


Abstract
The human organic anion transporter 2 (hOAT2, SLC22A7) mediates the sodium-independent uptake of numerous drugs, including cephalosporins, salicylates, dicarboxylates, and prostaglandins, and is mainly expressed in hepatocytes. Because the regulation of hOAT2 expression is poorly understood, we characterized cis-acting elements in the 5′-flanking region that regulate hOAT2 transcription. A consensus binding motif for the hepatocyte nuclear factor-4α (HNF-4α), arranged as a direct repeat (DR)-1, is located at nucleotides –329/-317 relative to the transcription initiation site. This element specifically binds HNF-4α in electrophoretic mobility shift assays. A luciferase-linked hOAT2 promoter fragment containing the HNF-4α binding site was transactivated upon cotransfection of an HNF-4α expression vector in Huh7 cells, whereas site-directed mutagenesis of the DR-1 element abolished activation by HNF-4α. Short interfering RNAs inhibiting endogenous HNF-4α expression markedly reduced endogenous expression of hOAT2 in Huh7 cells. Because HNF-4α is a known target for bile acid-mediated repression of gene transcription, we studied whether chenodeoxycholic acid (CDCA) suppresses hOAT2 gene expression by inhibiting HNF-4α-mediated transactivation. Treatment of Huh7 cells with CDCA or the synthetic farnesoid X receptor (FXR) agonist GW4064 decreased mRNA and protein levels and also nuclear binding activity of HNF-4α. The FXR-inducible transcriptional repressor small heterodimer partner inhibited transactivation of hOAT2 promoter constructs and of endogenous hOAT2 expression by HNF-4α. We conclude that the hOAT2 gene is critically dependent on HNF-4α and that bile acids repress the hOAT2 gene by inhibiting HNF-4α. Hepatic uptake of hOAT2 substrates may thus be decreased in disease conditions associated with elevated intracellular levels of bile acids.
The members of the organic anion transporter (OAT) family of proteins play important roles in the absorption, distribution, and elimination of numerous endogenous substances, xenobiotics, and their metabolites (Burckhardt and Wolff, 2000; Sekine et al., 2000; Marzolini et al., 2004; You, 2004). The OAT transporters differ structurally from the members of the organic anion-transporting polypeptide (OATP) family (Hagenbuch and Meier, 2004). Although the OATPs typically transport larger and more lipophilic organic anions, the OATs are efficient transporters of small hydrophilic organic anions. The transport substrates for OATs include anionic drugs such as diuretics, antibiotics, nonsteroidal anti-inflammatory drugs, antiviral nucleoside analogs, and antitumor drugs (Burckhardt and Wolff, 2000; Sekine et al., 2000; Marzolini et al., 2004). The five phylogenetically related transporters called hOAT1–5 belong to the solute carrier gene superfamily SLC22A in humans. All members of the OAT protein family contain 12 putative transmembrane domains, and a long hydrophilic loop containing multiple predicted N-glycosylation sites is believed to form between transmembrane domains 1 and 2 (Burckhardt and Wolff, 2000; Sekine et al., 2000; You, 2004). Furthermore, the OAT proteins contain several potential phosphorylation sites for protein kinase C, as well as for protein kinase A, casein kinase II, and tyrosine kinases. The significance of potential phosphorylation events in regulating the transport function of OATs is largely unknown.
The 6.8-kilobase human OAT2 gene (gene symbol SLC22A7) is located on chromosome 6q21.1-2 and contains 10 exons and nine introns (Kok et al., 2000). An alternatively spliced transcript lacking the second exon has been described previously (Sun et al., 2001). The hOAT2 mRNA is expressed most abundantly in the liver but also in the kidney (Sun et al., 2001). The rat Oat2 protein is exclusively localized to sinusoidal, but not bile canalicular, membranes of hepatocytes (Simonson et al., 1994). The human OAT2 mediates sodium-independent uptake of p-aminohippurate, cephalosporins, salicylates, tetracyclines, and methotrexate (Sekine et al., 2000; Sun et al., 2001; Khamdang et al., 2003; Marzolini et al., 2004).
