


Abstract
Our previous studies have suggested a role for AMP-activated protein kinase (AMPK) in the induction of CYP2B6 by phenobarbital (PB) in hepatoma-derived cells (Rencurel et al., 2005). In this study, we showed in primary human hepatocytes that: 1) 5′-phosphoribosyl-5-aminoimidazol-4-carboxamide 1-β-d-ribofuranoside and the biguanide metformin, known activators of AMPK, dose-dependently increase the expression of CYP2B6 and CYP3A4 to an extent similar to that of PB. 2) PB, but not the human nuclear receptor constitutive active/androstane receptor (CAR) ligand 6-(4-chlorophenyl)imidazol[2,1-6][1,3]thiazole-5-carbaldehyde, dose-dependently increase AMPK activity. 3) Pharmacological inhibition of AMPK activity with compound C or dominant-negative forms of AMPK blunt the inductive response to phenobarbital. Furthermore, in transgenic mice with a liver-specific deletion of both the α1 and α2 AMPK catalytic subunits, basal levels of Cyp2b10 and Cyp3a11 mRNA were increased but not in primary culture of mouse hepatocytes. However, phenobarbital or 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene, a mouse CAR ligand, failed to induce the expression of these genes in the liver or cultured hepatocytes from mice lacking hepatic expression of the α1 and α2 subunits of AMPK. The distribution of CAR between the nucleus and cytosol was not altered in hepatocytes from mice lacking both AMPK catalytic subunits. These data highlight the essential role of AMPK in the CAR-mediated signal transduction pathway.
Induction of drug-metabolizing enzymes and drug transporters by drugs and other chemicals is an adaptive response of mammals and other organisms to increase the removal of potentially toxic endobiotics and xenobiotics. Phenobarbital (PB) represents a class of inducers in which the effect on detoxification is part of a pleiotropic response that includes liver hypertrophy, tumor promotion, and induction or repression of multiple genes, in addition to genes coding for enzymes or transporters that regulate drug disposition. The molecular mechanism of the induction response remains incompletely understood. The induction of human cytochrome P450 CYP2B6 and its rat and mouse orthologs CYP2B1 and Cyp2b10 by PB is mediated by the nuclear receptor constitutive active/androstane receptor (CAR, NR1I3) (Honkakoski et al., 1998). In untreated primary mouse hepatocytes, CAR is retained in the cytoplasm within a protein complex of chaperones and cochaperones, such as the 90-kDa heat shock protein and a protein called cytoplasmic CAR retaining protein (CCRP) (Kobayashi et al., 2003). Exposure to xenobiotics such as PB causes CAR to dissociate from this complex and to transfer into the nucleus, where it forms a heterodimer with retinoid X receptor and binds to cognate DNA sequences of target genes (Kawamoto et al., 1999). This process is influenced by phosphorylation and dephosphorylation of unknown proteins (Hosseinpour et al., 2005). In human hepatoma cells, such as HepG2, CAR is found exclusively in the nucleus and is constitutively active, resulting in CYP2B6 gene expression in cells not exposed to any inducers (Sueyoshi et al., 1999). Coexpression of exogenous CCRP with exogenous CAR in HepG2 cells confirms the cytoplasmic retention of CAR by CCRP, but no nuclear transfer is observed upon drug treatment (Kobayashi et al., 2003). This suggests that additional proteins or reactions are required for drug induced CAR cytoplasmic-nuclear translocations, which are missing or nonfunctional in HepG2 cells. Because hepatoma cells do not reciprocate the physiological activation of CAR or its binding to DNA, we decided to focus on CAR in primary human and mouse hepatocytes.
In the liver of fasted mice, CYP2b10 and mCAR expression are induced (Maglich et al., 2004). AMP-activated protein kinase (AMPK) functions as an energy sensor and is activated when cells experience energy depleting stresses such as nutrient starvation (Carling, 2004). AMPK is also activated by pharmacological manipulations. 5′-Phosphoribosyl-5-aminoimidazol-4-carboxamide 1-β-d-ribofuranoside (AICAR) is a cell-permeant adenosine analog commonly used to activate AMPK. Although activation of AMPK because of an increase in the AMP/ATP ratio is the best-described mechanism, some studies suggest alternative mechanisms of AMPK activation.
AMPK is a heterotrimeric complex ubiquitously expressed and consists of a catalytic subunit, α, and two regulatory subunits, β and γ (Woods et al., 1996). All three subunits have been identified, and each subunit is encoded by two (α1,α2, β1,β2) or three genes (γ1, γ2, γ3) Formation of the trimeric complex is necessary for optimal kinase activity. Changes in the cellular energy state activate AMPK through mechanisms involving an AMP allosteric regulation and phosphorylation by an upstream kinase on threonine residue 172 within the α subunit (Stein et al., 2000). This upstream kinase has recently been identified in liver as LKB1 (Woods et al., 2003).
In a previous study, we showed that PB can activate AMPK in a HepG2-derived hepatoma cell line, WGA, and that activation of AMPK by AICAR induced CYP2B6 (Rencurel et al., 2005). However, the activation of AMPK was observed only at high concentrations of PB (above 1 mM). Questions thus remain as to whether 1) AMPK activation by PB occurs only in transformed cells or 2) only at high concentrations of the inducer. Thus, it has remained unclear whether activation of AMPK is a necessary component of the PB signaling pathway in fully differentiated liver cells either in vitro and or in vivo. We therefore investigated the relationship between AMPK activation and induction of cytochromes P450 in primary human hepatocytes. Moreover, the recent development of transgenic mice with a deletion of AMPK catalytic subunits α1 and α2 in the liver provides a unique experimental model to address the role of AMPK in PB induction of CYP2b10 and CYP3a11 gene expression in vivo and in vitro. In primary human hepatocyte culture, we now show a concentration-dependent activation of AMPK by PB. Through ablation of AMPK activity by 1) pharmacological approaches, 2) overexpressing a dominant-negative form of AMPK, or 3) genetic ablation of AMPK α1 and α2 genes in mice. We here provide unequivocal evidence that AMPK activation is a necessary step in the induction of CYP2B6 and Cyp2b10 by PB in both human and mouse hepatocytes.
Materials and Methods
Reagents. Phenobarbital (sodium salt) was purchased from Fluka (Buchs, Switzerland). All other chemicals were from Sigma (Buchs, Switzerland). Cell culture media, fetal bovine serum, other tissue culture reagents, and TRIzol reagent were from Invitrogen (Carlsbad, CA). Antibodies raised against AMPK α1 and α2 subunits and phospho-acetyl-CoA-carboxylase were purchased from Upstate Biotechnology (Lucerne, Switzerland).
