


Abstract
The binding and function of muscarinic acetylcholine receptors can be modulated allosterically. Some allosteric muscarinic ligands are “atypical”, having steep concentration-effect curves and not interacting competitively with “typical” allosteric modulators. For atypical agents, a second allosteric site has been proposed. Different approaches have been used to gain further insight into the interaction with M2 receptors of two atypical agents, tacrine and the bispyridinium compound 4,4′-bis-[(2,6-dichloro-benzyloxy-imino)-methyl]-1,1′-propane-1,3-diyl-bispyridinium dibromide (Duo3). Interaction studies, using radioligand binding assays and the allosteric ligands obidoxime, Mg2+, and the new tool hexamethonium to antagonize the allosteric actions of the atypical ligands, showed different modes of interaction for tacrine and Duo3 at M2 receptors. A negatively cooperative interaction was observed between hexamethonium and tacrine (but not Duo3). A tacrine dimer that exhibited increased allosteric potency relative to tacrine but behaved like a typical allosteric modulator was competitively inhibited by hexamethonium. M2/M5-receptor mutants revealed a dependence of tacrine and Duo3 affinity on different receptor epitopes. This was confirmed by docking simulations using a three-dimensional model of the M2 receptor. These showed that the allosteric site could accommodate two molecules of tacrine simultaneously but only one molecule of Duo3, which binds in different mode from typical allosteric agents. Therefore, the atypical actions of tacrine and Duo3 involve different modes of receptor interaction, but their sites of attachment seem to be the “common” allosteric binding domain at the entrance to the orthosteric ligand binding pocket of the M2-receptor. Additional complex behavior may be rationalized by allosteric interactions transmitted within a receptor dimer.
A rapidly increasing number of G protein-coupled receptors have been discovered to be sensitive to allosteric modulation (Christopoulos and Kenakin, 2002). Potentially favorable features for a clinical application of such modulation include the enhancement of the binding of endogenous ligands (but also exogenous agonists and antagonists), absolute subtype selectivity of action, and self-limiting effects on receptor function (see Christopoulos and Kenakin, 2002 for review).
Over the past decade, allosteric interactions at muscarinic acetylcholine receptors have been intensively studied (e.g., Ellis, 1997; Mohr et al., 2003, Birdsall and Lazareno, 2005). All five muscarinic receptor subtypes are sensitive to allosteric modulation (Ellis et al., 1991), but many allosteric modulators have their highest affinity for the M2 subtype (e.g., Ellis et al., 1991; Buller et al., 2002; Jakubík et al., 2005), a fact that has been used to gain insight into the binding topology of allosteric ligands. The effects of many of these (“typical”) allosteric agents have been shown to be mediated via a “common allosteric site” of the M2 receptor (Ellis and Seidenberg, 1992; Lanzafame et al., 1997; Tränkle and Mohr, 1997; Ellis and Seidenberg, 2000; Tränkle et al., 2003). Site-directed mutagenesis has revealed, for some “common site” allosteric agents, that the amino acids M2 177Tyr and M2 423Thr (Buller et al., 2002; Voigtländer et al., 2003) fully account for the M2/M5 selectivity of the ligands. According to a three-dimensional model of the M2 receptor, the allosteric site is located at the entrance of the ligand binding pocket in a cleft-like vestibule that is linked by a narrow corridor with the orthosteric, acetylcholine binding site (Voigtländer et al., 2003).

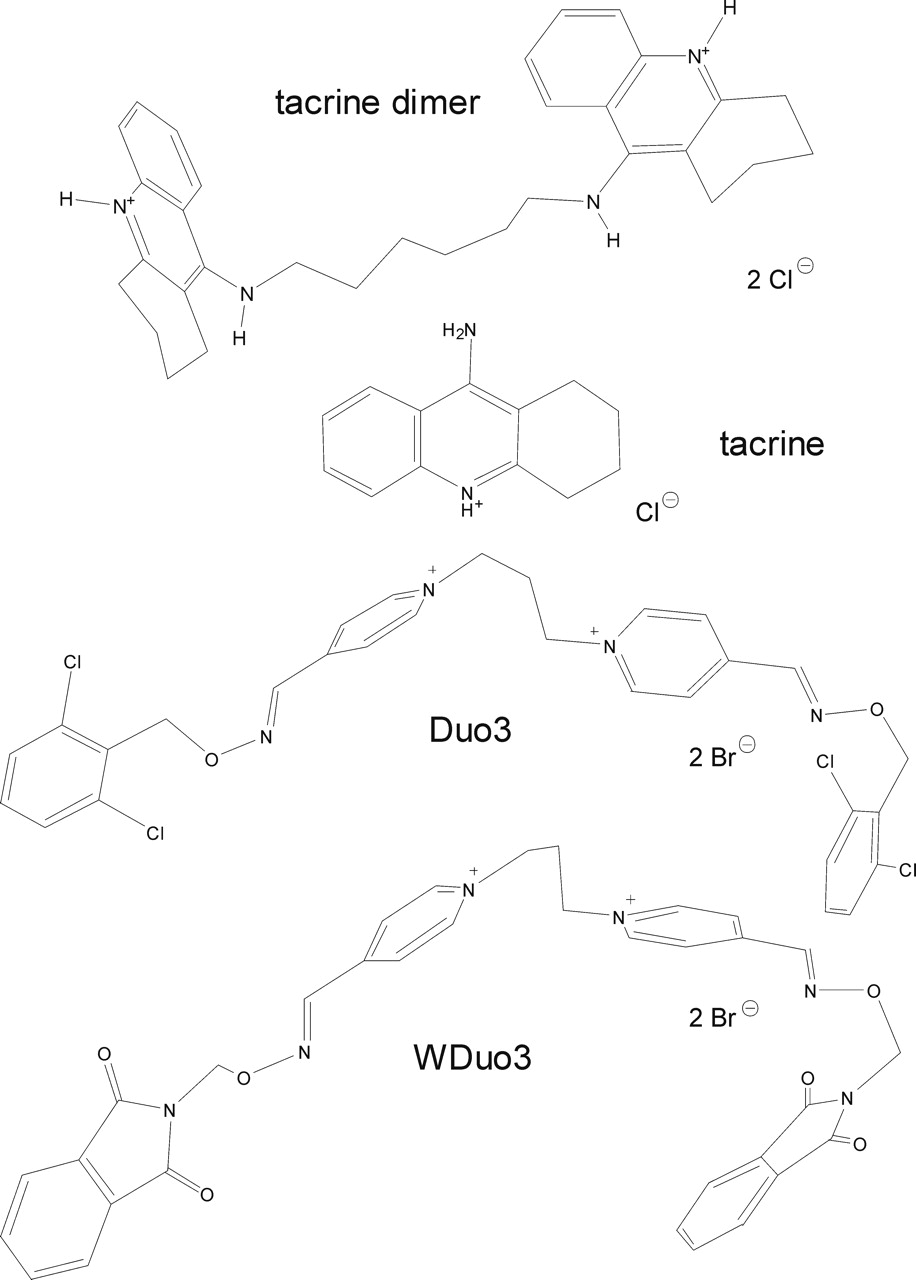
Structures of the allosteric ligands. The molecules are shown in an energetically favorable conformation.
There is another group of muscarinic allosteric agents termed “atypical” allosteric modulators. Tacrine (Flynn and Mash, 1989; Potter et al., 1989) and Duo3 (Fig. 1; Tränkle and Mohr, 1997) represent archetypal members of this group (Fig. 1). These agents have concentration-effect curves with slope factors greater than 1 (Potter et al., 1989; Tränkle and Mohr, 1997). Furthermore, the competitive ligand at the common allosteric site, obidoxime (Ellis and Seidenberg, 1992), inhibits the actions of Duo3 only weakly and in a noncompetitive fashion (Tränkle and Mohr, 1997). In addition, the affinity of Duo3 is much less sensitive to the buffer composition compared with typical agents that are also biscationic but competitive with obidoxime (Tränkle et al., 1996; Schröter et al., 2000). Based on these data, it was suggested that another allosteric site might be involved in the action of Duo3 (Tränkle and Mohr, 1997) as has also been suggested for derivatives of staurosporine and WIN 62,577 (Lazareno et al., 2000, 2002).
