treated with IL-1β (1 ng/ml) for 2 and 6 h. Nuclei were harvested, and RNA was prepared for RT-PCR analysis of unspliced IL-8 (IL-8 A) and GAPDH (GAPDH B) RNA using 32 and 34 cycles, respectively. Representative agarose gels are shown. 1 kb, 1-kilobase ladder (Invitrogen, Carlsbad, CA). RT+ refers to amplification reaction performed on cDNA generated in the presence of reverse transcriptase. RT-refers to the absence of reverse transcriptase in the cDNA synthesis step. Water controls did not reveal a product and are not shown. C, A549 cells were treated as in A. Nuclei and cytoplasmic lysates were collected, and RNA was prepared from each. After cDNA synthesis, TaqMan PCR was used to examine unspliced IL-8 and GAPDH RNA in nuclear samples and IL-8 and GAPDH mRNA in cytoplasmic samples. In each case, data (n = 5-6), expressed as a ratio of IL-8/GAPDH, are plotted as a percentage of the IL-1β stimulation at 2 h as means ± S.E.M. D, A549 cells were treated with combinations of IL-1β (1 ng/ml), dexamethasone (1 μM), or RU24858 (10 μM) and harvested after 6 h. Nuclear RNA was prepared and analyzed for unspliced IL-8 (IL-8 A) and GAPDH (GAPDH B) using real-time TaqMan RT-PCR. Data (n = 8), expressed as a ratio of IL-8/GAPDH, are plotted as a percentage of the IL-1β stimulation as means ± S.E.M. *, P < 0.05; ***, P < 0.001.
Steroid-Dependent Repression of Inflammatory Gene mRNA Expression Is Blocked by Actinomycin D. In previous studies, the repression of IL-1β-induced steady-state COX-2 mRNA was profoundly repressed by dexamethasone despite only a 40% reduction in COX-2 transcription rate and even when added 2 h after the IL-1β stimulus (Newton et al., 1998). This repression was in part attributed to destabilization of COX-2 mRNA, and such effects are widely reported (Ristimaki et al., 1996; Newton et al., 1998; Lasa et al., 2001). Therefore, IL-8 mRNA expression was examined after treatment with IL-1β and dexamethasone, which was added 2 h after the IL-1β (Fig. 4A shows the treatment protocol). Unstimulated cells revealed low or undetectable levels of IL-8 mRNA, which were substantially elevated by2hof treatment with IL-1β and at time points thereafter (Fig. 4B). Steady-state mRNA levels reached a peak 5 (2 + 3) h after the addition of IL-1β and decreased thereafter. The addition of actinomycin D, an inhibitor of RNA polymerase II, 2 h after the IL-1β stimulus (t = 0) revealed that the IL-1β-induced IL-8 mRNA was relatively stable, and little decay was observed in these experiments (Fig. 4B). In marked contrast, the addition of dexamethasone initially had no effect on steady-state IL-8 mRNA levels induced by IL-1β, but after 1 h, any further increases in IL-8 mRNA were halted, and mRNA expression decreased over the next 3 h (Fig. 4B). Thus, simple blockade of RNA polymerase II-dependent transcription is insufficient for repression of IL-8 mRNA. This, along with the reporter, nuclear run-on, and nuclear RNA data, strongly implicates a requirement for additional post-transcriptional mechanisms of repression by glucocorticoids. In context of the current study, we wished to know whether the dexamethasone-dependent repression of steady-state IL-8 mRNA was dependent on ongoing gene expression. A549 cells were therefore treated with IL-1β for 2 h before the addition of various combinations of dexamethasone and actinomycin D and harvesting 6 h later (Fig. 4C). As above, IL-1β strongly induced IL-8, and this was not significantly affected by actinomycin D. Again, dexamethasone produced a robust repressive effect, and this was totally prevented by simultaneous addition of actinomycin D.