Generation of AMPKα1/α2LS-/- Knockout Mice. The liver-specific knock out of both α subunits of AMPK has been described previously (Guigas et al., 2006). In brief, to generate deletion of both catalytic subunits in the liver (α1/α2LS-/-), liver-specific AMPKα2-null mice were first generated by crossing floxed AMPKα2 mice (Viollet et al., 2003) and Alfp Cre transgenic mice expressing the Cre recombinase under the control of albumin and α-fetoprotein regulatory elements. A liver-specific AMPKα2 deletion was then produced on an AMPKα1-/- general knockout background by crossing liverspecific α2-/- mice with AMPKα1-/- general knockout mice (Jorgensen et al., 2004).
Recombinant Adenoviruses. Adenovirus encoding constitutively active α1 AMPK subunit (ad-CA-α1312) or dominant-negative mutant α1 AMPK (ad-DNα1) were prepared as described previously (Diraison et al., 2004). Adenoviruses encoding constitutively active α2 AMPK subunit (ad-CA-α2312) or β-galactosidase were also amplified as described previously (Foretz et al., 2005). Adenovirus encoding human CAR in fusion with enhanced green fluorescence protein (ad-hCAR-GFP) was a kind gift of Dr. Ramiro Jover (Hospital La Fe, Valencia, Spain). AMPK adenoviruses also express, under control of a distinct cytomegalovirus promoter. Viral particles were purified by cesium chloride density centrifugation, and human and mouse hepatocytes were infected 12 h after seeding with a multiplicity of infection of 30 to 100.
Culture of Primary Human Hepatocytes. Primary human hepatocytes were isolated from the resected liver tissue of consented patients undergoing liver surgery. Human hepatocytes were enzymatically dissociated from human liver samples using a two-step enzymatic microperfusion technique with collagenase and kept on ice in suspension (Strain, 1994). Hepatocytes were subsequently seeded on plastic dishes coated with rat-tail collagen (25 μg/cm2)at a density of 130,000 viable cells/cm2 and cultured in Dulbeco's minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 1 μM dexamethasone.
After overnight culture, the medium was replaced by serum-free Williams' E medium supplemented with 100 nM hydrocortisone, 50 U/ml penicillin, 50 μg/ml streptomycin, and solution ITS + 1, containing insulin (5 μg/ml), transferrin (2.75 μg/ml), and selenium (25 ng/ml) (Sigma, Buchs, Switzerland). The medium also was supplemented with 250 μg/ml bovine serum albumin and 2.35 μg/ml linoleic acid. Twenty-four hours after serum deprivation, the human hepatocytes were kept in serum-free medium and exposed to various chemicals for ≤16 h as indicated in the figure legends.
Preparation and Culture of Primary Mouse Hepatocytes. Liver cells were prepared by the two-step collagenase method (Berry and Friend, 1969) from postabsorptive male mice (25-30 g) after anesthesia with ketamine/xylazine (1 mg/100 g of body weight). Hepatocytes were seeded on dishes coated with rat-tail collagen type 1 and cultured overnight in M199 supplemented with 1% Ultroser G (Biosepra SA, Cergy-Saint-Christophe, France) and (50 U/ml penicillin/50 μg/ml streptomycin. After overnight culture, the medium was replaced by a serum free Williams' E medium. Twenty-four hours after serum deprivation, cells were exposed to chemicals for 16 h, or as indicated in the figure legends, and were maintained in serum-free medium.
Real-Time PCR Assays. One microgram of total RNA was reverse-transcribed and used in real-time PCR assays for quantification of different target genes on an ABI PRISM 7700 sequence detection system (Applied Biosystems, Foster City, CA). Expression levels of these genes were normalized against 18s rRNA for human samples and normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA for mouse samples. The primers sequences are described in Table 1.
Primers and probes sequences used in quantitative real-time PCR (TaqMan)
Western Blot Analysis. Cultured hepatocytes were washed in ice-cold PBS and harvested in a 300-μl/6-cm dish of extraction buffer (100 mM KCl, 25 mM HEPES, 7.5 mM MgCl2, and 20% glycerol, pH 7.4) supplemented with protease inhibitor cocktail tablets (Roche, Rotkreuz, Switzerland) containing dithiothreitol (4 mM), aprotinin (2 mg/ml), and β-mercaptoethanol (1 mM). The cell suspension was sonicated for 5 s, and cellular debris was removed by centrifugation (1000g for 10 min at 4°C).
Thirty micrograms of total cellular protein were separated by Tris-Tricine glycerol/SDS-polyacrylamide gel electrophoresis and blotted onto nitrocellulose. The following primary antibodies were employed: anti-AMPK α1 subunit and anti-AMPK α2 subunit, antiphospho-ACC (Cell Signaling, Allschwill, Switzerland) and anti-Myc (clone 9E10; Sigma, Buchs, Switzerland). Secondary antibodies antirabbit IgG and anti-mouse IgG conjugated to horseradish peroxidase were employed for chemiluminescence immunodetection. Blots were developed using ECL reagent (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and exposure to X-ray films (X-OMAT Kodak; Sigma).
AMP-Activated Kinase Activity Assay. The AMPK assay was performed using the SAMS peptide phosphorylation assay kit from Upstate Biotechnology according to the manufacturer's protocol. In brief, cells were cultured in serum-free medium for 16 h before drug exposure. Chemicals were added straight to cell culture dishes, and cells were incubated for 1 h at 37°C. Culture medium was quickly removed, and cells were washed once with ice-cold PBS and harvested in Tris-HCL (50 mM, pH 7.5), EGTA (1 mM), EDTA (1 mM), dithiothreitol (1 mM), sodium fluoride (50 mM), sodium pyrophosphate (5 mM), benzamidine (1 mM), soybean trypsin inhibitor (4 μg/ml), phenylmethylsulfonyl fluoride (1 mM), mannitol (250 mM), and protease inhibitor tablets (Roche). Cellular debris was removed by centrifugation at 10,000g at 4°C for 20 min and the supernatant snap-frozen in liquid nitrogen. Samples were stored at -70°C before AMPK activity assays.
Proteins in the supernatant were concentrated by polyethylene glycol PEG 8000 precipitation, and the AMPK reaction was performed for 10 min at 30°C with 20 μM SAMS peptide, 10 μCi of [γ-32P]ATP and a 10-μg protein sample. The reaction mixture was then spotted on P81 phosphocellulose paper (Upstate Biotechnology), which was washed with 0.75% phosphoric acid and acetone, and the radioactivity of phosphor SAMS peptide was quantified by scintillation counting.