Changes in the expression level of hOAT2 could influence the rate of hepatic clearance of hOAT2 substrates from sinusoidal blood. However, the mechanisms that control the expression of hOAT2 in human liver have not been studied. Our aim, therefore, was to characterize the transcriptional regulation of the hOAT2 gene. Computer analyses of the 5′-flanking region of the hOAT2 gene identified a consensus binding site for the liver-enriched hepatocyte nuclear factor HNF-4α (nuclear receptor 2A1) between nt –329 and –317 upstream of the transcription initiation site. In this study, we characterize the role of HNF-4α in transactivating the hOAT2 promoter and as a target for suppression of hOAT2 gene transcription by bile acids. Bile acid-mediated suppression of hOAT2 gene expression could serve to decrease the hepatocellular uptake of drugs and xenobiotics in disease conditions associated with increased intrahepatic concentrations of potentially toxic bile acids.
Materials and Methods
Chemicals. The synthetic farnesoid X receptor (FXR) ligand GW4064 was a gift from Dr. Daniel Berger (GlaxoSmithKline, Uxbridge, Middlesex, UK). Deoxyadenosine 5′-[α-32P]triphosphate (6000 Ci/mmol) was purchased from Amersham Biosciences Inc. (Otelfingen, Switzerland). All restriction enzymes were from Roche Diagnostics (Rotkreuz, Switzerland), the Pfu Turbo DNA polymerase was from Invitrogen (Basel, Switzerland), and T4 DNA Ligase was from Stratagene (La Jolla, CA). All other chemicals were purchased from Sigma-Aldrich (Buchs, Switzerland), unless stated otherwise.
Plasmids. Based on the available SLC22A7 sequence (GenBank accession no. AL583834), fragments of the 5′-flanking sequence of the human hOAT2 gene were amplified by PCR using primers that contained internal restriction sites (Table 1). The resulting PCR products were digested with the appropriate restriction enzymes and subcloned into the luciferase vector pGL3basic (Promega Catalys, Wallisellen, Switzerland). Point mutations within the putative HNF-4α binding site (nucleotides –329/–317) were introduced into the hOAT2(–679/+54)luc reporter construct using the QuikChange site-directed mutagenesis kit (Stratagene). The hOAT2(–679/ +54mut1)luc construct contained two base changes in both DR-1 repeats, and hOAT2(–679/+54mut2)luc contained three base changes centering around the core adenine of the DR-1 element. The sequence of all luciferase reporter constructs was confirmed by DNA sequencing. The mammalian expression plasmids pCMX-SHP and pcDNA3-HNF-4α were obtained from Dr. David Mangelsdorf (University of Texas Southwestern Medical Center at Dallas, Dallas, TX) and Dr. David Moore (Baylor College of Medicine, Houston, TX), respectively.
Sequences of oligonucleotides used for cloning, mutagenesis, EMSA probes, and real-time PCR
Engineered restriction enzyme sites are underlined and the corresponding restriction enzyme is given in parentheses behind the sequence.
Cell Culture. The human hepatoma cell lines Huh7 and HepG2 (LGC Promochem, Molsheim Cedex, France) were cultured in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The human ovarian cancer cell line PA-1 (American Type Culture Collection) was maintained in Eagle's modified essential medium (Invitrogen) containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Reporter Assays. Cells transiently transfected for reporter assays were seeded in 48-well plates at a density of 1 × 105 cells/well. Cells were transfected with 350 ng of the indicated luciferase constructs and 50 ng of the indicated expression plasmids using Fu-GENE 6 reagent (Roche Diagnostics), with a ratio of 3 μl of FuGENE 6 per 1 μg of DNA. To normalize the total amount of DNA transfected, pcDNA3.1 vector (Invitrogen) was added where appropriate. As a control for transfection efficiency, 50 ng of a pSV-β-galactosidase reporter plasmid (Promega Catalys) was cotransfected. 24 h after transfection, cells were harvested in passive lysis buffer (Promega Catalys), and luciferase activities were determined using the Luciferase assay system (Promega Catalys) in a Lumat LB 9507-2 luminometer (Berthold Technologies, Regensdorf, Switzerland). β-Galactosidase activities were quantified by a chlorophenol red-β-d-galactopyranoside-based colorimetric assay in a microplate reader (Molecular Devices, Sunnyvale, CA). All transfection experiments were performed at least three times, and results are expressed as mean values ± standard deviations.