To further explore this effect, A549 cells were treated with dexamethasone or RU24858 in the presence or absence of actinomycin D, and the expression of IL-8 and COX-2 was examined by semiquantitative reverse-transcriptase polymerase chain reaction RT-PCR (Fig. 5). In these experiments, IL-1β induced the expression of both IL-8 and COX-2, and this was repressed by the simultaneous addition of dexamethasone (1 μM) or RU24858 (10 μM) (Fig. 5). The addition of either steroid at 2 h after the IL-1β resulted in a similar level of mRNA repression, and this effect was prevented by the presence of actinomycin D (Fig. 5). The addition, of actinomycin D alone showed no significant effect on the expression of IL-8 or COX-2. Given the pivotal nature of this experiment, all cDNA samples were subsequently reanalyzed using TaqMan real-time PCR, and essentially identical data were obtained (Supplemental Fig. S6A). In addition, the existence of feedback process resulting in the loss of steady-state IL-8 mRNA levels from 5 (2 + 3) h onward (Fig. 4B) increases the possibility of confounding factors. Therefore the above experiment was also conducted at a shorter time point (i.e., after only3hof steroid/actinomycin D treatment and only 5 h in total). Again, a qualitatively similar result was obtained (Supplemental Fig. S6B) and suggests that inhibition by both dexamethasone and RU24858 requires ongoing transcription at all time points after the addition of steroid.

Effect of dexamethasone on steady-state IL-8 mRNA. A, schematic representation of the experiments in B and C. A549 cells were treated with IL-1β (1 ng/ml) or not stimulated for 2 h before the addition of combinations of dexamethasone (1 μM) and actinomycin D (10 μg/ml) (Act D) and then harvested. B, cells were treated as indicated and harvested at the indicated times (after Dex/Act D addition) for Northern blot analysis of IL-8 and GAPDH. Representative blots are shown. After densitometric analysis, data from four such experiments were expressed as a percentage of the IL-1β after 2-h stimulation (i.e., t = 0) and are plotted as means ± S.E.M. Note that data for the 3-h time point is n = 2. C, cells were treated as indicated in A and B. Two hours after the IL-1β stimulus, combinations of dexamethasone and actinomycin D were added, and the cells were harvested after a further 6 h for Northern blot analysis of IL-8 and GAPDH. Representative blots are shown. After densitometric analysis, data (n = 4) were expressed as a percentage of IL-1β and are plotted as means ± S.E.M. ***, P < 0.001.
Effect of Dexamethasone and RU24858 on mRNA Decay. The above data strongly implicate additional cytoplasmic mechanisms of repression to account for the full repressive effects of glucocorticoids. To specifically test this hypothesis, classic actinomycin D chase experiments were conducted (Fig. 6). In the previous analyses (Figs. 4 and 5), the addition of actinomycin D at the same time as either dexamethasone or RU24858 resulted in no repression of COX-2 or IL-8 mRNA. Therefore, to examine the possibility of steroid-dependent changes in mRNA stability, A549 cells were first stimulated with IL-1β for 2 h before the addition of dexamethasone or RU24858 (Fig. 6A). To allow time for steroid-dependent gene expression, the classic chase experiment was commenced by the addition of actinomycinD1h after the steroids (t = 0). RNA was then harvested, and expression of IL-8, COX-2, and GAPDH monitored in cDNA by TaqMan PCR (Fig. 6B). In each case, the addition of dexamethasone resulted in more rapid initial decay rates of both COX-2 and IL-8 mRNA (Fig. 6B and Table 2). In addition, dexamethasone led to the loss of a greater fraction of the initial starting mRNA compared with IL-1β-treated samples. In each case, RU24858 resulted in an intermediated effect, which for COX-2 was not significantly different from either the dexamethasone or the IL-1β-treated decay rates. In the case of IL-8, the initial decay rate produced by RU24858 was significantly more than for IL-1β alone but was less than that produced by IL-1β plus dexamethasone (Fig. 6B and Table 2).
IL-1β-induced COX-2 and IL-8 mRNA decay: effect of dexamethasone and RU24858
Using the mRNA decay data presented in Fig. 6., the slope for each set of experimental values was calculated by linear regression analysis (GraphPad Prism) using values for t = 0, 0.25, 0.5, and 1 h. In each case, this provides an initial slope in which active mRNA degradation appeared to be occurring. Data (n = 7-8) for each initial slope are presented as means ± S.E. Significance or nonsignificance (N.S.) between these values is indicated.
Steroid-Dependent Repression of Inflammatory Gene mRNA Expression Is Attenuated by Cycloheximide. In complementary experiments, A549 cells were incubated with various combinations of IL-1β, the protein synthesis inhibitor cycloheximide, dexamethasone, and RU24858 before incubation for 4 h and semiquantitative RT-PCR analysis of steady-state mRNA levels (Fig. 7). The addition of steroid had no effect on basal levels of IL-8 and COX-2 because these were essentially undetectable. Alone, cycloheximide had little or no effect on IL-8 but resulted in increased expression of COX-2. IL-1β resulted in robust increases in IL-8 and COX-2 mRNA, and this was repressed by the simultaneous addition of either dexamethasone or RU24858. This steroid-dependent repression was significantly reduced in the presence of cycloheximide, suggesting a requirement for ongoing protein synthesis.