Measurement of ATP Concentration in Primary Hepatocytes. Primary human hepatocytes were seeded in a 96-well plate as described above. Cells were exposed for 1 h with different inducers as stated in the figure legend. ATP concentration was estimated by the luciferase activity using the ATP bioluminescence assay kit (Roche Applied Bioscience, Rothkreuz, Switzerland). Results are expressed as percentage of control cells not exposed to any drugs.
Immunocytochemistry. Hepatocytes were cultured on glass coverslips coated with rat-tail collagen (25 μg/cm2). Mouse hepatocytes were infected 12 h after seeding in a serum-free medium with Ad-hCAR-GFP at an MOI of 30 to 100. Twelve hours after infection, cells were exposed to chemicals for 6 h then washed twice at room temperature with PBS and fixed for 20 min in 4% (w/v) paraformaldehyde. Cells were visualized in Mowiol mounting medium with a 40× objective (1.40 numerical apertures) by using a Leica TCS NT confocal laser scanning microscope (Leica, Wetzlar, Germany).
Ethical Issues. This work was carried out in accordance with the Declaration of Helsinki and/or with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. Primary human hepatocytes used in this study were from patients undergoing liver surgery who gave consent in accordance with standard procedures approved by an institutional review board, (approval 1.05.01.30.-17).
Results
Induction of CYP2B6 and CYP3A4 by Phenobarbital, Rifampicin, and CITCO in Cultured Primary Human Hepatocytes. Human hepatocytes in primary culture are considered the “gold standard” in vitro model to study drug induction of cytochrome P450 gene expression. Our system of primary human hepatocyte culture was therefore first tested for inducibility of CYP2B6 and CYP3A4 mRNA expression by three typical inducers [i.e., phenobarbital (PB), rifampicin (Rif) and CITCO (6-(4-chlorophenyl)imidazol[2,1-6][1,3]thiazole-5-carbaldehyde)]. Exposure of cells for 16 h to PB (500 μM) and Rif (10 μM) led to an increase of CYP2B6 mRNA levels by 9.97 ± 2.44-fold (PB) and 13.26 ± 1.35-fold (Rif), respectively (Fig. 1A), and of CYP3A4 mRNA levels by 12.18 ± 2.48-fold (PB) and 25.30 ± 3.37-fold (Rif) (Fig. 1B). CITCO at 50 and 100 nM induced CYP2B6 expression by 2.9 ± 0.5-fold and 3.3 + 0.43-fold and CYP3A4 mRNA expression by 3.5 ± 0.38-fold and 3.6 ± 0.41-fold, respectively. These results validate our model of primary human hepatocytes culture to study the regulation of CYP2B6 and CYP3A4. Our previous study in a hepatoma-derived cell line showed induction of CYP2B6 expression in response to AICAR (Rencurel et al., 2005). In the present study, using primary human hepatocytes, the two AMPK activators AICAR and metformin (Sabina et al., 1985; Hawley et al., 2002) both induced CYP2B6 and CYP3A4 expression in a dose-dependent manner (Fig. 2, A-C). This indicates that classic AMPK activators increase expression of drug metabolizing enzymes such as CYP2B6 and CYP3A4. In addition, induction of CYP2B6 and CYP3A4 by PB and AICAR is additive suggesting involvement of different mechanism of induction (Fig. 2B-D).
Phenobarbital Activates AMPK in Human Hepatocytes. We then tested whether drug inducers such as PB, CITCO, and rifampicin were able to change the AMPK activity in primary human hepatocytes. Activity was assessed in cells exposed for 1 h to PB, AICAR, CITCO, Rif, and the mouse CAR ligand TCPOBOP (Fig. 3A). As expected, a strong increase of AMPK activity was observed with AICAR, and, most importantly, PB strongly activated AMPK (Fig. 3A) in a concentration dependent manner, the highest activity being reached at 1 mM PB (Fig. 3B). PB was as potent as AICAR. It is noteworthy that no changes in AMPK activity were observed with CITCO (50 and 100 nM), Rif (10 μM), and TCPOBOP (250 nM) (Fig. 3A). Western blot analysis of crude hepatocyte lysates revealed no changes in expression of both AMPK α1 and α2 catalytic subunits after treatment with PB or AICAR (Fig. 3D). To confirm AMPK activation by PB, phosphorylation of acetyl-CoA carboxylase at serine 79 was visualized by Western blot using a phospho-Ser79-specific antibody. An increase in the ACC phosphorylation because of AMPK activation is clearly shown (Fig. 3D). Finally, we investigated whether PB activation of AMPK is associated with a decrease in ATP concentration. A significant decrease by 40% of ATP levels was determined in cells after 1-h exposure to 0.5 mM PB (Fig. 3C). We conclude that activation of AMPK by PB may be mediated by a decrease in ATP concentration. We are presently exploring the mechanism by which PB and PB-like inducers may cause this decrease in ATP.