Tacrine hydrochloride is a comparatively small monocationic compound (Fig. 1) whose binding is known to be sensitive to obidoxime (Ellis and Seidenberg, 1992), but the mechanism of its antagonism with muscarinic receptors has not yet been elucidated. Studies on M2/M5-chimeric receptors (Ellis and Seidenberg, 2000) are compatible with the idea that 177Tyr and 423Thr of the M2 receptor are involved in the binding of tacrine, but this has not been directly investigated. The epitope dependence of Duo3 binding has not been studied.
Our study is aimed at gaining further insight into the molecular events and interactions underlying the allosteric actions of the atypical agents, Duo3 and tacrine, in comparison with the typical allosteric agent WDuo3 (Fig. 1). Three structurally different allosteric ligands, obidoxime, hexamethonium (Eglen et al., 1989), and Mg2+ (Burgmer et al., 1998), have been used to probe for differences in their interactions with WDuo3, Duo3, and tacrine. Obidoxime and hexamethonium resemble the central chains of Duo3 and WDuo3 (Fig. 1) and of alkane-bisammonio-type allosteric agents (Mohr et al., 2003), respectively. Interactions with a newly synthesized tacrine dimer (Fig. 1) were also investigated. Point-mutated M2 receptors and chimeric M2/M5-receptors have been used to explore the different binding modes of the chemically related typical and atypical allosteric ligands. Docking simulations using a model of the M2 receptor (Jöhren and Höltje, 2002; Voigtländer et al., 2003) suggested that the allosteric site can easily accommodate two molecules of tacrine simultaneously. Duo3 also fits into the cleft-like allosteric site but does not come as close as typical allosteric agents to the entrance of the orthosteric site.
Our findings suggest that some atypical allosteric agents may interact with the common allosteric domain of the M2 receptor protein but in an atypical fashion.
Materials and Methods
Materials
[3H]N-methylscopolamine ([3H]NMS; specific activity, 70–82 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). The synthesis of the radiolabeled compound [3H]dimethyl-W84 (specific activity, 154–168 Ci/mmol) was carried out by GE Healthcare (Little Chalfont, Buckinghamshire, UK) using the method described by Tränkle et al., 1998. Atropine sulfate and (-)-scopolamine methylbromide were obtained from Sigma Chemicals (München, Germany). Obidoxime dichloride was generously provided by Merck KG (Darmstadt, Germany). The allosteric modulators dimethyl-W84, Duo3, and WDuo3 were synthesized as described elsewhere (Mohr et al., 2003). Hexamethylene-linked bis-tacrine: tacrine hydrochloride (5.0 mM; 1.17 g) was suspended in 100 ml of dry tetrahydrofuran and cooled to -70°C (ethanol dry ice). n-Butyl lithium (30 mM) was added and stirred for 15 min at -70°C. Dibromohexane (2.5 mM, 0.61 g) was then added and the solution was stirred for 2 h at -70°C. The temperature was then allowed to rise to -30°C. Water (10 ml) was added, and the solution was stirred for a further 30 min at 20°C. The solvent was evaporated and the residue was diluted with 50 ml of water. The solid was filtered off and dried. The crude product was refluxed for 30 min in 20 ml of ethyl alcohol and filtered while hot. The filtrate was crystallized at + 4°C to yield 0.72 g (60%) of a colorless solid. Mp 225°C (dec). 1H NMR- and mass spectroscopy data are in accord with that of Hu et al. (2002).
W84 is commercially available from AXXORA Deutschland (Grünberg, Germany). Ham's F-12 medium, Dulbecco's modified Eagle's medium, fetal calf serum, penicillin G, streptomycin, glutamine, G418, trypsin-EDTA-solution, and HEPES were purchased from Sigma-Aldrich Chemie (Steinheim, Germany). Sodium butyrate was from Acros Organics (Geel, Belgium).
Receptor Mutagenesis and Expression
The human M2/M5 chimeric receptors used in this study have been described previously (Wess et al., 1992, Ellis and Seidenberg, 2000); schematic sketches of the chimeric receptors are shown in Fig. 6. The exact sequences are as follows: CR1: hM2, 1 to 69; hM5, 77 to 532; CR2: hM5, 1 to 76; hM2, 70 to 155; hM5, 163 to 532; CR3: hM5, 1 to 162; hM2, 156 to 300; hM5, 336 to 532; CR6, hM2, 1 to 69; hM5, 77 to 445; hM2, 391 to 466. In addition, two M2 receptors were used in which single amino acids were mutated: M2 423Thr→His and M2 177Tyr→Gln plus 423Thr→His (Voigtländer et al., 2003). Plasmids containing the human M2 or M5 wild-type or mutated receptor genes were purified from bacterial cultures and transiently transfected into COS-7 cells by calcium phosphate precipitation. Cells were harvested 72 h after transfection by scraping into 5 mM Na/K/Pi buffer, pH 7.4.
Membrane Preparation
Porcine Cardiac Membranes. Membranes were prepared as described previously (Tränkle and Mohr, 1997). All steps were carried out at 4°C. In brief, ventricular tissue (40 g) of porcine hearts, obtained from the local slaughterhouse, was homogenized in a 0.32 M sucrose solution and centrifuged for 11 min at 300g (2000 rpm in a Beckman rotor 35; Beckman Coulter, Fullerton, CA). The supernatant was centrifuged for 40 min at 80,000g (32,000 rpm in a Beckman rotor 35), and the resulting pellet resuspended in 4 mM Na2HPO4, 1 m MKH2PO4, pH 7.4 (4 ml/g original tissue wet weight). Aliquots (0.5 ml) were shock-frozen in liquid nitrogen and stored at -80°C, and the protein content (3.9 ± 0.7 mg/ml) was measured according to Lowry et al. (1951) with human serum albumin as a standard.
COS-7 Membrane Preparations. COS-7 cells were grown in Dulbecco's modified Eagle's medium containing 100 units/ml penicillin G, 0.1 mg/ml streptomycin, 1 mM l-glutamine, and 10% fetal calf serum until confluent and harvested after addition of trypsin-EDTA by scraping in 4 ml of ice-cold Na/K/Pi-buffer (5 mM). Cells were homogenized by means of a Polytron homogenizer (PT 10-35; Kinematica AG, Basel, Switzerland) three times at “level 6” for 10 s on ice. Membranes were centrifuged (Avanti J25 centrifuge, JA 25.50 rotor; Beckman Coulter) at 40,000g (18,000 rpm, 30 min, 4°C) and the pellets were resuspended in Na/K/Pi-buffer (5 mM). Membranes were stored in 1-ml aliquots at -80°C until use.
CHO Cell Membranes. CHO cells expressing human hM2 receptors were prepared for binding studies using [3H]dimethyl-W84 as described previously (Tränkle et al., 2003). In brief, Chinese hamster ovary (CHO) cells stably transfected with the human M2-receptor gene (generously provided by Dr. N. J. Buckley, University of Leeds, Leeds, UK) were grown in a medium consisting of nutrient mixture Ham's F-12, 10% fetal calf serum, 100 units/ml penicillin G, 0.1 mg/ml streptomycin, 1 mM l-glutamine, and 0.2 mg/ml of G418 in a humidified atmosphere at 37°C and 5% CO2. Sixteen hours before cell harvesting (cell confluence approximately 80%) cells were treated with 5 mM sodium butyrate. The cells were lysed and harvested by scraping in homogenization buffer (20 mM HEPES and 10 mM Na4EDTA, pH 7.4, 4°C). Cells were homogenized with a Polytron homogenizer (twice at level 6 for 6 s). The membranes were pelleted by centrifugation at 40,000g (18,000 rpm, 10 min, 4°C) in an Avanti J25 centrifuge with a JA 25.50 rotor (Beckman, Palo Alto, CA) and washed twice in storage buffer (20 mM HEPES, 0.1 mM Na4EDTA, pH 7.4, 4°C). The final pellets were resuspended and stored as a membrane suspension in storage buffer (approximately 1.4 mg of protein/ml) at -80°C.