Evidence for a Requirement of Steroid-Inducible Genes in the Repression of Inflammatory Gene Protein Expression. In previous studies, dexamethasone was reported to block the IL-1β-induced expression of COX-2 protein, even when added substantially after the IL-1β (Newton et al., 1998). To examine the possible role of steroid-inducible genes in the repression of COX-2 and IL-8 protein expression, A549 cells were treated with IL-1β for 6 h before the addition of dexamethasone, RU24858 or actinomycin D (Fig. 8A). The addition of either dexamethasone or RU24858 6 h after the IL-1β stimulus resulted in a marked repression of COX-2 (Fig. 8B). The repression of IL-8 was considerably weaker (only 25-30% repression) (Fig. 8C), presumably because significant quantities of IL-8 protein had already been produced by the time the steroids were added after the IL-1β stimulus. It is noteworthy that the presence of actinomycin D with both dexamethasone and RU24858 (at 6 h) resulted in a total loss of the inhibitory properties in respect of COX-2 and IL-8 protein expression.

Repression of inflammatory gene mRNA expression by dexamethasone and RU24858 is prevented by actinomycin D. A549 cells were stimulated with IL-1β (1 ng/ml), or not stimulated (NS). Dexamethasone (1 μM) (Dex) or RU24858 (10 μM) was either added simultaneously with the IL-1β (t =-2), or 2 h after the IL-1β (t = 0) in the presence or absence of actinomycin D (10 μg/ml) (Act D) (as shown in Fig. 3B). Cells were then harvested 6 h later (i.e., a total of 8 h after the IL-1β), and RNA was prepared. Semiquantitative RT-PCR analysis for COX-2, IL-8, and GAPDH was performed. Each experiment carried two IL-1β-stimulated samples, and these were averaged and set to 100% for data presentation. Data (n = 8 for COX-2 and 7 for IL-8) were normalized to GAPDH, expressed as a percentage of the averaged IL-1β, and are plotted as means ± S.E.M. *, P < 0.05; **, P < 0.01.

Effect of dexamethasone and RU24858 on IL-1β-induced COX-2 and IL-8 mRNA stability. A549 cells were stimulated with IL-1β (1 ng/ml) for 2 h before no further treatment (○) or were treated with dexamethasone (1 μM) (Dex) (▪) or RU24858 (10 μM) (•). After a further1h(t = 0), actinomycin D (10 μg/ml) (Act D) was added. Cells were harvested for total RNA at 0, 0.25, 0.5, 1, and 2 h after the addition of actinomycin D. A, schematic depicting the above experimental protocol. B, after RNA extraction and cDNA synthesis, real-time TaqMan PCR was used to analyze the expression of COX-2, IL-8, and GAPDH. In each case, relative mRNA levels were obtained by reference to a standard curve generated by serial cDNA dilution. Relative COX-2 and IL-8 mRNA levels were normalized to GAPDH and were expressed as a percentage of the value at t = 0 for each treatment group. Data (n = 7-8) are plotted as means ± S.E.M. Values from data at t = 0, 0.25, 0.5, and 1 h were used to calculate the initial slope for each treatment, and these are to be found in Table 2.
Effects of Dexamethasone and RU24858 on Steroid-Inducible Genes. In the above sections, the repression of inflammatory gene expression by dexamethasone and the dissociated steroid RU24858 is prevented by actinomycin D or cycloheximide. This suggests a requirement for steroid-dependent gene expression but raises a considerable issue in respect of the action of RU24858, which is largely incapable of classic GRE-dependent transcription (Fig. 2A). However, in addition to the simple or classic GRE, GR may induce transcriptional responses via interactions with numerous other transcription factors (Newton, 2000). Because the effects of dissociated steroids on such forms of transactivation are not generally tested, these could, in theory, be important in the current responses. However, because the functional relevance of nonclassic or complex GREs is unclear with respect to glucocorticoid-inducible gene expression, we elected to directly test dexamethasone-inducible genes for possible inducibility by RU248585.