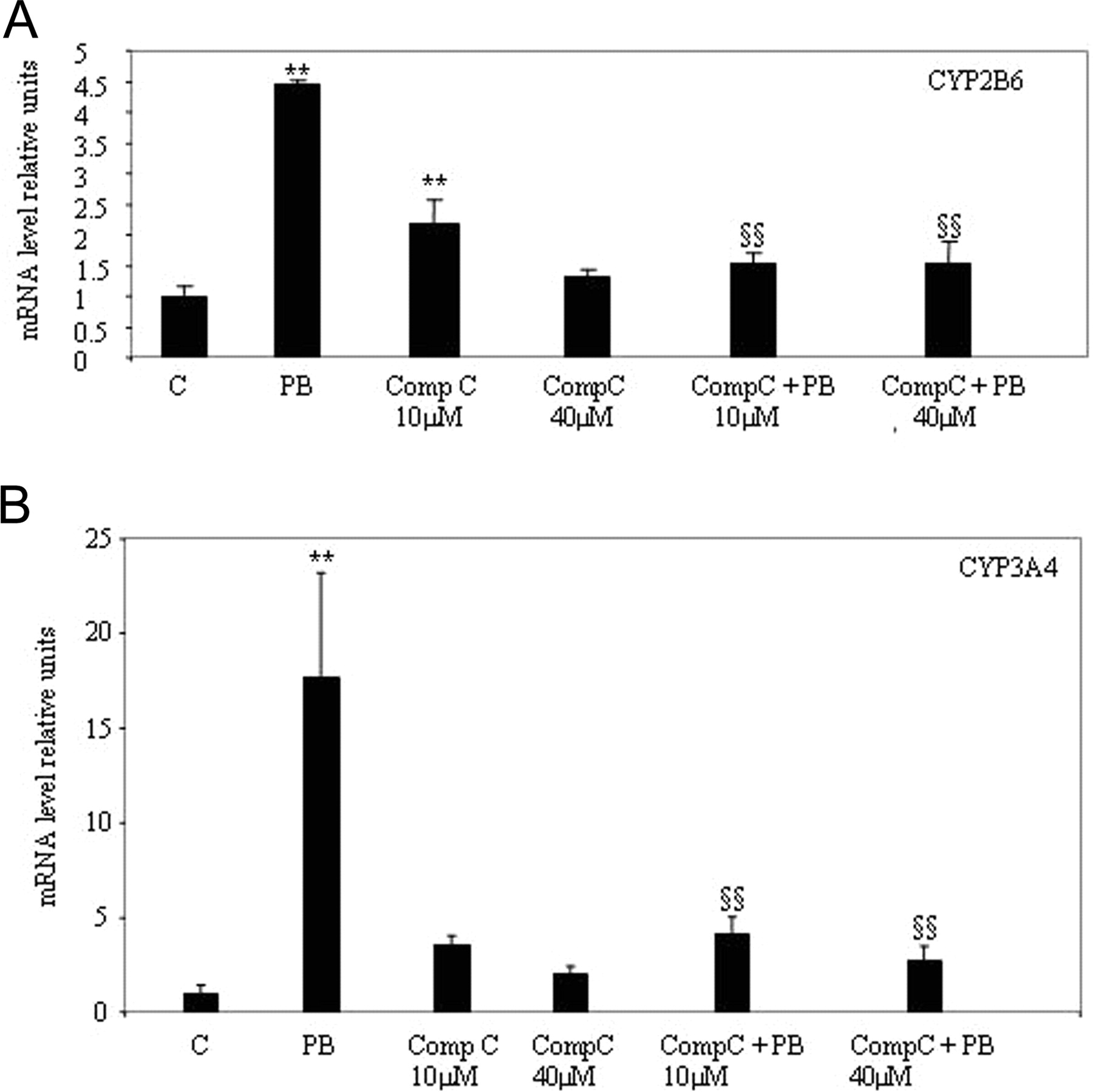
Pharmacological Inhibition of AMPK Activity by Compound C Lowers PB Induction. To further investigate the functional effects of AMPK activation, we examined the effect of Compound C, a selective AMPK inhibitor, in primary human hepatocytes (Zhou et al., 2001). Compound C (10 and 40 μM) was added to the culture medium 30 min before addition of PB. Both concentrations of compound C tested were able to blunt PB induction of CYP2B6 expression, whereas compound C alone at a concentration of 10 μM induced CYP2B6 (Fig. 4A). This inhibitory effect of Compound C was also observed on PB induction of CYP3A4 expression (Fig. 4B). We thus conclude that inhibition of AMPK activity markedly reduces PB induction.
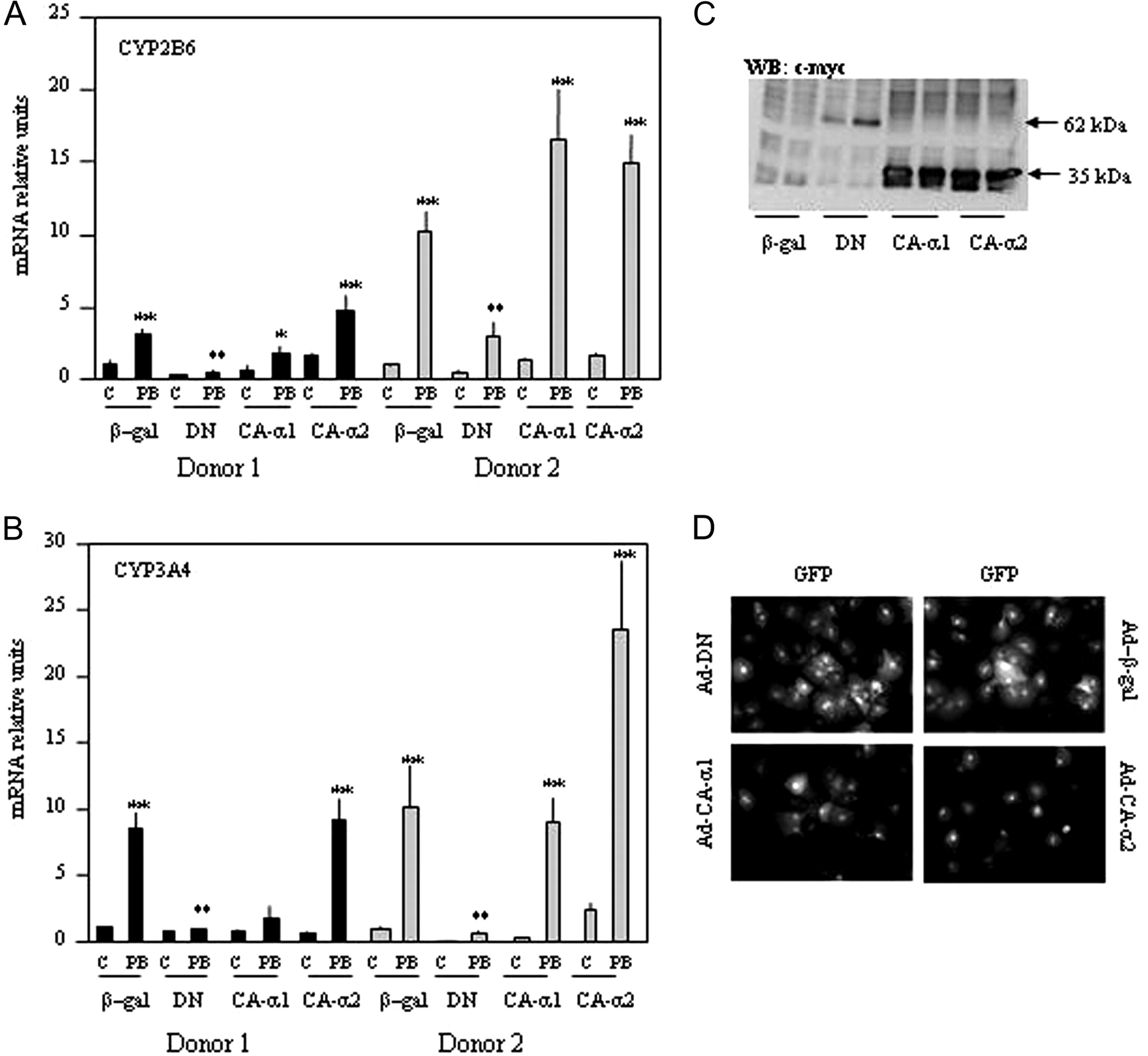
Dominant-Negative Forms of AMP-Kinase Blunt the PB Response. It was previously shown that a truncation of AMPK at residue 312 yields a peptide that no longer associates with the β and γ subunits but retains significant kinase activity (Crute et al., 1998). Moreover, replacement of threonine 172 within the α subunit by an aspartic acid mimics the effect of phosphorylation at this site (Stein et al., 2000). The AMPK mutants used in the present study provide a constitutively active form of AMPK. By contrast, mutation of aspartate 157, an essential residue for MgATP binding within the α1 subunit to alanine, yields an inactive kinase but does not have any effect on the binding to the β and γ subunits (Stein et al., 2000). Inactive α1 subunit (DNα1) acts as a dominant-negative inhibitor by competing with the native α subunits for binding with β and γ, which is essential for AMPK activity. In primary rat hepatocytes, expression of the inactive α1 subunit was able to inhibit both α1- and α2-containing complexes to a similar extent (Woods et al., 2000). Adenoviral infection of primary human hepatocytes caused more than 50% to express the AMPK mutant constructs as visualized by enhanced green fluorescence protein (Fig. 5D).