RNA Isolation, Reverse Transcription, and Real-Time PCR. Huh7 cells were seeded in six-well plates, and after reaching 80% confluence were treated with 50 μM chenodeoxycholic acid (CDCA), 200 nM FXR agonist GW4064, or the vehicle DMSO for 24 h. Where RNA was prepared from transfected Huh7 cells, 4 μg/well of the indicated expression constructs was transfected. Total RNA was extracted 24 h after the transfection using TRIzol reagent (Invitrogen). One microgram of each RNA preparation was reverse transcribed to cDNA by random priming (reverse transcription system; Promega Catalys). Five microliters of each resulting cDNA, from a final reaction volume of 50 μl, was used for real-time PCR, which was performed on an ABI PRISM 7700 sequence detection system (Applied Biosystems, Rotkreuz, Switzerland) using the SYBR Green PCR Master Mix (QIAGEN, Hombrechtikon, Switzerland) and the primers listed in Table 1. Constitutively expressed 18S rRNA was used as internal standard for sample normalization. The relative levels of HNF-4α, hOAT2, and small heterodimer partner (SHP) mRNAs were calculated using the comparative threshold cycle (ΔΔCT) method.
Transfection of Cells with siRNAs. Huh7 cells were transfected in six-well plates at 70% confluence. Per transfection, 5 μl of TransIT-TKO transfection reagent (Dharmacon, Lafayette, CO) was mixed with 250 μl of serum-free OptiMEM (Invitrogen), followed by addition of either the SMARTpool short interfering RNA (siRNA) targeting HNF-4α (M-00346-01; Dharmacon) or siCONTROL nontargeting siRNA 1 (Dharmacon) to a final concentration of 25 nM. After a 20-min incubation at room temperature, the siRNA mixtures were added to the 1250 μl of RPMI 1640 medium supplemented with 10% FBS contained in each well. Cells were harvested 24 h after transfection, and total cellular RNA and protein were extracted using TRIzol.
Preparation of Protein Extracts. To prepare nuclear extracts from 100-mm dishes, confluent Huh7 cells were washed twice with ice-cold phosphate-buffered saline (PBS), scraped off the plates, and centrifuged for 10 s at 12,000 rpm/4°C. Cell pellets were resuspended in 400 μl of ice-cold buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and 1 mM DTT) supplemented with Complete protease inhibitors (Roche Diagnostics), and incubated on ice for 5 min. Next, 5 μl of 10% (v/v) Igepal CA-630 was added, followed by a 20-s centrifugation at 12,000 rpm/4°C. The pellets containing the nuclei were resuspended in 40 μl of buffer C (20 mM HEPES, pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 1 mM DTT), supplemented with Complete protease inhibitors. The soluble nuclear proteins were extracted by rotating at 4°C for 1 h. The debris was removed by centrifugation for 10 min at 14,000 rpm/4°C. The supernatants containing the nuclear proteins were analyzed for protein concentration with the bicinchoninic acid protein assay (Pierce Chemical, Perbio Science, Lausanne, Switzerland) and stored at –80°C until use.
To prepare whole cell extracts, cells on 100-mm dishes were washed with ice-cold PBS, lysed by a 5-min incubation in 250 μl of ice-cold radioimmunoprecipitation assay buffer [50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% (v/v) Igepal CA-630, 0.5% (w/v) sodium deoxycholate, 1 mM EDTA, 0.1% (w/v) SDS, and 10% (v/v) glycerol], supplemented with Complete protease inhibitors. The debris was removed by centrifugation for 30 min at 14,000 rpm/4°C. Protein concentrations of the whole cell extracts were determined with the bicinchoninic acid protein assay, and the samples were stored at –80°C until use.