A prior Affymetrix microarray (Hu95Av2 and B chips) study in A549 cells identified some 300 genes as being dexamethasone-inducible (data not shown). Importantly, a number of these dexamethasone-inducible genes are also potentially anti-inflammatory in effect (Table 3). For example, MKP-1 switches off MAPK signaling (Clark and Lasa, 2003), GILZ represses AP-1-dependent transcription (Mittelstadt and Ashwell, 2001), and regulator of G-protein signaling (RGS)-2 reduces Gq-linked signaling (Heximer, 2004). In addition, the metabolic genes AMP N and MET 1X were also identified as being dexamethasone-inducible.
Dexamethasone-inducible genes in A549 cells
A549 cells were either not treated or treated with dexamethasone (1 μM) for 6 and 18 h. RNA was prepared, and microarray analysis was performed using recommended Affymetrix methodology. Data (n = 3), analyzed using the P-fold algorithm, are presented as relative -fold induction (R) versus not treated at each time point. P values are indicated

Repression of inflammatory gene mRNA expression by dexamethasone and RU24858 is prevented by cycloheximide. A549 cells were stimulated with IL-1β (1 ng/ml) or were not stimulated (NS). Dexamethasone (1 μM) or RU25858 (10 μM) was added simultaneously with the IL-1β in the presence or absence of cycloheximide (10 μg/ml) (CHX). Cells were then harvested 4 h later, and RNA was prepared. Semiquantitative RT-PCR analysis for COX-2, IL-8, and GAPDH was performed. Data (n = 4) were normalized to GAPDH, expressed as a percentage of IL-1β, and are plotted as means ± S.E.M. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
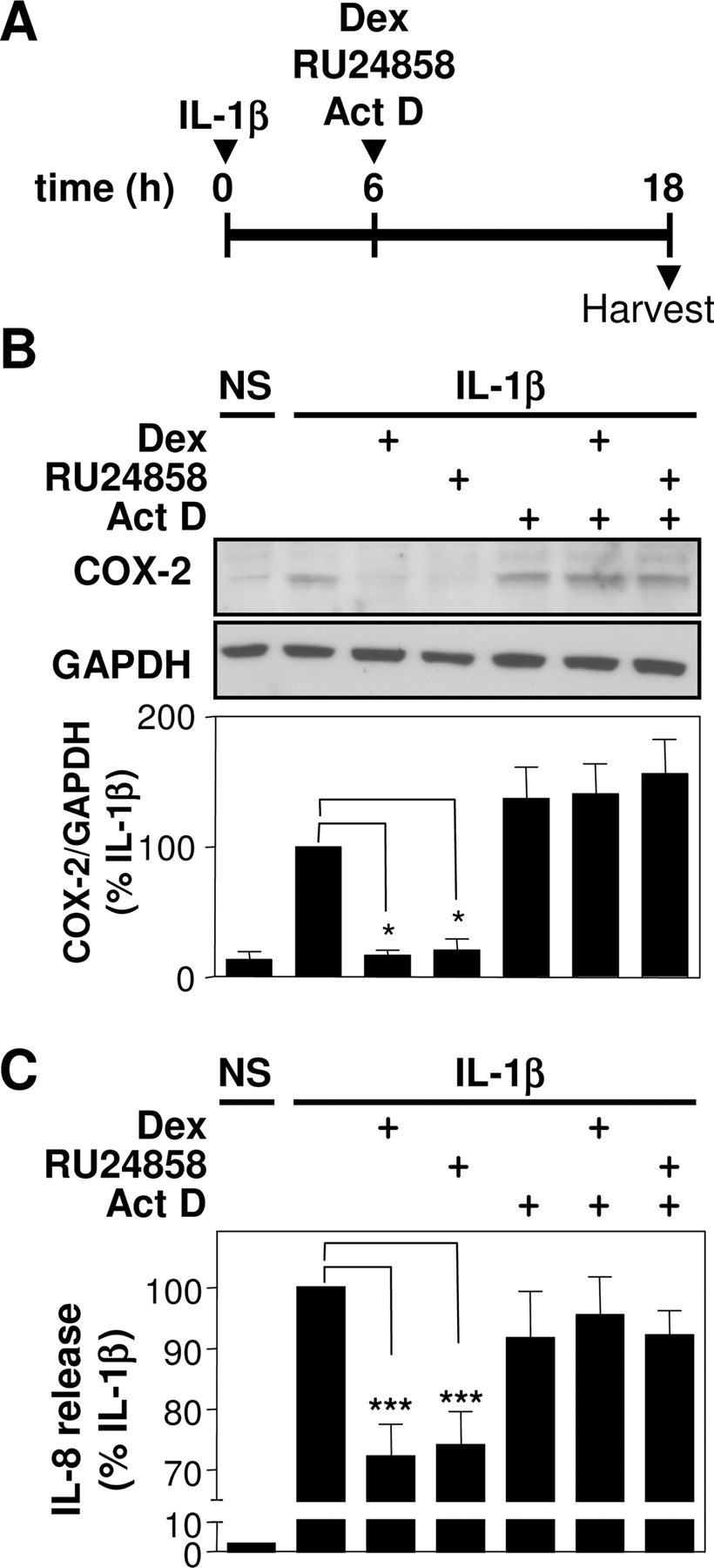
Repression of inflammatory gene protein expression by dexamethasone and RU24858 is prevented by actinomycin D. A, A549 cells were stimulated with IL-1β or not stimulated (NS), and combinations of dexamethasone (1 μM) (Dex), RU24858 (10 μM), and actinomycin D (10 μg/ml) (Act D) were added 6 h later before harvesting at 18 h after the IL-1β.B, total cellular lysates were subject to Western blot analysis for COX-2 and GAPDH. Representative blots are shown. Optical density data (n = 6), normalized to GAPDH and expressed as a percentage of IL-1β, are plotted as means ± S.E.M. C, supernatants were harvested and subjected to ELISA for IL-8. Data (n = 8) were expressed as a percentage of IL-1β and are plotted as means ± S.E.M. ***, P < 0.001.
A549 cells were therefore treated with dexamethasone or RU24858 for various periods of time, and the expression of MKP-1, RGS-2, GILZ, AMP N, and MET 1X was examined by semiquantitative RT-PCR (Fig. 9). In each case, mRNA expression was up-regulated by dexamethasone, and this largely correlated with the array data (Fig. 9 and Table 3). The expression of MKP-1, GILZ, AMP N, and MET 1X, but not RGS2, were also up-regulated by RU24858. Thus, although RU24858 is dissociated by reference to a classic GRE response, this compound can nevertheless induce the expression of glucocorticoid-inducible genes. Because these genes may contribute to both the anti-inflammatory or side effect profile of this steroid, these data provide an explanation for the ability to block the repressive effects of RU24858 with transcriptional and translational blockers.