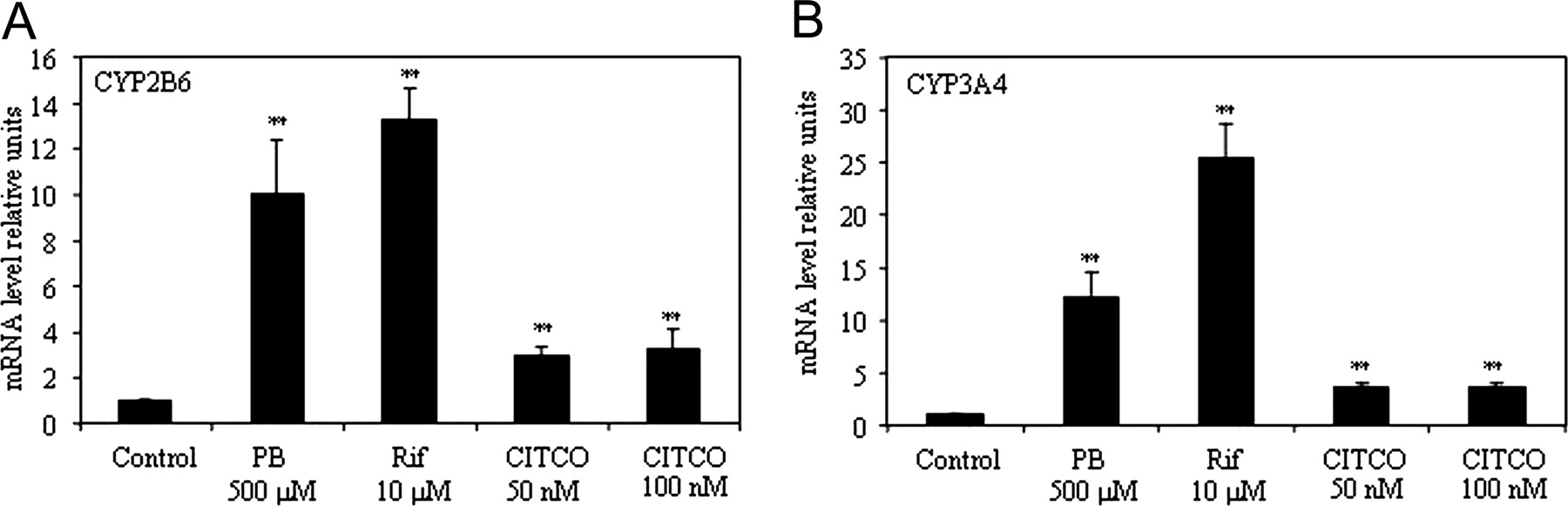
Regulation of CYP2B6 and CYP3A4 in primary human hepatocyte cultures. Human hepatocytes were exposed for 16 h to PB, Rif, or CITCO with the concentrations indicated, and CYP2B6 (A) and CYP3A4 (B) mRNAs were quantified by real-time PCR as described in Materials and Methods. Data are expressed as relative units compared with control cells not exposed to any drugs and corrected to 18s rRNA. Results are means of three different donors ± S.D., with each determination performed in triplicate. (**, p < 0.01).
Western (immuno)blot analysis confirmed the expression of ad-CA-α1, ad-CA-α2, and ad-DNα1 with anti-c-Myc antibody, which recognizes the Myc epitope tag contained in the three mutants (Fig. 5C). An adenovirus expressing β-galactosidase (ad-βgal) was used as control. Twelve hours after infection, cells were induced with 0.5 mM PB for 16 h and mRNA coding for CYP2B6 quantified by real time RT-PCR. Figure 5, A and B, shows PB induction of CYP2B6 and CYP3A4 expression in ad-βgal infected hepatocytes confirming that adenoviruses do not alter the PB response.

AICAR and metformin induce CYP2B6 and CYP3A4 in primary human hepatocyte cultures. Primary human hepatocytes were exposed for 16 h to different concentrations of metformin (Met) and AICAR as indicated, and CYP2B6 (A) and CYP3A4 (C) mRNA were quantified by RT-PCR as described under Materials and Methods. Hepatocytes were exposed for 16 h to 500 μM PB alone or in combination with different concentration of AICAR. CYP2B6 (B) and CYP3A4 (D) mRNA were quantified by real-time PCR. Results are means of three different donors ± S.D. with each determination done in triplicates. (**, p < 0.01). Values obtained with combinations of AICAR and PB treatment are compared with values obtained from cells treated with PB alone (§, p < 0.05; §§, p < 0.01)

Phenobarbital activates AMPK in primary human hepatocyte cultures and lowers ATP. Primary human hepatocytes in culture were exposed for 1 h to different concentrations of PB (A and B), AICAR (A and B), Rif (B), CITCO (B), and TCPOBOP (B), as indicated. AMP kinase activity was measured as described under Materials and Methods, and results are expressed as -fold stimulation compared with values measured in control cells (Control) not exposed to any drugs. Results are means ± S.D. of three to four different human hepatocyte cultures, and each measurement was performed in triplicate. C, ATP concentration was measured in primary human hepatocytes by luciferase activity as described under Materials and Methods after 1-h exposure to PB. Results are expressed as -fold stimulation compared with values obtained in nontreated cells (Control). **, p < 0.01. D, human hepatocytes were exposed for 1 h to PB (0.5 mM) or AICAR (2 mM), and expression of AMPK α1, AMPK α2, and phosphorylated acetyl-CoA-carboxylase (p-ACC) was estimated by Western blot using specific polyclonal antibodies.