Immunoblotting. Twenty micrograms of nuclear or whole-cell extracts was separated on a 10% SDS polyacrylamide gel and electroblotted onto nitrocellulose membranes (Hybond ECL; Amersham Biosciences). Membranes were blocked for 1 h in 5% (w/v) nonfat milk in PBS-T [0.1% (v/v) Tween 20 in PBS]. After blocking, the membranes were probed with an antibody against HNF-4α (H-171; Santa Cruz Biotechnology, Inc., LabForce, Nunningen Switzerland) at a concentration of 1 μg/ml in 1% bovine serum albumin/PBS-T. After three washes with PBS-T, the horseradish peroxidase-conjugated secondary goat anti-rabbit antibody (Pierce Chemical) was added at a concentration of 5 ng/ml in 3% (w/v) nonfat milk/PBS-T for 1 h. Blots were washed three more times with PBS-T and twice with PBS, followed by detection with the SuperSignal West Femto maximum sensitivity substrate (Pierce Chemical) and exposure on Hyperfilm ECL (Amersham Biosciences). To verify equal loading of the protein samples, the blots were stripped with Restore Western blot stripping solution (Pierce Chemical), reblocked, and reprobed for constitutively expressed Ku-70 antigen. The Ku-70 probing and detection were performed as described above, except that the Ku-70 antibody (C-19; Santa Cruz Biotechnology, Inc.) was used at a concentration of 50 ng/ml, and a horseradish peroxidase-conjugated donkey anti-goat antibody (Santa Cruz Biotechnology, Inc.) was used as secondary antibody at a concentration of 100 ng/ml.
Electrophoretic Mobility Shift Assays (EMSAs). Oligonucleotides used in EMSAs (Table 1) were designed to have 5′-GATC overhangs when annealed, allowing radioactive labeling by fill-in reactions. Fifty nanograms of annealed oligonucleotides were labeled in a 20-μl reaction containing 200 U of Superscript II RNase H– Reverse Transcriptase (Invitrogen), 1× First-Strand buffer (Invitrogen), 10 mM DTT, 250 nM dGTP/dCTP/dTTP, and 2 μCi of [α-32P]dATP (Amersham Biosciences). Unincorporated nucleotides were removed using Microspin G-25 columns (Amersham Biosciences Inc.). Ten micrograms of protein from whole cell extracts was used for individual DNA binding reactions. Approximately 50,000 cpm (0.5–1.0 ng) of the radioactive probes were used per reaction. Protein-DNA complexes were formed in binding buffer [20 mM Tris-HCl, pH 8.0, 60 mM KCl, 2 mM MgCl2, 12% (v/v) glycerol, 0.3 mM DTT, and 87.5 ng/ml poly(dI-dC) · poly(dI-dC)] in a total volume of 20 μl for 10 min at 30°C, with gentle shaking. In supershift experiments, 1 μg of the HNF-4α (H-171; Santa Cruz Biotechnology, Inc.) antibody was added to the extracts 1 h before the addition of the probe, and incubated at 4°C. In competition experiments, the indicated molar excess of the competing oligonucleotides was added simultaneously with the radioactive probe. Immediately after the binding reactions, the samples were loaded onto a pre-electrophoresed 5% (acrylamide/bis, 30:1) native acrylamide gel and run at 200 V in 0.5× Tris borate-EDTA for 1.5 h. After the runs, the gels were fixed in 10% (v/v) acetic acid for 10 min, rinsed with distilled water, dried down onto Whatman DE81 paper under vacuum, and exposed to Kodak BioMax MR-1 film (Sigma-Aldrich) at –70°C.
Results
Characterization of thehOAT2Promoter in Cell Lines. To study the function of the hOAT2 promoter in vitro, we subcloned DNA fragments of the human OAT2 promoter upstream of a luciferase reporter gene in the pGL3basic vector. Promoter constructs hOAT2(–3430/+54)luc, hOAT2(–1294/+54)luc, and hOAT2(–679/+54)luc and the promoterless vector pGL3basic were used to transfect the two human hepatoma cell lines Huh7 and HepG2 and the human ovarian teratocarcinoma cell line PA-1. As shown in Fig. 1, baseline promoter activity of all constructs containing regions from the proximal hOAT2 promoter were higher in liver-derived cell lines compared with the nonliver cell line PA-1. Low baseline activity of the hOAT2 promoter was also observed in the nonliver HeLa cell line, derived from cervical carcinoma (data not shown). Previous Northern blot analysis showed that the hOAT2 mRNA is expressed most abundantly in the liver (Sun et al., 2001). The data presented in Fig. 1 imply that liver-enriched transcription factors present in Huh7 and HepG2 cells, but absent in PA-1 cells, are required for baseline activity of the hOAT2 promoter and may determine the tissue distribution of the hOAT2 mRNA. Both in Huh7 and HepG2 cells, the promoter elements rendering high levels of reporter activity were contained within the proximal 679 base pairs upstream of the transcription start site. We thus searched for putative response elements for transcription factors within this region (Fig. 2A). By computer analysis [Genomatix MatInspector (www.genomatix.de) and NUBIScan; Podvinec et al., 2002] we identified a strong predicted binding site for the homodimeric nuclear receptor HNF-4α between nucleotides –329 and –317 relative to the hOAT2 transcription start site. This sequence, 5′-AGGACAaAGGGCA-3′, conforms well to the consensus site determined for HNF-4α (Fig. 2B) by a PCR-based binding site selection (Fraser et al., 1998) and is arranged as a DR-1 (direct repeat of hexameric elements separated by one base pair), which is the preferred configuration for DNA-binding by HNF-4α (Fig. 2B). It is interesting that upon aligning the rat Oat2 promoter sequence (GenBank accession no. L30107), we could identify a strong putative binding site for HNF-4α in the corresponding location relative to the transcription start site of the rat rOat2 gene (Fig. 2B). Given that the rat and human proximal OAT2 promoters are otherwise conserved to only a low degree, the clear interspecies conservation of the DR-1 element further supports its role as a functional element.