[3H]NMS Binding Assays
Interaction Studies on Cardiac Membranes. Porcine cardiac membranes (200–450 μg of protein/ml) were incubated with 0.2 nM [3H]NMS in 5 mM Na/K/Pi-buffer, pH 7.4, at 23°C. Nonspecific binding was determined in the presence of 1 μM atropine. Specific binding of [3H]N-methylscopolamine under control conditions was characterized by a pKD of 10.04 ± 0.16 and Bmax of 104 ± 12 fmol/mg of protein (means ± S.E.M., n = 12). To measure the kinetics of radioligand dissociation, the assays were prepared in a larger volume. Membranes were preincubated with radioligand for 30 min, before the time course of [3H]NMS dissociation was initiated by adding 1 μM atropine. Test compounds were added together with atropine, either alone or combined with an additional allosteric ligand. At specified time intervals, 1-ml aliquots were removed and filtered rapidly (glass fiber filter no. 6; Schleicher and Schüll, Dassel, Germany). The filters were washed twice with 5 ml of ice-cold incubation buffer and placed into scintillation vials, and the radioactivity was determined in the presence of 5 ml of Ready Protein (Beckman Coulter) by liquid scintillation counting in a Beckman LS 6500.
Binding Studies with Membranes from Cultured Cells.COS-7 cell membranes. Equilibrium binding and kinetic experiments with [3H]NMS and using membranes from COS-7 cells were performed in a 1-ml volume in reaction tubes and filtered using a 48-place Brandel cell harvester (Brandel Inc., Gaithersburg, MD) and GF/B filters (Whatman, Maidstone, UK), preincubated with 0.1% PEI for 30 min). After separation of the membranes, filters were washed twice with 5 ml of ice-cold 40 mM Na/K/Pi-buffer. For two-point kinetic dissociation experiments (Kostenis and Mohr, 1996), membranes were preincubated with [3H]NMS for 30 min. Thereafter, aliquots of the mixture were added to excess unlabeled atropine (final concentration, 3 μM) in buffer, alone, or in the presence of an allosteric agent over a total period of up to 120 min followed by simultaneous filtration of all samples.
[3H]Dimethyl-W84 Binding Assay
Binding experiments using the allosteric radioligand [3H]dimethyl-W84 were carried out as described previously (Tränkle et al., 2003). In brief, all handling of [3H]dimethyl-W84 (specific activity, 154–168 Ci/mmol) was carried out in 20 mM NaCl/0.01% bovine serum albumin. [3H]Dimethyl-W84 (1.5–2.0 nM) and membranes containing CHO hM2 receptors (100 μg of protein/ml) were incubated in HEPES buffer (10 mM HEPES, 20 mM NaCl, and 0.01% bovine serum albumin, pH 7.4 at 23°C). [3H]Dimethyl-W84 binding experiments were carried out in a 0.3-ml volume in 1.2-ml deep-well plates (ABgene, Epsom, Surrey, UK) at 23°C for 2 h. Nonspecific [3H]dimethyl-W84 binding was determined in the presence of 10 μM gallamine. Specific binding of [3H]dimethyl-W84 under control conditions was characterized by a pKD of 8.47 ± 0.04 (KD = 3 nM) and Bmax of 511 ± 45 fmol/mg of protein (mean ± S.E.M., n = 22). The receptor bound radioligand was filtered on a Tomtech 96-well Mach III Harvester (PerkinElmer Wallac, Turku, Finland) and the filter (Filtermat A; PerkinElmer Wallac) was washed once (0.8 ml of 100 mM NaCl, 4°C, 1.7 s) and dried in a microwave oven. Thereafter, scintillation wax (Meltilex A; PerkinElmer Wallac) was melted for 1 min at 90°C onto the filtermat using a Dri-Block DB-2A (Techne, Duxford, Cambridge, UK). The filters were placed in sample bags (PerkinElmer Wallac), and filter-bound radioactivity was measured using a Microbeta Trilux-1450 scintillation counter (PerkinElmer Wallac).
Homology Modeling
The model is based on the X-ray structure of bovine rhodopsin (Protein Data Bank code 1F88/1HZX; Palczewski et al., 2000) and the sequence of the human M2 receptor (SwissProt code P08172; Bonner et al., 1987). Transmembrane regions of the M2 receptor were detected using so-called pinpoints, identified by Baldwin et al. (1997). The extracellular and intracellular loops were created employing a combination of different methods: secondary structure prediction and/or application of a loop search routine based on homology aspects as implemented in the Homology module of Insight II 2000 (Accelrys Inc., San Diego, CA). Three-dimensional coordinates for the N and C termini were built in analogy to the X-ray structure of bovine rhodopsin. A detailed description of the modeling procedure has been given elsewhere (Jöhren and Höltje, 2002).
Docking
To dock the ligands into their binding sites within the loop region of the receptor model, FlexX (Kramer et al., 1999) was used. As a prerequisite, a three-dimensional structure of the target protein is needed. In the case reported here, a validated protein model was used. In addition, the active site has to be defined accurately. A particularly important feature of FlexX is that the ligands are treated as flexible molecules. This is accomplished by a three-step procedure. First, a basic part of the molecule, “the base fragment”, is selected and placed inside the binding pocket. Thereafter, the complete molecule is constructed in a stepwise manner. For the search of various placements of the base fragment inside the binding pocket, two algorithms are used. One superimposes triples of interaction centers of a base fragment with triples of compatible interaction points in the active site. If a base fragment has fewer than three interaction centers or if the number of placements is too low, another algorithm, called line matching, is implemented. This procedure matches pairs of interaction centers with pairs of interaction points. Because of geometry ambiguity, multiple placements are generated by rotation around the axis defined by the interaction points and centers. Both placement algorithms generate a large number of solutions, which are reduced by clash tests and clustering. Ranking of the docking results is performed employing a modified scoring function (Böhm, 1994). Algorithms and scoring functions are described in more detail elsewhere (Böhm, 1994; Rarey et al., 1997; Kramer et al., 1999).
Data Analysis
The data of individual experiments were analyzed by nonlinear regression analysis using Prism software (ver. 4.0; GraphPad Software, San Diego, CA). The dissociation data were fitted to a monoexponential decay; biexponential curve-fitting did not yield better results (F test, P ≥ 0.05; data not shown). Concentration effect curves for the reduction of the observed rate constant of dissociation, kobs, were analyzed using a four-parameter logistic function (except for hexamethonium; see Fig. 2 and legend). The parameters “inflection point” and “slope factor” n were variables, the upper plateau of the curve was the control value of kobs and was set k0 = 100%. We tested whether the lower plateau of the curve yielded a better fit with kobs as a variable >0 compared with kobs = 0% (F test). If this was not the case, the lower plateau of the curve was fixed at kobs = 0%. In addition we tested whether the slope factors of the curves were different from unity (i.e., n = 1) by statistically comparing the fits obtained by nonlinear regression analysis, using a partial F test. A p value < 0.05 was taken as the criterion for significance. In the case of hexamethonium, a two-site fit was used, in which it was assumed that each site of the fit exhibited a slope factor of unity (n = 1).
Analysis of Interactions between Allosteric Ligands. The interaction of one allosteric ligand with a second allosteric agent at [3H]NMS-occupied receptors was analyzed using a method described by Lazareno and Birdsall (1993). The procedure can be regarded as a condensed form of the Schild method (Arunlakshana and Schild,1959) to analyze the action of an antagonist. It requires the simultaneous analysis of two sets of data. The first dataset is the concentration-effect curve of the test allosteric agent, A, alone. The second dataset is an antagonist concentration-effect curve for the attenuation of the action of a single, fixed concentration of A by varying concentrations of a second allosteric antagonist, B. In a complementary fashion, we measured a concentration-effect curve for A in the presence of a fixed concentration of B to determine whether B causes a parallel shift of the concentration-effect curve for A. In the current study, the observed effect monitored was the retardation of [3H]NMS dissociation by the test allosteric agent in the absence and presence of B (e.g., hexamethonium).
To analyze the antagonist action of one allosteric ligand on the binding of a second allosteric ligand, the effect of the first ligand on [3H]NMS dissociation was eliminated from the analysis by normalization (for details, see Results). Using hexamethonium as an example, the dose-response curves for the test allosteric modulator alone (a), the test allosteric modulator in the presence of a high concentration of hexamethonium (b), and the effect of hexamethonium on [3H]NMS dissociation (c) in the presence of a fixed concentration of test modulator were simultaneously fitted using two independent variables, the concentrations of the test modulator A and of the additional allosteric antagonist B (e.g., hexamethonium). This analysis was based on the following equation (Lazareno and Birdsall, 1993) using Sigma Plot for Windows (version 8.0; SPSS Inc., Chicago, IL):  [A] is the concentration of the allosteric agent, Emax and basal denote the maximum and the minimum effects of A, respectively, n is the slope factor of the curve (corresponding to the Hill slope factor) for A alone, EC0.5,control indicates the concentration at which A produces a half-maximal effect, [B] is the concentration of the antagonist, KB denotes the equilibrium affinity constant of B, and s corresponds to the Schild slope factor.