The dominant-negative mutant of α1 AMPK (DN) was able not only to lower the basal expression of CYP2B6 and CYP3A4 genes but also to completely block the PB induction of these two genes. It is noteworthy that the constitutively active mutant, CA-α2, did not significantly change basal levels of the corresponding mRNAs but tended to potentiate the effects of PB effect on CYP2B6 in both donors. More results that are heterogeneous appear with the CA-α1 where no variations in PB induction of CYP2B6 were observed, and even a lower PB induction of CYP2B6 and CYP3A4 was present in donor 1. These results strongly suggest that AMPK activation is required for PB induction of CYP2B6 and CYP3A4. The specific role played by each AMPK catalytic subunit in such an effect is under further investigation in our laboratory.

Pharmacological inhibition of AMPK activity with compound C lowers phenobarbital induction of CYP2B6 and CYP3A4 in human hepatocytes. Hepatocytes were exposed for 16 h to 500 μM PB or compound C (10-40 μM). Compound C was added to cells 30 min before addition of PB when both chemicals were used in combination (Comp C+PB). CYP2B6 (A) and CYP3A4 (B) mRNA were quantified by real-time PCR and are expressed as relative units corrected to 18s rRNA. Results represent cultures from two different donors (mean ± S.D.) with each determination done in triplicate. Comparison with values in control cells (C) not exposed to drugs (**, p < 0.01). Comparison with values in cells exposed to PB (§§, p < 0.01)
Induction of Cyp2b10 and Cyp3a11 Expression by Phenobarbital Depends on AMPK Expression in Mouse Liver and Mouse Hepatocytes. To test further the importance of in vivo AMPK in drug induction, we tested induction of Cyp2b10 and Cyp3a11 by PB and TCPOBOP in AMPK α1/α2 liver-specific knockout mice (α1/α2LS-/-). As expected, i.p. injection of PB (100 mg/kg body weight) and TCPOBOP (3 mg/kg body weight) in wild-type C57BL/6 mice massively induced Cyp2b10 and Cyp3a11 expression in the liver (Fig. 6B). To our surprise, a strong increase of Cyp2b10 (100-fold) and Cyp3a11 (6-fold) expression occurred in untreated α1/α2LS-/- mice. The PB and TCPOBOP induction of Cyp2b10 and Cyp3a11 was decreased from 155- to 1.54-fold for Cyp2b10 and from 13.6- to 2.42-fold for Cyp3a11 in α1/α2LS-/- knockout mice. To address the question of whether the strong up-regulation of Cyp2b10 and Cyp3A11 observed in vivo in untreated animals is a direct or indirect consequence of the ablation of AMPK catalytic subunits, we chose to perform primary culture of hepatocytes from wild-type and α1/α2LS-/- knockout mice. Primary mouse hepatocytes from wt animals exhibit an induction of Cyp2b10 after exposure to PB (500 μM), TCPOBOP (250 nM), metformin (1 mM), or AICAR (500 μM) (Fig. 6A). Lack of expression of AMPK catalytic subunits α1 and α2 in primary mouse hepatocytes, resulted in the abolition of Cyp2b10 induction by either PB, TCPOBOP (TCP), metformin (Metf), or AICAR (Fig. 6A). It is noteworthy that contrary to the in vivo situation, the basal expression of Cyp2b10 and Cyp3a11 was not higher in hepatocytes from α1/α2LS-/- mice compared with hepatocytes from wt mice. This implies a role of circulating factors responsible of high basal Cyp2b10 and Cyp3a11 gene expression observed in vivo. Moreover, these results provide further evidence for the important role of AMPK in Cyp2b10 and Cyp3a11 induction by PB AICAR and TCPOBOP.
The Absence of AMPK Catalytic Subunits Has No Effect on CAR Cytoplasmic-Nuclear Transfer Induced by PB. The capacity of PB to trigger CAR cytoplasmic/nuclear transfer was tested in primary mouse hepatocytes expressing the human CAR in fusion with enhanced GFP. After 6-h exposure to PB, CAR was clearly located predominantly in the nuclei of hepatocytes from wild-type mice. An apparent staining close to the plasma membrane and/or cytoskeleton network was also observed in untreated (Control) and PB-treated hepatocytes from both wild-type and α1/α2LS-/- mice (Fig. 7). Unexpectedly, the cytoplasmic/nuclear shuttling of CAR upon PB treatment was not altered by the absence of AMPK α1 and α2 catalytic subunits. However, we noted that CAR-GFP fluorescence was located in condensed region of the nucleus in α1/α2LS-/- mice hepatocytes after PB treatment, a phenomenon not observed in hepatocytes from wild-type mice. It is therefore unlikely that the deficiency in the induction of Cyp2b10 and Cyp3a11 by PB observed in α1/α2LS-/- mouse hepatocytes is caused by an alteration in CAR translocation into the nucleus.
Discussion
Despite numerous attempts, the molecular mechanism by which PB exerts its effects on gene expression are still incompletely understood, and no intracellular protein target of PB has been identified. We propose here a new mechanism of drug induction, activation of AMPK, which may ultimately explain some of the diverse effects of PB in human and mouse hepatocytes such as its effect on the transcription of cytochrome P450 genes. We have reported previously that pharmacological activation of AMPK leads to induction of CYP2B6 in a hepatoma-derived cell line (WGA) (Rencurel et al., 2005). However, concentrations of PB above 1 mM were required for CYP2B6 induction and AMPK activation in WGA cells; these concentrations are associated with toxic effects in normal hepatocytes. Thus, the importance of this mechanism in normal hepatocytes was questioned. We therefore chose two experimental systems, primary culture of human hepatocytes and cultures of hepatocytes from AMPK α1/α2 liver-specific KO mice (α1/α2LS-/-) to investigate the role of AMPK in drug induction. We validated our human hepatocyte culture system by testing classic inducers on CYP2B6 and CYP3A4 gene expression and observed robust and reproducible, dose-dependent induction of CYP2B6 and CYP3A4 when cells were exposed to PB and rifampicin for 16 h. It is noteworthy that, compared with PB, CITCO, a human CAR agonist (Maglich et al., 2003), was not a potent CYP2B6 inducer. This could be due to the shorter time of exposure than those described by Maglich et al. (2003) and to its partially different mode of action (i.e., direct activation of CAR) (Maglich et al., 2003).