Analysis of baseline activity of reporter-linked hOAT2 promoter deletion variants in human liver-derived cell lines (HepG2 and Huh7) and in an ovarian cancer cell line (PA-1). Numbers at the left indicate the most upstream (–) and the most downstream (+) nucleotide included in the promoter constructs and are relative to the hOAT2 transcription start site. Reporter activities obtained for the promoterless construct pGL3basic in each cell line are set to 1, and activities of the hOAT2 promoter constructs are shown relative to this.

A, proximal promoter sequence of the hOAT2 gene. The start and end positions of the hOAT2 promoter deletions –679/+54 and –256/+54, as well as the transcriptional and translational start sites are indicated with arrows. The DR-1 element (–329/–317) identified in this study is shown framed. B, comparison of the hOAT2 DR-1 motif with the consensus element for the binding of HNF-4α (Fraser et al., 1998), as well as with the conserved DR-1 element in the rat Oat2 promoter. Also shown are the mutated variants of the hOAT2 DR-1 element with a disrupted HNF-4α binding motif. These same mutants were used in the functional analysis shown in Fig. 3.
The HumanOAT2Promoter Is Transactivated by HNF-4α through the DR-1 Element. To investigate whether the potential binding site for HNF-4α is a functional HNF-4α response element, we cotransfected Huh7 cells with hOAT2 promoter constructs in the absence or presence of an expression vector for HNF-4α. As shown in Fig. 3, the hOAT2(–679/+54)luc but not the hOAT2(–256/+54)luc construct was strongly transactivated by coexpression of HNF-4α. This is consistent with the location of the DR-1 element at nt –329 to –317, contained only in the hOAT2(–679/+54)luc construct.

HNF-4α transactivates the hOAT2 promoter construct containing the intact DR-1 motif at nt –329/–317. Huh7 cells were cotransfected with the indicated reporter-linked hOAT2 promoter variants, as well as the promoterless reporter vector pGL3basic. With each reporter construct, either a CMV promoter-driven expression plasmid for HNF-4α or an empty pcDNA3.1 vector (control) was cotransfected. To calculate the factor of induction by HNF-4α, the reporter activities obtained for pGL3basic either in the presence or absence of the HNF-4α expression plasmid were set to 1.
Next, we generated point mutations in the conserved residues within the DR-1 element. The hOAT2(–679/ +54mut1)luc variant contains two base changes in both of the hexameric repeats of the DR-1 element, and the hOAT2(–679/+54mut2)luc construct three base changes centered around the core base of the DR-1 element, as indicated in Fig. 2B. Both mutated variants of the DR-1 motif are predicted to be unable to bind HNF-4α (Fraser et al., 1998). Compared with the wild-type construct hOAT2(–679/ +54)luc, the reporter constructs harboring either of the mutated DR-1 elements are largely refractory to activation by coexpressed HNF-4α, confirming that the DR-1 motif is the HNF-4α responsive element within the hOAT2 promoter.