[A] is the concentration of the allosteric agent, Emax and basal denote the maximum and the minimum effects of A, respectively, n is the slope factor of the curve (corresponding to the Hill slope factor) for A alone, EC0.5,control indicates the concentration at which A produces a half-maximal effect, [B] is the concentration of the antagonist, KB denotes the equilibrium affinity constant of B, and s corresponds to the Schild slope factor.
Curvilinear Schild data from antagonist studies were fitted according to Lazareno and Birdsall (1995), applying the following equation:  KB is the equilibrium affinity constant for the binding of allosteric agent B at the NMS-liganded receptor. α is the cooperativity factor for the interaction between A and B at NMS-occupied M2 receptors (α > 1, α < 1, α = 1 indicating positive, negative and neutral cooperativity, respectively). Fits applying eq. 1 with a Schild slope of unity were tested versus eq. 1 with a variable Schild slope (F test); if the latter fitted the data better, this fit was compared with a fit to eq. 2. Because eqs. 1 and 2 possess the same number of variables, the equation that produced the lower sum of squares determined by nonlinear regression analysis was designated the better fit to the data.
KB is the equilibrium affinity constant for the binding of allosteric agent B at the NMS-liganded receptor. α is the cooperativity factor for the interaction between A and B at NMS-occupied M2 receptors (α > 1, α < 1, α = 1 indicating positive, negative and neutral cooperativity, respectively). Fits applying eq. 1 with a Schild slope of unity were tested versus eq. 1 with a variable Schild slope (F test); if the latter fitted the data better, this fit was compared with a fit to eq. 2. Because eqs. 1 and 2 possess the same number of variables, the equation that produced the lower sum of squares determined by nonlinear regression analysis was designated the better fit to the data.
Results
Sensitivity of M2 Receptors to Allosteric Antagonism. The hypothesis of a common allosteric site in M2 muscarinic acetylcholine receptors was derived from experiments in which the orthosteric site of the receptors was occupied by the conventional orthosteric antagonist [3H]NMS. Under these conditions, the binding of allosteric agents is reflected by an allosteric modulation (usually reduction) of the observed dissociation rate constant of [3H]NMS. Structurally different allosteric agents have been shown to share an equal sensitivity to the competitive antagonist action of obidoxime on [3H]NMS dissociation, a finding that is compatible with obidoxime and the allosteric ligands binding competitively (Ellis and Seidenberg, 1992; Tränkle and Mohr, 1997). Herein, we introduce hexamethonium as an allosteric antagonist tool in addition to obidoxime that has the potential to provide useful additional information.
[3H]NMS dissociation from porcine heart M2 receptors under control conditions occurred monoexponentially (data not shown) as reported previously (Tränkle et al., 1996) with t1/2,control = 4.30 ± 0.15 min (means ± S.E.M, n = 105). Under all conditions examined, [3H]NMS dissociation remained monoexponential in the presence of hexamethonium and WDuo3, Duo3, tacrine, and the tacrine dimer (Fig. 1). This finding validates the use of two-point estimations of rate constants, as performed in the mutagenesis experiments (see Sensitivity of Binding of the Allosteric Ligands to M2/M5-Receptor Mutations). As shown in Fig. 2, hexamethonium interacts with the [3H]NMS-occupied M2 receptor in an allosteric fashion but with submaximal efficacy; i.e., at high concentrations, it does not totally abolish the dissociation of [3H]NMS from the receptor. The concentration-effect curve for the allosteric effect of hexamethonium on the rate of dissociation of [3H]NMS, illustrated in Fig. 2, levels off at 15 ± 3%; i.e., the maximal inhibition of the [3H]NMS dissociation rate constant is ∼7-fold. The pEC0.5, slope factor, and lower plateau for a global fit of the combined data were 4.11 ± 0.08, -0.57 ± 0.06, and 0.12 ± 0.04, respectively. Nonlinear regression analysis revealed that a two-site model yielded a significantly better fit to the data compared with a one-site model (F test, p < 0.05). The parameters characterizing the high-affinity (H) and low-affinity (L) components were pEC0.5,high = 5.03 ± 0.23, capacity %H = 47 ± 0.1%; pEC0.5,low = 3.55 ± 0.21, capacity %L = 53 ± 0.1% (mean ± S.E.M, n = 42).
The antagonist behavior of hexamethonium on the allosteric actions of tacrine and Duo3 in porcine cardiac M2 receptors is illustrated in Fig. 3, and the parameters obtained from the data analysis are compiled in Table 1. The concentration effect curves for the retarding actions of tacrine and Duo3 on [3H]NMS dissociation in the absence of hexamethonium in Fig. 3, A and B, respectively, are shown by ○. Their allosteric potencies (Duo3, pEC0.5,control = 6.08 ± 0.04; tacrine, pEC0.5,control = 5.28 ± 0.03; n = 16 for both) and their steep curve slopes (Duo3, n = 1.92 ± 0.26; tacrine, n = 1.82 ± 0.18) were in accord with published data (Tränkle et al., 1996). Antagonism by increasing concentrations of hexamethonium was measured at submaximally effective concentrations of tacrine (20 μM) and Duo3 (2 μM), which slowed [3H]NMS dissociation by approximately 90%. The rate constant k0 for these combination experiments was always normalized to the allosteric action of hexamethonium alone to compensate for the hexamethonium induced slowing of [3H]NMS dissociation; in other words, kobs observed in the presence of hexamethonium alone was set at k0 = 1.0. A 6-fold slowing of [3H]NMS dissociation was produced by the highest concentration of hexamethonium used (10 mM).
Parameters characterizing the actions of the indicated allosteric agents and allosteric antagonists at porcine heart M2 receptors with the orthosteric site blocked by NMS

Concentration-dependent effect of hexamethonium on the apparent rate constant of [3H]NMS dissociation from porcine cardiac M2 receptors. Ordinate, ratio of the observed rate constant of [3H]NMS dissociation in the presence of allosteric agent + atropine (kobs) to the rate constant of [3H]NMS dissociation in the presence of atropine alone (k0). Data points are derived from complete dissociation curves. Illustrated are the mean values ± S.E.M. of 42 experiments. Error bars are only shown when they exceed the size of the symbols. Curve fitting was based on a two-site model.
As can be seen from Fig. 3, hexamethonium inhibited the allosteric actions of Duo3 and tacrine in a concentration-dependent manner (Fig. 3, •). The antagonism by hexamethonium resulted in an acceleration of [3H]NMS dissociation in the presence of the fixed concentrations of Duo3 and tacrine. The increase in observed rate constant varied from 2.6-fold (0.1 mM hexamethonium) to 7.4-fold (3 mM hexamethonium) for Duo3 and 1.9-fold (0.1 mM hexamethonium) to 6.7-fold (10 mM hexamethonium) for tacrine.