The two AMPK activators, AICAR and metformin, induced CYP2B6 and CYP3A4 expression in a dose-dependent manner in primary human hepatocytes, a hallmark of drug induction. An additive effect of PB and AICAR on CYP2B6 expression was observed, suggesting the involvement of distinct mechanism of action of these two drugs. AICAR is the most commonly used method of activating AMPK; recently, however, Guigas et al. (2006) highlighted an AICAR effect independent of AMPK activation on glucose phosphorylation. To clearly distinguish AMPK-dependent and AMPK-independent effects on cytochrome P450 induction, we turned to primary hepatocytes from α1/α2LS-/- mice. In these hepatocytes, no induction of Cyp2b10 was observed with PB, TCPOBOP, metformin, or AICAR, demonstrating the essential role of AMPK in the induction of CYP2b10. However, minor induction of Cyp3a11 by AICAR was still present in hepatocytes from α1/α2LS-/- mice, perhaps reflecting the existence of an AMPK-independent mechanism by which AICAR regulates this gene, possibly acting as a ligand of pregnane X receptor.
The activation of AMPK by phenobarbital is unique in the sense that other drug inducers tested so far, such as the CAR ligand/activator CITCO, the mouse CAR ligand/activator TCPOBOP, and the pregnane X receptor ligand/activator rifampicin did not change AMPK activity in human hepatocytes. This suggests that PB and PB-like inducers affect transcription of cytochrome P450 genes by a unique mechanism.

Exogenous expression of dominant-negative and constitutively active mutants of AMPK in primary human hepatocytes affects phenobarbital induction. Primary human hepatocytes were infected with adenovirus containing β-galactosidase (β-gal), AMPK α1 dominant-negative mutant (DN), AMPK constitutively active α1or α2 subunits (CA-α1; CA-α2). Twenty-four hours after infection, cells were exposed for 16 h to 0.5 mM PB. Expression of CYP2B6 (A) and CYP3A4 (B) was quantified by real-time PCR, and results were expressed as -fold stimulation compared with cells infected with ad-βgal (β-gal) and not exposed to drug (C = controls). Results are expressed as relative units corrected to 18s rRNA comparison with values in control cells (C) not exposed to drugs and infected with ad-βgal (β-gal) (*, p < 0.05; **, p < 0.01). C, gene expression in primary human hepatocytes was tested by Western blot. Twenty-four hours after infection, cells were lysed, and 30 μg of total cell lysate was separated in 10% acrylamide gel. The Myc tag of AMPK mutants was detected using a specific monoclonal antibody. The 62-kDa band corresponds to the full-length α1 dominant-negative mutant (DN) and the 35-kDa band to the truncated constitutive active α1 and α2 mutants (CA-α1 and CA-α2) as described under Results. D, the efficiency of infection was estimated by immunocytochemistry in primary human hepatocytes 24 h after infection. Each adenoviral construct expressed GFP under the control of a cytomegalovirus promoter as described under Materials and Methods.
The classic view of AMPK activation is a decrease in cellular energy charge as reflected by the increase in the AMP/ATP ratio. PB is able to significantly lower ATP concentration in hepatocytes within an hour, a finding in agreement with our previous study in hepatoma cells (Rencurel et al., 2005). The lowering effect of PB on ATP in hepatoma cells was observed only at high concentrations (above 1 mM), a level also required for CYP2B6 induction in these cells (Rencurel et al., 2005). The capacity of PB to induce CYP2B6 expression may thus be related to its efficiency to lower ATP and consequently to activate AMPK. Another interesting observation comes from the CYP2B6 induction by metformin in primary human hepatocytes. The biguanide metformin (N1,N1-dimethylbiguanide) is prescribed to lower fasting blood glucose in patients with non-insulin-dependent diabetes mellitus, yet its primary site of action is still uncertain. Shaw et al. (2005) described the inhibitory effect of metformin on hepatic glucose production to be dependent on the expression of LKB1 kinase. Experiments in LKB1-/- mice have indeed established that LKB1 is the principal AMPK kinase in the liver (Shaw et al., 2005). Although the molecular mechanisms through which metformin affects cellular energy homeostasis remain disputed, it seems increasingly likely that metformin may act, at least in large part, by inhibiting respiratory chain complex 1 (Owen et al., 2000), hence suppressing mitochondrial ATP synthesis. It is not yet known whether PB activates AMPK via a similar mechanism in the liver, but if so, this could explain the known blood glucose-lowering effect of PB in patients with non-insulindependent diabetes mellitus (Sotaniemi and Karvonen, 1989).
The most compelling evidence for the role of AMPK in PB-type induction is reflected in our experiments, which demonstrate that liver-specific deletion of the AMPKα subunit genes in the mouse abolishes the drug induction of CYP2b10 and CYP3a11. It is surprising that markedly higher basal levels of the mRNA of these genes were observed. By contrast, no increase of Cyp2b10 and Cyp3a11 basal expression was observed in primary hepatocytes cultured from the livers of α1/α2LS-/- mice. Such a discrepancy between in vivo and in vitro findings suggests that circulating factors in response to the metabolic changes in the liver might have caused the increased basal expression of Cyp2b10. The streptozotocin-induced diabetic mouse model exhibits a high Cyp2b10 basal expression level that was corrected by insulin treatment to lower hyperglycemia (Sakuma et al., 2001). Guigas et al. (2006) observed a low glucokinase expression in hepatocytes from α1/α2LS-/- mouse, which is associated with low glucose phosphorylation and low glucose uptake. A low rate of glucose phosphorylation was described earlier in primary rat hepatocytes from alloxaninduced diabetic rats (Bontemps et al., 1978) and in fasted animals, where Cyp2b10 and CAR expression is induced (Maglich et al., 2004). We therefore speculate that a circulating factor related to the low carbohydrate turnover in hepatocytes from α1/α2LS-/- could be responsible for the high basal expression of Cyp2b10, and we are currently testing this hypothesis.