HNF-4α Binds to the DR-1 Element within the hOAT2 Promoter. To confirm that HNF-4α binds to the DR-1 element between nt –329 and –317 within the hOAT2 promoter, EMSA was performed using whole-cell protein extracts prepared from Huh7 cells and a double-stranded oligonucleotide that contained a consensus DR-1 binding motif for HNF-4α (Table 1) as a radiolabeled probe. Oligonucleotides used for competition assays contained the consensus binding site for HNF-4α, the wild-type DR-1 element from the hOAT2 promoter, or the mutated DR-1 element from the hOAT2 promoter corresponding to the mut1 used in reporter assays described above (Table 1). As shown in Fig. 4, the consensus HNF-4α and the hOAT2 wild-type DR-1 elements were efficient competitors, although hOAT2 DR-1 (wt) was less potent than the ideal binding motif (con) for HNF-4α (Fig. 4, lanes 3 and 4). The weaker binding of the hOAT2 DR-1 element may be attributable to the nonoptimal base guanine in position 4 of the second hexameric repeat—a position where thymidine, or to a lesser degree cytosine (Fig. 2B), would be preferred by HNF-4α. However, this competition experiment confirms that the functionally HNF-4α-responsive DR-1 element in the hOAT2 promoter also directly binds HNF-4α in cellular extracts. As expected, the mut1 variant of the DR-1 motif, shown to be functionally unresponsive to HNF-4α, was also inefficient in competing off HNF-4α binding (Fig. 4, lane 5). The specificity of the DNA-protein complex was confirmed by supershift analysis using an HNF-4α-specific antibody (Fig. 4, lane 1). Identical results to those shown in Fig. 4. were obtained when using protein extracts from HepG2 cells or in vitro-translated recombinant HNF-4α protein in electrophoretic mobility shift assays (data not shown).

HNF-4α binds to the DR-1 element (–329/–317) in the hOAT2 promoter. Electrophoretic mobility shift assays were performed using whole cell extracts from Huh7 cells and a consensus binding site for HNF-4α as a radiolabeled probe. In lane 1, the HNF-4α-specific antibody produces a supershift, confirming the identity of the DNA-protein complex shown in lane 2. A 50-fold molar excess of unlabeled consensus HNF-4α oligonucleotide abolished the binding of HNF-4α to the radiolabeled probe (con, lane 3). As shown in lane 4, the double-stranded oligonucleotide