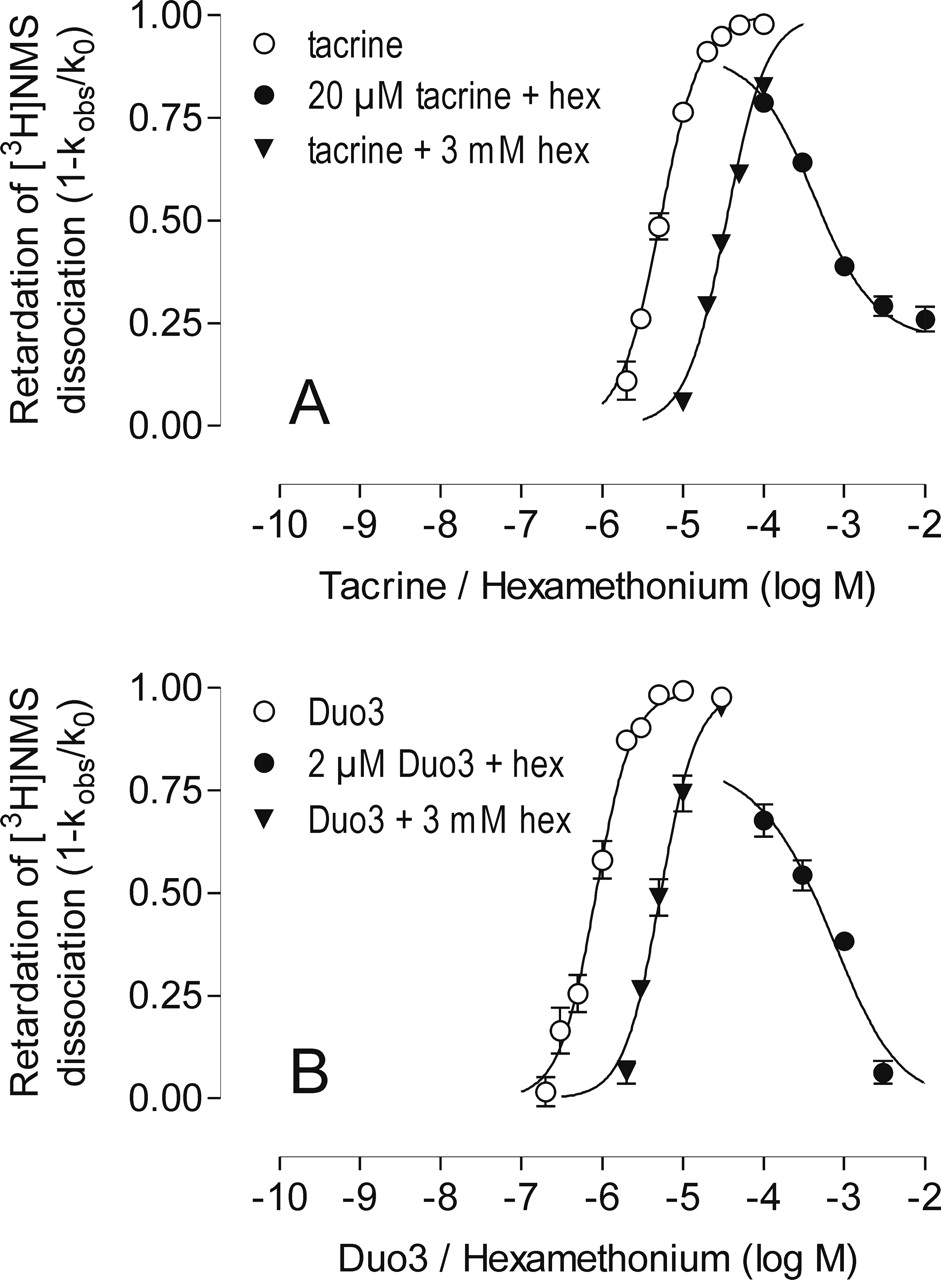
Retardation of [3H]NMS dissociation from porcine cardiac M2 receptors by increasing concentrations of tacrine (A) and Duo3 (B) alone (○) or in the presence of a fixed concentration of hexamethonium (▴). The antagonist action of hexamethonium on the effect of a fixed concentration of the respective test compound (•) is also shown. Ordinate, inhibition of [3H]NMS dissociation (1 - kobs/k0, compare legend to Fig. 2); k0 for the experiments in the presence of hexamethonium was always normalized to the allosteric action of hexamethonium alone. Abscissa, log concentration of the respective allosteric agent (retardation curves) or of hexamethonium (inhibition curve). Experiments were carried out in Na/K/Pi-buffer using porcine cardiac membranes. Simultaneous nonlinear fitting of the three curves of A and B, respectively, was carried out according to Lazareno and Birdsall (1993). For details, see Materials and Methods. Mean values of 8 to 18 independent experiments yielding complete dissociation curves are shown.
To check whether the antagonism by hexamethonium results in parallel shifts of the concentration-response curves, a second complete curve for each allosteric agent was measured in the presence of a high concentration of hexamethonium (3 mM; Fig. 3, ▴); the top plateaux of these curves were not different from a level of 100% inhibition of [3H]NMS dissociation (F test, p > 0.05). All sets of three curves were included in the respective global nonlinear regression analyses to characterize the type of antagonism. The results of the global nonlinear regression analysis of the data shown in Fig. 3 were used to display an array of curves shifted in parallel to the right (data not shown), which allowed dose-ratios (DR = EC0.5,antagonist/EC0.5,control) to be calculated, where EC0.5,control and EC0.5,antagonist are the concentrations of allosteric agent to induce a half-maximal inhibition of [3H]NMS-dissociation in either the absence or the presence, respectively, of the antagonist (in this example, hexamethonium). Schild plots of the data are shown in Figs. 4, A and B (⋄), to illustrate the different modes of interaction between these ligands and hexamethonium. For comparison, the interaction of hexamethonium with the conventional allosteric agent WDuo3 was also measured (Fig. 4C, ⋄; Table 1).
WDuo3 exhibited the simplest interaction pattern with hexamethonium. The potency of WDuo3 in the absence of hexamethonium (pEC0.5,control = 8.30 ± 0.30) and a slope not different from unity (n = 1.04 ± 0.06; mean ± S.E.M., n = 9) were in line with published data (Tränkle et al., 1996; Tränkle and Mohr, 1997). The global fit yielded a Schild slope for the interaction of hexamethonium with WDuo3 that was not significantly different from unity (F test, p > 0.05) indicating a formally competitive interaction with a calculated log KB of 4.32 ± 0.09.
The interaction of tacrine with hexamethonium (Figs. 3A and 4A) was best described by eq. 2, suggesting a cooperative interaction between these ligands, log α = -0.92 ± 0.03, and a log KB for hexamethonium of 3.93 ± 0.06 (mean ± S.E.M., n = 16). The interaction of Duo3 with hexamethonium (Fig. 3B) was best described by eq. 1 with a Schild factor s = 0.68 ± 0.06, which was different from unity (F test, p < 0.05), and a pA2 = 3.59 ± 0.19 (means ± S.E.M., n = 16). Thus, the hexamethonium/Duo3 interaction was noncompetitive.
The antagonist actions of obidoxime and Mg2+ on the allosteric interactions of tacrine, Duo3, and WDuo3 with [3H]NMS were also studied. The findings are illustrated in Fig. 4. Obidoxime (Fig. 4, ▵) has been shown previously to be a rather weak and noncompetitive antagonist against Duo3 (pA2 = 3.00 ± 0.08; s = 0.51 ± 0.04), identifying Duo3 as an atypical allosteric agent (Tränkle and Mohr, 1997). Tacrine was also sensitive to obidoxime (Ellis and Seidenberg, 1992), but the present data additionally reveal that the interaction does not seem to be competitive (s = 0.63 ± 0.02; see Table 1). In other words, tacrine also behaves like an atypical allosteric agent. As shown previously (Tränkle and Mohr, 1997), the conventional allosteric agent WDuo3 is antagonized by obidoxime in a competitive fashion (logKB = 4.16 ± 0.07; s not different from unity) and has therefore been classified as a common site allosteric agent.
Magnesium ions (Fig. 4, •) inhibited the allosteric actions of Duo3 on [3H]NMS dissociation only at very high concentrations, whereas the actions of tacrine were considerably more sensitive to Mg2+. To our surprise, the type of antagonism between tacrine and Mg2+ (analogous to the tacrine/hexamethonium interaction) was clearly cooperative (log α = -0.77 ± 0.09; log KB = 2.64 ± 0.04). WDuo3 was antagonized by Mg2+ in an apparently competitive fashion; the potency of Mg2+ (log KB = 3.20 ± 0.06) was comparable with the potency of Mg2+ against the common site allosteric agent W84 (log KB = 3.40 ± 0.09; Burgmer et al., 1998).
Taken together, according to their sensitivities to the three allosteric antagonists, WDuo3 can be classified as a common site allosteric agent, whereas Duo3 and tacrine behave atypically. It is noteworthy that hexamethonium and Mg2+ are negatively cooperative with tacrine; i.e., tacrine seems to have the propensity to bind simultaneously with these antagonists to the M2 receptor.