Discussion
The GR is modular, with functional activities localized to specific domains (Rhen and Cidlowski, 2005). However, whereas the ligand binding domain and the DNA binding domains are necessary for both transactivation of classic GRE-dependent transcription and transrepression (e.g., of AP-1), mutations in these domains may differentially affect each function. For example, the GR A458T mutation used to generate the GRdim/dim mouse renders the receptor incapable of classic GRE-dependent transcription, but both anti-inflammatory activity and the ability to transrepress are retained (Reichardt et al., 1998, 2001). Likewise, the generation of dissociated GR ligands that are transrepression-competent, defective at classic transactivation, yet retain anti-inflammatory effects seems to support the view that transrepression and not transactivation accounts for the main anti-inflammatory properties of glucocorticoids (Heck et al., 1994; Vayssiere et al., 1997). Likewise, RU24858 is dissociated in A549 cells and represses IL-1β-induced expression of COX-2 and IL-8. At first, this result seems to agree with the above dogma and is supported by the correlation between the EC50 values for inhibition of NF-κB and these functional outputs. Despite this, the slightly reduced efficacy of RU24858 on IL-8 and COX-2 expression leaves open the possibility of a minor repressive role for classic GRE-dependent responses. However, the fact that activation of GRE-dependent transcription by dexamethasone occurred at a higher concentration (EC50 = 93 nM) than that observed for repression of COX/PGES (EC50 = 3.78), or IL-8 (EC50 = 2.55 nM) also argues against a major role for classic GRE-dependent transcription. Importantly, the finding that IL-8 transcription rate, the activation of NF-κB-dependent transcription and the analysis of IL-8 nuclear mRNA intermediates show no more than 40 to 60% repression by steroids, plus similar data in respect of COX-2 transcription rate (Newton et al., 1998), casts considerable doubt on transcriptional repression being the sole, or even main, mechanism for glucocorticoid-dependent repression. Indeed, we show a significant role for post-transcriptional events in mediating the glucocorticoid-dependent repression of IL-8 and COX-2, and this is consistent with a substantial body of data documenting critical roles for post-transcriptional repression (Newton, 2000; Stellato, 2004).