Liver-specific deletion of AMPK α1 and α2 subunits in mouse impairs induction of Cyp2b10 and Cyp3a11 in vivo and in vitro. Primary hepatocytes from wild type (wt, black bars) mice or from mice with liver-specific deficiencies of AMPK catalytic subunit α1 and α2(α1/α2LS-/-, gray bars) were exposed for 16 h to 500 μM PB, 500 μM TCPOBOP (TCP), 1 mM metformin (Metf) or 500 μM AICAR. Expression of Cyp2b10 (A, top) and Cyp3a11 (A, bottom) was quantified by real-time PCR, and results were expressed as -fold stimulation compared with nondrug-exposed cells (C = controls) from wild-type mice (wt). Results represent two different cell preparations. (**, p < 0.01 unpaired Student's t test). Mice in which AMPK α1 and α2 were deleted in liver (α1/α2LS-/-, gray bars) and wild-type mice (wt, black bars) were injected i.p. with saline solution (C = controls), PB or TCPOBOP (TCP) and sacrificed 16 h later as described under Materials and Methods Cyp2b10 (B, top) and Cyp3a11 (B, bottom) expression was quantified by RT-PCR and standardized to GAPDH. Results are expressed as -fold stimulation compared with values obtained in saline-injected animals (C = controls) of each group of Wt and α1/α2LS-/- mice. The inset results are expressed as -fold induction compared with values obtained in saline-injected (C) wild-type animals (wt). Results are expressed as means ± S.D., *, p < 0.05; **, p < 0.01 determined by unpaired Student's t test.
The blunted induction of Cyp2b10 and CYP3a11 by PB in hepatocyte from α1/α2LS-/- mice was not related to an alteration of CAR nuclear translocation upon drug treatment. It is well documented that CAR translocates into the nucleus upon drug treatment to form an active transcriptional complex with the retinoid X receptor (Zelko et al., 2001). Moreover, the translocation and activation of CAR is influenced by phosphorylation/dephosphorylation events (Kawamoto et al., 1999; Yoshinari et al., 2003). For instance, the protein phosphatase 2A inhibitor okadaic acid blocks CAR translocation in mouse hepatocytes exposed to either PB or TCPOBOP (Kawamoto et al., 1999). In addition, okadaic acid induces expression of CYP2B6 in HepG2 cells that were engineered to express the mouse CAR isoform. Again in HepG2 cells, residue serine-202 of mouse CAR was shown to be phosphorylated, and this modification was shown to be important for the retention of CAR in the cytoplasm (Hosseinpour et al., 2005). On the other hand, despite this preliminary evidence that the serine-202 dephosphorylation of mCAR by protein phosphatase 2A affects the cytoplasmic-nuclear transfer of CAR, the identity of the kinase responsible of serine-202 phosphorylation is unknown (Hosseinpour et al., 2005). Our own experiments suggest that AMPK does not phosphorylate serine-202 of mouse CAR or the corresponding serine-192 of human CAR (M. Matis and F. Rencurel, unpublished observation). In hepatocytes from α1/α2LS-/- mice, hCAR-GFP was located in condensed regions of the nucleus, whereas a more homogenous staining was observed in nucleus of wild-type mice hepatocytes after PB treatment. These condensed regions are comparable with nuclear speckles described previously in COS-7 cells in which CAR and the cofactor peroxisome proliferator-activated receptor γ coactivator-1α (PGC1α) were coexpressed {Shiraki, 2003 #117}. Nuclear speckles contain proteins involved in pre-mRNA splicing but also several kinases (e.g., CLK/STI, PRP4) and phosphatases (e.g., protein phosphatase 1) which can phosphorylate/dephosphorylate components of the splicing machinery (Handwerger and Gall, 2006). Although little or no transcription takes place in nuclear speckles, a set of proteins involved in transcriptional regulation is associated with speckles from where they are shuttled to transcription sites. PB induction of Cyp2b10 was not altered in PGC1α liver-specific knockout mice (Handschin et al., 2005). If and how PGC1α interacts with CAR thus remains an open question. A sequestration of CAR in or near nuclear speckles may control its activity by keeping the nuclear receptor removed from or near the transcription sites. Our data therefore suggest the existence of another control step of CAR signaling independent of CAR intracellular location.

Human CAR cytoplasmic/nuclear transfer induced by phenobarbital is not altered by the deletion of AMPK α1 and α2 catalytic subunits in primary mouse hepatocytes. Primary hepatocytes from wild-type (Wt) mice or from mice in which the AMPK catalytic subunits α1 and α2 were deleted in the liver (α1/α LS-/-2) were infected with adenovirus coding for human CAR in fusion with enhanced green fluorescence protein (ad-hCAR-GFP) as described under Materials and Methods Twelve hours after infection, cells were exposed to PB for 6 h; control mice were not exposed to any drugs. Human CAR-GFP was visualized in cells in Mowiol mounting medium with a 40× objective (1.32 numerical aperture) by using a Leica TCS NT confocal laser scanning microscope. White scale bar, 10 μm.
In conclusion, we demonstrate an essential role of AMPK in induction of CYP2B6 and CYP3A4 by PB in human hepatocytes and of CYP2b10 and CYP3a11 in mouse hepatocytes. This highlights yet another interesting effect of this anticonvulsant and hypnotic drug that also has antidiabetic properties. The role of AMPK in drug induction obviously needs further investigation, and its role in the control of CAR activity is of particular interest. Because AMP-activated kinase is a target for the development of drugs for type II diabetes and obesity, its role in the induction of drug metabolism deserves close attention.
Acknowledgments
We thank Dr. Houchaima Ben Tekaya for help with immunocytochemistry.
Footnotes
-
This work was supported by the Swiss National Science Foundation (to F.R.) and by programme grants from the Wellcome Trust (to G.A.R.) and the Medical Research Council, a Postdoctoral Fellowship from the Juvenile Diabetes Research Fund International (to G.d.S.X.), and a Wellcome Trust Advanced Fellowship (to I.L.), a Wellcome Trust Research Leave Fellowships (to G.A.R.). M.F. was supported by a postdoctoral fellowship from the European Commission (grant LSHM-CT-2004-005272/exgenesis). B.V. and F.R. were supported by an exchange program from EGIDE association (PAI Germaine de Staël).
-
ABBREVIATIONS: PB, phenobarbital; CAR, constitutive active/androstane receptor; CCRP, cytoplasmic CAR retaining protein; AMPK, AMP-activated protein kinase; AICAR, 5′-phosphoribosyl-5-aminoimidazol-4-carboxamide 1-β-d-ribofuranoside; PCR, polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PBS, phosphate-buffered saline; GFP, green fluorescent protein; CITCO, 6-(4-chlorophenyl)imidazol[2,1-6][1,3]thiazole-5-carbaldehyde; Rif, rifampicin; RT, reverse transcription; PCR, polymerase chain reaction; TCPOBOP,1,4 bis[2-(3,5-dichloropyridyloxy)]benzene; PGC1α, peroxisome proliferator-activated receptor γ coactivator-1α.
- Received July 31, 2006.
- Revision received September 19, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}