Effects of a Dimerized Tacrine. To investigate whether the allosteric site has room for more than one molecule of tacrine, two molecules of tacrine were linked by a hexamethylene chain, yielding the “tacrine dimer” (Fig. 1). If the allosteric site can accommodate one molecule of the dimer instead of two molecules of tacrine, then the dimer should have an increased affinity compared with tacrine and a normal instead of a steep concentration-effect relationship. [3H]NMS-dissociation experiments using porcine cardiac M2 receptors revealed that this was the case: dimerization increased the allosteric potency 20-fold (pEC0.5 = 6.56 ± 0.08, means ± S.E.M., n = 18) compared with tacrine (pEC0.5 = 5.27 ± 0.01, means ± S.E.M., n = 3, Fig. 5A), with the slope factor decreasing (ntacrine = -1.86 ± 0.08), as predicted by the hypothesis, to a value (ntacrine dimer = -1.14 ± 0.15) that was not significantly different from unity (F test, p > 0.05). Hexamethonium was used to antagonize the allosteric action of the dimer (Fig. 5A, inset) because it is cooperative with tacrine and has a higher potency than Mg2+. Global nonlinear regression analysis of the data for the antagonist action of hexamethonium against the dimer (3 μM) and the data for the concentration-effect curve of the dimer in the presence of 3 mM hexamethonium indicated that eq. 1 was sufficient to describe the data; s = 0.78 ± 0.14 was not different from unity (F test, P > 0.05) and pKB = 3.48 ± 0.06 (means ± S.E.M., n = 18). Thus, in contrast to tacrine, the tacrine dimer acted in a formally competitive manner with hexamethonium in porcine cardiac M2 receptors.
To monitor the binding of the tacrine-dimer and tacrine to M2 receptors whose orthosteric site is not occupied by NMS, receptors were labeled with the allosteric radioligand [3H]dimethyl-W84 (Fig. 5B). In this assay, we used membranes of CHO cells stably expressing human M2 receptors, because measurement of [3H]dimethyl-W84 binding (Fig. 5B) is facilitated by the higher receptor density than that found in porcine cardiac membranes (Fig. 5A) (Tränkle et al., 2003). Control experiments did not reveal a difference in allosteric potency between porcine cardiac M2 and CHO hM2 receptors (data not shown). The tacrine-dimer (pKi = 6.94 ± 0.04, means ± S.E.M., n = 5) inhibited [3H]dimethyl-W84 binding with approximately 50-fold higher affinity than tacrine (pIC50 = 5.21 ± 0.03; Tränkle et al., 2003). Furthermore, the inhibition curve of the dimer had a slope factor (n = -1.05 ± 0.09) not different from unity, whereas the slope of the tacrine curve (n = -1.41 ± 0.15) is significantly greater than unity, as reported previously (Tränkle et al., 2003). Thus dimerization of tacrine considerably increases the binding affinity compared with tacrine, both at the [3H]NMS-occupied and the unoccupied M2 receptors, suggesting that the allosteric site may have room for more than one tacrine molecule. In principle, the finding does not allow one to differentiate whether the full length of the dimer is bound within the site or whether binding of only the additional hexamethylene chain accounts for the observed increase in binding affinity. Structure-activity-relationships in W84 derivatives suggest that the hexamethylene middle chain would not fully explain the observed increase in affinity (Mohr et al., 2004). Furthermore, dimerization eliminates the atypical features of tacrine action, in that the concentration-effect curves of the dimer have slopes not different from unity, and the interaction with the antagonist hexamethonium changes from cooperative to competitive. It should be noted that the experiments shown in Fig. 5B were conducted under somewhat different buffer conditions from those shown in Fig. 5A and elsewhere in this manuscript to allow [3H]dimethyl-W84 binding to be accurately measured (Tränkle et al., 2003). Because allosteric actions are sensitive to the buffer conditions, the resulting affinities obtained from the data in the two panels are not strictly comparable.
Sensitivity of Binding of the Allosteric Ligands to M2/M5-Receptor Mutations. To shed light on possible differences in the receptor epitopes involved in the binding selectivity between M2 and M5 receptors of the atypical ligands Duo3 and tacrine, compared with the conventional ligands WDuo3 and the tacrine dimer, we investigated the allosteric actions of these ligands on a number of human M2/M5 chimeras (Ellis and Seidenberg, 2000) and two mutants of the human M2 receptor, M2 423Thr→His (Buller et al., 2002) and the double point mutant M2 177Tyr→Gln + 423Thr→His (Voigtländer et al., 2003). All receptors were expressed in transiently transfected COS-7 cells.

Antagonism by obidoxime, hexamethonium, and Mg2+ of the effects of tacrine (A), Duo3 (B), and WDuo3 (C) displayed in the form of Schild-plots. The results of the global nonlinear regression analysis, as shown in Fig. 3, were used to display an array of curves shifted in parallel to the right (not shown) which allowed dose ratios DR = EC0.5,antagonist/EC0.5,control to be calculated, where EC0.5 is the concentration of allosteric agent that induces a half-maximal inhibition of the [3H]NMS dissociation rate constant in the presence (EC0.5,antagonist) and in the absence of antagonist (EC0.5,control), respectively. Likewise, the results of the global analysis allowed lines with Schild slopes s of unity for WDuo3 and s = 0.68 for Duo3 to be constructed (see Table 1). In the case of tacrine, the curve was based on the ternary complex model of allosteric interactions and drawn according to Lanzafame et al. (1996). For details, see text.
We have shown previously that, for some typical, common site allosteric agents, two amino acids, M2 177Tyr and M2 423Thr, may account entirely for the M2/M5-selectivity in receptors whose orthosteric site was occupied by NMS (Voigtländer et al., 2003). The starting point for the identification of these amino acids was studies on chimeric M2/M5 muscarinic receptors.

A, inhibition of [3H]NMS dissociation by tacrine (○) and the tacrine dimer (•) at muscarinic M2 receptors in porcine cardiac membranes. Ordinate, ratio of the observed rate constant of [3H]NMS dissociation in the presence of allosteric agent + atropine (kobs) to the rate constant of [3H]NMS dissociation in the presence of atropine alone (k0). Abscissa, log concentrations of the allosteric agents. Inset, Schild plot of the antagonist action of hexamethonium on the allosteric action of the tacrine dimer on [3H]NMS dissociation from porcine cardiac M2 receptors. Data were analyzed as in Fig. 3 and displayed as described in the legend to Fig. 4. B, inhibition of specific [3H]dimethyl-W84 binding by tacrine and the tacrine dimer in membranes from CHO cells expressing human M2 receptors. Nonspecific [3H]dimethyl-W84 binding was defined in the presence of 10 μM gallamine. Specific binding of [3H]dimethyl-W84 (1.5 nM) in the absence of inhibitor was constrained to 100%. For comparison, data obtained with tacrine from Tränkle et al. (2003) was included in B as a dashed line. Error bars are shown when they exceed the size of the symbols. Curve fitting was based on a one-site model. Mean values ± S.E.M. of three to eight experiments are shown; dissociation experiments were carried out in duplicate whereas binding experiments were performed as triplicate determinations.
The chimeric receptors represent the M5 receptor, with parts of the amino acid sequence being replaced by the corresponding amino acids of the M2 receptor. Thus, stretches of amino acids in M2 that increase the binding affinity of allosteric agents above the level of M5 can be identified. All of these mutants and chimeras are depicted in Fig. 6.
The allosteric inhibition of [3H]NMS dissociation was used as the measure of receptor binding of the test compounds. For all four modulators, the observed affinity was higher at M2 than at M5 receptors. The M2/M5 subtype selectivity was large in the case of WDuo3 (140-fold) and considerably lower for the tacrine dimer (16-fold), Duo3 (15-fold), and for tacrine (13-fold) (Table 2). Another notable feature of the inhibition curves of [3H]NMS dissociation from M5 receptors is that these ligands were only capable of generating a maximum 2- to 4-fold slowing of the rate constant. This contrasts with the ability of the same ligands to essentially abolish [3H]NMS dissociation from M2 receptors. Of the chimeras used in this study, only CR6 consistently had this aspect of the M2 behavior for the ligands examined. This suggests the importance of a region of the o3 loop, the N terminus, and possibly TM1/TM6/TM7 in regulating [3H]NMS dissociation from [3H]NMS-receptor-allosteric ligand complexes. Another surprising result is that the potency of Duo3 to inhibit [3H]NMS dissociation is higher at the human M2 receptor expressed in COS-7 cells than at the porcine heart M2 receptor. The reason for this is not known.