Dexamethasone-inducible genes may be induced by RU24858. Cells were stimulated with dexamethasone (1 μM) (Dex) or RU24858 (10 μM) for 2, 6, or 18 h. Cells were harvested, and RNA was prepared for semiquantitative RT-PCR analysis of the indicated genes. A, representative ethidium bromide-stained gels showing expression of MKP-1, RGS2, GILZ, AMP N, MET IX, and GAPDH are provided. B, After optical density determination (TotalLab software), data (n = 4) normalized to GAPDH are presented as -fold induction ± S.E.M.
In considering the contribution of transrepression and transactivation to the anti-inflammatory properties of glucocorticoids, it is worth remembering that GR dimerization and activation of classic GRE-dependent transcription is not the only mechanism of GR-dependent transactivation (Newton, 2000). Thus, GR may interact, cross-talk, or even synergize with signal transducer and activator of transcription 3, signal transducer and activator of transcription 5, CATT enhancer binding protein family members, and other nuclear hormone receptors to bring about transcriptional activation (Kordula and Travis, 1996; Stocklin et al., 1996; Boruk et al., 1998; Newton, 2000; Savory et al., 2001; Cassuto et al., 2005). Even interactions between GR and AP-1 components or on NF-κB reporters may lead to transcriptional activation (Diamond et al., 1990; Hofmann and Schmitz, 2002). Such effects are clearly not mediated via classic GREs and, critically for the present discussion, are not generally evaluated in the context of GR mutants, dissociated GR ligands, or the induction of glucocorticoid-inducible genes. Therefore, there exists a formal possibility that nonclassic forms of GR transactivation are important in glucocorticoid-dependent repression of inflammatory genes. Furthermore, numerous prior studies have documented that the ability of glucocorticoids to repress inflammatory gene expression is lost in the presence of either transcriptional or translational blockers (Ristimaki et al., 1996; Newton et al., 1998; Chang et al., 2001; Lasa et al., 2001; Korhonen et al., 2002). Such observations are difficult to reconcile with models for anti-inflammatory glucocorticoid action that predominantly invoke classic transrepression of NF-κB and AP-1. In these schemes, GR is believed, either by direct interaction or indirectly by recruitment of corepressors or histone deacetylases, to interfere with transcriptional activation of inflammatory genes, and this does not require steroid-dependent gene expression. In contrast, the dexamethasone- and RU24858-dependent repression of IL-8 and COX-2 steady-state mRNA and protein synthesis requires both transcription and translation because repression is both actinomycin D- and cycloheximide-sensitive. Thus, despite not activating classic GRE-dependent transcription, our data clearly suggest that repression by dexamethasone and RU248585 requires GR-dependent gene expression.
In past years, the existence of steroid-inducible genes, for example lipcortin 1 (annexin 1), β2-adrenergic receptor, secretory leukocyte protease inhibitor, IL-1 receptor antagonist, and others (Newton, 2000), although undoubtedly making an anti-inflammatory contribution, was not generally considered sufficient to explain the repression of inflammatory gene expression. More recently, microarray-based approaches have documented the existence of numerous glucocorticoid-inducible genes (Rogatsky et al., 2003). Thus, glucocorticoid-inducible genes, such as MKP-1, RGS-2, and GILZ, show potentially anti-inflammatory properties and offer a real possibility that substantive anti-inflammatory effects may be attributed to glucocorticoid-dependent gene induction. To examine this possibility with respect to RU24858, the expression of MKP-1, RGS-2, and GILZ were examined. In this analysis, the expression of MKP-1 and GILZ were both induced by RU24858 and dexamethasone, whereas RGS-2 was not induced. These data confirm the possibility that RU24858 can induce anti-inflammatory glucocorticoid-inducible genes, presumably via mechanisms other than at a classic GRE. Furthermore, this supports the idea that such effects could be important in the repression of inflammatory genes such as COX-2 and IL-8. This statement also finds considerable support from the work of Rogatsky et al. (2003), in which microarray profiling was used to describe the induction of multiple genes by dexamethasone. Critically for the current argument, mutations in either of the two GR activation domains (AF1 and AF2) or the GRdim mutant resulted in loss of responsiveness for different groups but not all dexamethasone-inducible genes (Rogatsky et al., 2003). Thus, different mechanisms of transactivation are responsible for the induction of these different genes, and this is consistent with the possibility that GR ligands differentially activate these functions to induce discrete subsets of glucocorticoid-inducible genes.