Potency of the allosteric agents to slow the rate of [3H]NMS dissociation from the indicated wild-type and mutant human muscarinic receptors as a measure of the agents' binding affinity

Concentration-effect curves for the allosteric retardation of [3H]NMS dissociation (A–F) induced by WDuo3 (left) and Duo3 (right) at the indicated wild-type and mutant receptors, using membranes from transiently transfected COS-7 cells. After prelabeling the receptors with 0.2 nM [3H]NMS, dissociation was measured in the absence or presence of the allosteric modulator (see Materials and Methods). Ordinate, ratio of the observed rate constant of [3H]NMS dissociation in the presence of allosteric agent + atropine (kobs) to the rate constant of [3H]NMS dissociation in the presence of atropine alone (k0). Abscissa, log concentrations of the allosteric agents. Indicated are mean values ± S.E.M of three to seven separate experiments performed as duplicate determinations. G, schematic sketches of the wild-type and mutant receptors used in the current study.
The high affinity of WDuo3 for M2 receptors was reduced by a factor of 3 in M2 423Thr (Fig. 6A), whereas the double point mutation reduced the affinity of WDuo3 to the level of its M5 affinity, suggesting a critical role of M2 177Tyr. Thus, WDuo3 behaves in the same manner as the common site allosteric agents W84, dimethyl-W84, and diallylcaracurine, (Voigtländer et al., 2003). In the case of Duo3, however, these point mutations hardly affected its affinity (Fig. 6B). The affinity of WDuo3 was clearly raised compared with M5 only in the CR3 chimera (Fig. 6, C and E). This is the only chimera to contain the M2 177Tyr.
In contrast, Duo3 gained affinity in CR1 and also in CR2, CR3, and CR6 (Fig. 6, D and F). The gain in affinity seen in CR1 and CR2 indicates that the M2/M5-selectivity of Duo3, but not WDuo3, involves epitopes in the N-terminal region of the M2 receptor.
The affinity of tacrine for M2 was essentially unaltered in the M2 423Thr→His single point mutant but was clearly reduced in the double point mutant (Fig. 7A), although the affinity for the double point mutant did not fully reach that of M5. The affinity of tacrine was increased in CR3 and CR6 relative to M5 (Fig. 7C) and unchanged in CR1 and CR2 (Fig. 7E). These results match the findings obtained previously in the same chimeric receptors (Ellis and Seidenberg, 2000). Thus, the epitope dependence of tacrine resembles that of the common site agent WDuo3.
The binding of the tacrine-dimer was unchanged in the M2 423Thr→His mutant but decreased at the M2 double point mutant (Fig. 7B), thus resembling tacrine. Furthermore, similar to tacrine, the dimer gained affinity relative to M5 in CR3 and, to a lesser extent, in CR6 (Fig. 7D), but not in CR1 (Fig. 7F). In contrast to tacrine however, the dimer gained affinity in CR2 (Fig. 7F).
Taken together, the epitope dependencies of the atypical allosteric agents Duo3 and tacrine are different. The epitope dependence of tacrine resembles that of common site allosteric modulators, whereas the affinity of Duo3 is sensitive to epitopes in both CR1 and CR2 that contain the N-terminal elements of the M2 receptor protein.
Docking Simulations in the Model of the M2 Receptor. As reported previously, this model is based on the three-dimensional structure of bovine rhodopsin in the inactive state (Voigtländer et al., 2003). Docking simulations were carried out for the human M2 receptor whose orthosteric site is occupied with NMS [i.e., in which the receptor is fixed in an inactive state similar to the crystallized bovine rhodopsin (Palczewski et al., 2000) used as a template for the model]. WDuo3 is chemically closely related to the common site allosteric ligand W84. As might be predicted, this leads to a W84-like binding mode of WDuo3. For both ligands one of the lateral phthalimide residues is close to the epitopes 423Thr and 177Tyr (Fig. 8A). These amino acids line the end of the allosteric binding cleft at the place where, in the unliganded M2 receptor, a corridor leads to the orthosteric site (Jöhren and Höltje, 2002).
For tacrine, FlexX predicts two different placements for this rather small molecule: one next to 177Tyr and 423Thr between o2 and o3, and another close to o1 and the N terminus (Fig. 8B). The distance between the two locations is approximately 12 Å and is therefore large enough to accommodate the hexamethylene spacer in the structure of the tacrine-dimer. The docking geometry of tacrine-dimer found by FlexX is illustrated in Fig. 8C.
Although hexamethonium is a relatively small and very flexible ligand, there seems to be one preferred position inside the common allosteric binding site. In this binding mode hexamethonium forms cation-π interactions with 177Tyr (data not shown). Another less favorable docking geometry locates hexamethonium closer to the N terminus. In this binding mode, an additional tacrine molecule can be placed simultaneously inside the allosteric binding cavity (Fig. 8D).
For Duo3, the docking simulations yield a position in which the molecule does not come close to the end of the allosteric binding cleft. This contrasts with the docked positions of WDuo3 and W84 (Fig. 8, E and A, respectively).
Discussion
Current evidence points to there being at least two allosteric ligand binding sites on muscarinic receptors: the common allosteric site (or “gallamine” site), to which typical allosteric ligands bind (e.g., gallamine, alcuronium, W84, and strychnine), and the “WIN” site that binds staurosporine and WIN 62,577 (Birdsall and Lazareno, 2005). Such ligands, binding to their respective sites, interact in a “simple” fashion with the binding of ligands to the orthosteric site, obeying the predictions of the allosteric ternary complex model in equilibrium, kinetic, and functional studies. Interaction studies using gallamine or strychnine and an analog of WIN 62,577 indicate that there is often neutral cooperativity at the two allosteric sites (Lazareno et al., 2002). It is therefore possible to generate muscarinic receptor complexes with both allosteric sites simultaneously occupied by two different ligands.
Another class of allosteric ligand, termed atypical, of which tacrine (Potter et al., 1989), Duo3 (Tränkle and Mohr, 1997), and pentacyclic carbazolones (Gharagozloo et al., 2002) are examples, binds with positive cooperativity (slope factors >1, and generally ≤2), even with the orthosteric site occupied. This implies the possibility of both allosteric sites being occupied simultaneously by the same ligand, with positive cooperativity being generated between the binding of the ligand to the two sites.
A third class of ligand, exemplified by obidoxime and also shown in this study by hexamethonium, exhibit slope factors less than 1 for inhibiting [3H]NMS dissociation from M2 receptors. This indicates heterogeneous binding or homotropic negative cooperativity (in contrast to the homotropic positive cooperativity shown by atypical ligands). Furthermore, and crucial to the present study, obidoxime and hexamethonium, in contrast to typical allosteric ligands, do not fully inhibit [3H]NMS dissociation from M2 receptors. This means that [3H]NMS can dissociate from the M2 orthosteric site when the allosteric site(s) are occupied by these ligands. A similar behavior to obidoxime and hexamethonium is also shown by Mg2+ (Burgmer et al., 1998).
This study explores the complex pattern of behavior of these ligands at the allosteric sites using interaction studies, mutant receptors, and docking studies. Using cardiac M2 receptors, our studies investigated two atypical ligands, tacrine and Duo3 and two related ligands, WDuo3 and tacrine dimer, which are typical in their ability to inhibit [3H]NMS dissociation. Their interactions with the third class of allosteric ligands (probe ligands) were studied. Three different interactions were observed, a competitive effect (linear Schild plot, slope 1), cooperative (nonlinear, Schild plot reaching plateau) and “noncompetitive” (complex, linear Schild plot, slope <1). It should be noted that what might be interpreted as a competitive interaction may be an allosteric action with high negative cooperativity. In addition, the intercept at log (DR-1) = 0 gives the pA2 estimate (or pKB if the slope factor equals 1) of the probe ligands. Any differences in pA2 estimates of the probe ligands suggests the presence of multiple binding sites.

Allosteric effects of tacrine (left) and the tacrine dimer (right) at the indicated wild-type and mutated receptors. Experiments were carried out and analyzed as described in the legend to Fig. 6. Mean values ± S.E.M. of three to six separate experiments carried out in duplicate are shown.
All three probes (hexamethonium, Mg2+, and obidoxime) seemed to be competitive with WDuo3. In contrast, Duo3 was affected in a noncompetitive fashion by hexamethonium and obidoxime. Furthermore, Duo3 was less sensitive than WDuo3 to these probes and was almost insensitive to Mg2+. Duo3 is clearly binding differently from WDuo3.
Compared with Duo3, tacrine was affected by lower concentrations of obidoxime, but also in a noncompetitive way. In contrast to Duo3, however, tacrine was antagonized by hexamethonium and Mg2+ in an allosteric (negatively cooperative) fashion, with obidoxime and possibly hexamethonium being more potent against tacrine than against Duo3. Duo3 seems to be binding differently from tacrine. Hexamethonium and Mg2+ seem to be capable of binding to a receptor-NMS-tacrine complex.

Docking simulations using a human M2 receptor model in which the orthosteric site is occupied by NMS. View from the membrane. Protein: helices, red; o1 and N terminus, gray; o2, cyan; EDGE (residues 172–175 in M2), green; o3, yellow; disulfide bridge, magenta. 177Tyr/423Thr: carbon, green; nitrogen, dark blue; oxygen, red; hydrogen, white. Volumes of the binding sites: gray grid. Allosteric modulator/orthosteric NMS: carbon, white; nitrogen, dark blue; oxygen, red; hydrogen, cyan. A, WDuo3 and W84 inside the allosteric binding site (top) of the M2 receptor whose orthosteric site (bottom) is liganded with NMS. W84: carbon, orange; nitrogen, dark blue; oxygen, red; hydrogen, cyan. B to E, docking at the allosteric site of two tacrine molecules simultaneously (B), the tacrine dimer (C), hexamethonium (left) and tacrine (right) simultaneously (D), and Duo3 (E).
Studies employing mutated human M2 receptors expressed in COS-7 cells and docking simulations in a M2 receptor model have shown that M2 177Thr account for the M2/M5-selectivity of typical allosteric agents (Voigtländer et al., 2003). The present study shows that the M2/M5-selectivity of the typical allosteric agent WDuo3 can also be fully explained by these epitopes.
The binding of tacrine is also somewhat sensitive to these epitopes, despite tacrine's behavior as an atypical allosteric agent in the interaction studies. This apparent paradox was resolved by the docking simulations, which show that the allosteric binding cleft has sufficient room for a simultaneous binding of hexamethonium and tacrine (Fig. 8D). Docking also allows two molecules of tacrine to bind simultaneously in the allosteric binding cleft, as predicted by Potter et al. (1989), with one of these molecules being located near the epitopes M2 177Tyr and M 423 Tyr and M2 423Thr deep in the allosteric binding cleft (Fig. 8B) with the other molecule being bound near the entrance of the allosteric domain. Thus, the allosteric actions of tacrine involve common-site epitopes, but the interaction of tacrine with hexamethonium is atypical, because both agents may bind simultaneously to the allosteric site. Docking experiments further suggest that two molecules of hexamethonium may bind simultaneously but with different affinities within the allosteric binding cleft (data not shown). This could explain the biphasic effect of hexamethonium on [3H]NMS-dissociation (Fig. 2) via an interaction solely within the allosteric binding domain. The WIN compounds and the relatively bulky staurosporine may interact with the second binding location of tacrine but do not reach the location that accommodates the “first” tacrine molecule or one end of W84 and WDuo3.
The tacrine dimer explores whether the allosteric site can accommodate two molecules of tacrine linked by a hexamethylene spacer present in several typical bisquaternary modulators (Nassif-Makki et al., 1999). Similar tacrine dimers are potent inhibitors of acetylcholinesterase (e.g., Pang et al., 1996). Likewise, the affinities of the tacrine dimer for unoccupied and [3H]NMS-occupied cardiac M2 receptors are considerably higher (20–60-fold) than those of tacrine (Fig. 5), suggesting additional binding interactions. However, the positive homotropic cooperativity is absent in the dimer. The competitive interaction of the tacrine dimer with hexamethonium suggests that the allosteric site cannot accommodate both modulators simultaneously.
For Duo3, however, there is no evidence from interaction or from docking studies for the simultaneous binding of two molecules. According to the three-dimensional model, the long Duo3 molecule almost fills the allosteric binding cavity (Fig. 8E) but it does not reach as deep into the cavity as W84 or WDuo3 (Fig. 8A). We found a small (15-fold) COS-7-hM2/M5 subtype selectivity of Duo3 (Table 2) relative to that found in the case of WDuo3 (140-fold). Because the affinity for the M5 subtype is nearly identical for the two compounds (Table 2), the small M2/M5-selectivity of Duo3 is the consequence of its relatively low affinity for M2 receptors. According to the 3D model, Duo3 does not come as close as WDuo3 and W84 to the amino acids M2 177Tyr and M2 423Thr, which are important for the M2/M5 subtype selectivity of these typical allosteric agents. This finding is in accord with the fact that the affinity of Duo3 for M2 is only slightly reduced in the double point mutant. Furthermore, Duo3 stands out in that its affinity is increased over the level of M5 in all the chimeric receptors, with the greatest gain found in CR1. None of the other agents have increased affinity in CR1, which contains the N-terminal domain of the M2 receptor.
Taken together, the mutagenesis studies and the 3D model both suggest that Duo3 binds to the allosteric binding cavity but in an atypical mode. Yet this does not directly explain the atypical features of its allosteric action such as the apparently noncompetitive interplay with obidoxime and hexamethonium and its steep slope factors. These results do not support the former hypothesis that a distinct second allosteric site is involved in the atypical action of Duo3.
Although atypical, the postulated binding of Duo3 and tacrine within the common allosteric binding cavity does explain our previous observation that both agents did not affect the dissociation of the typical allosteric ligand [3H]dimethyl-W84 from M2 receptors (Tränkle et al., 2003); when the allosteric binding cavity is occupied by the radioligand, the binding site of the atypical agents is occluded.
It has been argued that atypical allosteric agents, including Duo3, might modulate receptor dimerization or generate a positive homotropic interaction transmitted via the receptor-receptor dimer interface (Gharagozloo et al., 2002; Tränkle et al., 2003). We cannot exclude this explanation at present. In fact, the “flat” curve of hexamethonium (and obidoxime) for inhibiting [3H]NMS dissociation (Fig. 2) could represent homotropic negative cooperativity across the receptor dimer and the steep curve for Duo3 inhibiting [3H]NMS dissociation represent the equivalent positive cooperativity. The apparent “noncompetitive” nature of the interaction of obidoxime and hexamethonium with tacrine and Duo3 (Table 1) could also be explained by complex interplay between a positive and negative cooperative system that is not accounted for in the model used to analyze the data. Such an interpretation does not exclude the possibility of two tacrine molecules bound/monomer (four per dimer) as suggested by the docking studies. The divergent binding modes of the atypical ligands (and one would predict obidoxime and hexamethonium) may be responsible for generating conformational changes at the receptor dimer interface that cause the positive and negative homotropic cooperativities.
In conclusion, this study shows for the first time that atypical muscarinic allosteric agents can have different binding modes, but that their atypical actions may, nevertheless, be mediated via the common allosteric binding domain of muscarinic receptors.
Acknowledgments
We thank Iris Jusen for expert technical assistance.
Footnotes
-
This work was supported by the Deutsche Forschungsgemeinschaft (Mo 821/1 to K.M, Tr 372/1 to C.T., and Ho1368/7 to U.H.), the Medical Research Council (to N.J.M.B.), by United States Public Health Service grant R01-AG05214 (to J.E.), and by a grant of the University of Bonn (to S.B.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.017707.
-
ABBREVIATIONS: M2 receptor, M2 subtype of the muscarinic acetylcholine receptor; Duo3, 4,4′-bis-[(2,6-dichloro-benzyloxy-imino)-methyl]-1,1′-propane-1,3-diyl-bis-pyridinium dibromide; WIN 62,577, 17-hydroxy-17-ethinyl-Δ4-androstano(3,2-b)pyrimido(1,2-a)benzimidazole; WDuo3, 1,3-bis[4-(phthalimidomethoxyimino-methyl)-pyridinium-1-yl] propane dibromide; tacrine dimer, 1,6-bis[amino-1,2,3,4-tetrahydroacridinyl]-hexane; W84, hexane-1,6-bis(dimethyl-3′-phthalimidopropyl-ammonium bromide; dimethyl-W84, N,N′-bis[3-(1,3-dihydro-1,3-dioxo-4-methyl-2H-isoindol-2-yl)propyl]-N,N,N′,N′-tetramethyl-1,6-hexanediaminium diiodide; NMS, N-methylscopolamine; CHO, Chinese hamster ovary.
- Received August 5, 2005.
- Accepted September 9, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}