Likewise, in vivo analysis of RU24858 revealed all of the main side effects of standard balanced glucocorticoids, suggesting that classic GRE-dependent transactivation does not, by itself, account for these side effects (Belvisi et al., 2001). Our analysis also supports this view as the glucocorticoid-inducible genes MET-1X and AMP N were induced by both dexamethasone and by RU24858. Assuming that other metabolic gene may also be induced, this provides an explanation for the emergence of side effects with RU24858 (Belvisi et al., 2001). In contrast, the nonsteroidal GR ligand ZK 216348 was obtained after functional cell-based screens for GR ligands that did not induce tyrosine amino transferase, yet were still able to repress IL-8 expression (Schacke et al., 2004). This approach resulted in GR ligands that were considerably more “dissociated” with respect to the ability to cause repression of IL-8 and other inflammatory genes but showed considerably reduced metabolic side effects.
In terms of different GR ligands differentially inducing GR-dependent responses, we have documented previously the ability of the antagonist, RU486, to cause repression of PGE2 release via effects predominantly occurring at the level of arachidonic acid release (Chivers et al., 2004). RU486 causes GR translocation yet is silent on both transrepression and transactivation (Chivers et al., 2004); therefore; it is possible that this could represent a nongenomic mechanism of glucocorticoid action, and this may explain the relatively potent effect of RU24858 on the repression of PGE2 release (EC50 = 53.9 nM). In addition, it is now clear that different GR ligands, presumably as a consequence of different conformational changes induced in respect of GR activation domains, are capable of differentially recruiting coactivators and corepressors (Adcock et al., 2004; Garside et al., 2004). Therefore, the relationship between the ligand, conformational changes produced by the ligand, and the recruitment (or not) of coactivators and repressors will have a profound effect on the transcriptional responses observed in different promoter contexts. Thus, different GR ligands have the potential to produce variable activational or repressional responses, depending on the exact nature of both the promoter and the complement of transcription factors, coactivators, and repressors that are present within the cell.
In conclusion, the repression of IL-8 and COX-2 by dexamethasone and RU24858 is not fully accounted for by transrepression (e.g., of NF-κB), and requires ongoing, steroid-dependent gene expression and post-transcriptional mechanisms of repression. Furthermore, and despite not activating a classic GRE-dependent reporter construct, RU24858 was nearly as effective as dexamethasone at inducing certain glucocorticoid-inducible genes, suggesting that such processes may not involve classic GRE-dependent transcription. Finally, because a number of these dexamethasone- and RU24858-inducible genes are potentially anti-inflammatory, we propose that glucocorticoid-dependent genes play a significant role the anti-inflammatory effects of glucocorticoids.
Acknowledgments
We thank the Sanofi-Aventis Genomic Center (Bridgewater, NJ) for conducting the Affymetrix microarray analysis.
Footnotes
-
J.E.C. was a collaborative Biotechnology and Biological Sciences Research Council student supported by Aventis Pharmaceuticals (now Sanofi-Aventis). R.N. is a Canadian Institutes of Health Research (CIHR) New Investigator and Alberta Heritage Foundation for Medical Research (AHFMR) scholar. This work was supported by both CIHR and the AHFMR. R.N. also received an unrestricted grant from GlaxoSmithKline.
-
J.E.C., W.G., and E.M.K. contributed equally to this work and are listed alphabetically.
-
ABBREVIATIONS: GR, glucocorticoid receptor; AMP N, aminopeptidase N; AP-1, activator protein 1; COX, cyclooxygenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GILZ, glucocorticoid-inducible leucine zipper protein; GRE, glucocorticoid response element; IL, interleukin; MET 1X, metallothionein 1X; MAPK, mitogen-activated protein kinase; MKP-1, mitogen-activated protein kinase phosphatase-1; NF-κB, nuclear factor κB; PG, prostaglandin; PGES prostaglandin E synthase; PCR, polymerase chain reaction; RGS2, regulator of G-protein signaling 2; RT-PCR, reverse transcriptase-polymerase chain reaction; SFM, serum-free medium; ELISA, enzyme-linked immunosorbent assay; RU486, mifepristone.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received April 19, 2006.
- Accepted September 19, 2006.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}