containing the intact DR-1 element from the hOAT2 promoter (wt) also efficiently competed with the radiolabeled probe for HNF-4α binding. A 50-fold molar excess of the oligonucleotide containing the same mutated variant of the hOAT2 DR-1 element as present in the hOAT2(–679/+54mut1)luc construct shown in Fig. 3 (mut) had no effect on the binding of HNF-4α to its consensus binding site (lane 5), further confirming that the DR-1 element in the hOAT2 gene binds HNF-4α specifically.
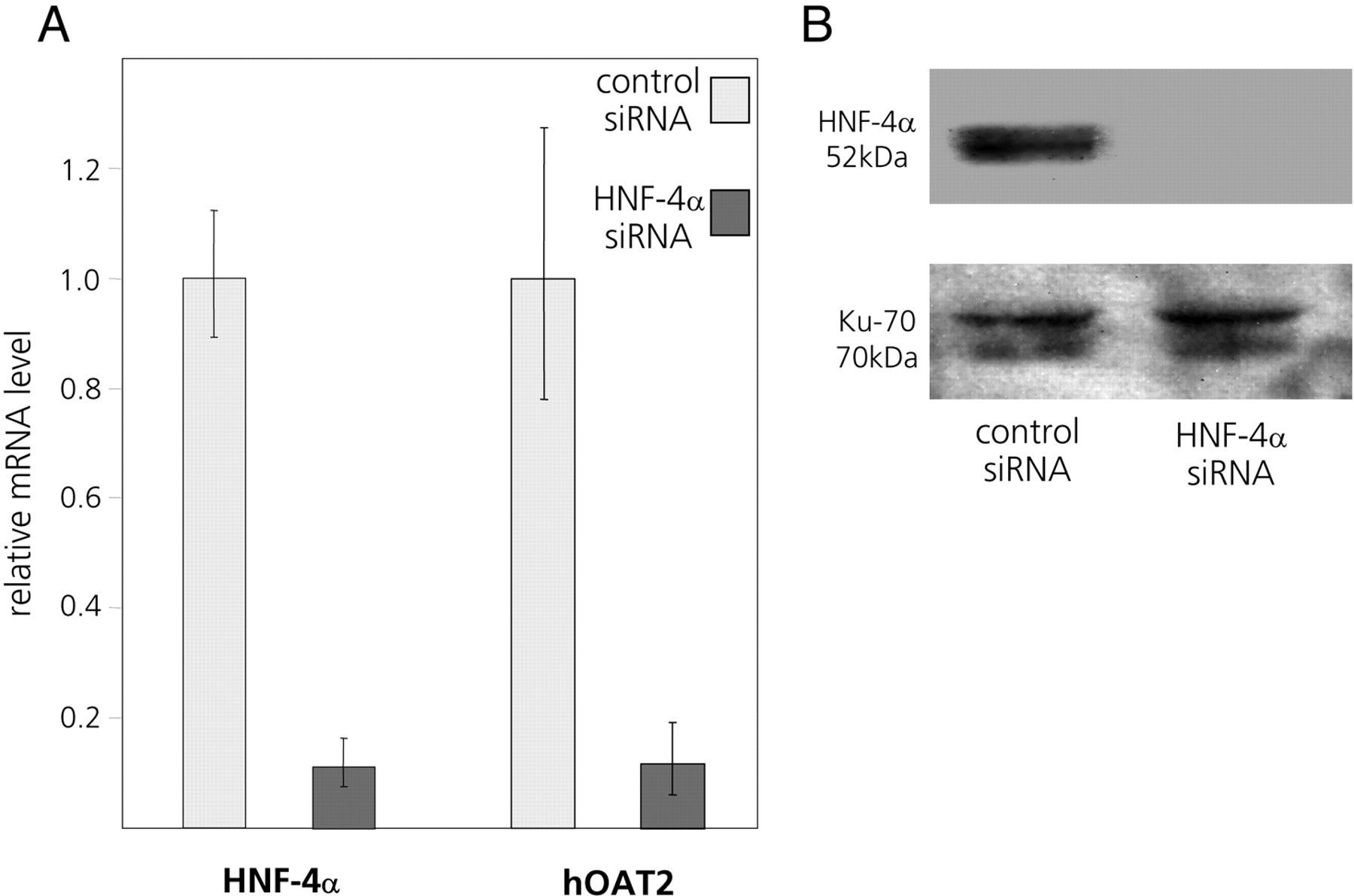
siRNA-Mediated Reduction in Cellular HNF-4α Levels Leads to Reduced Expression of the hOAT2 Gene. Having established that exogenous overexpression of HNF-4α activates the hOAT2 promoter in reporter-linked transient transfection assays, we next examined whether reducing the expression of endogenous HNF-4α decreases endogenous expression of the hOAT2 gene in cultured hepatoma cells. Huh7 cells were transfected with a pool of four HNF-4α-specific siRNAs. To control for possible nonspecific effects, we transfected an equal amount of control siRNAs designed so as not to specifically target any known human genes.
As shown in Fig. 5A, by transfecting the HNF-4α-specific siRNA pool, HNF-4α mRNA decreased to 10% compared with cells transfected with nontargeting siRNAs. HNF-4α protein was reduced to nondetectable levels (Fig. 5B). hOAT2 mRNA expression was reduced to a corresponding degree by the siRNAs targeting HNF-4α (Fig. 5A), confirming that HNF-4α is crucial for baseline expression of the hOAT2 gene. Of note, inhibition of endogenous hOAT2 gene expression in Huh7 cells by the HNF-4α-specific siRNAs is more pronounced than the reduction of the basal level of the reporter-linked hOAT2 promoter upon disruption of the DR-1 element (Fig. 3). This may imply that HNF-4α exerts further direct or indirect effects on the hOAT2 promoter via mechanisms not involving the DR-1 motif between nucleotides –329 and –317.

A pool of four HNF-4α-specific siRNAs reduces the endogenous mRNA levels of both HNF-4α and hOAT2 in Huh7 cells. A, transfection of siRNAs targeting HNF-4α efficiently reduces the levels of both HNF-4α and hOAT2 mRNA in Huh7 cells, compared with cells transfected with an equal amount of control siRNAs. B, immunoblot of protein samples isolated from siRNA-transfected Huh7 cells. siRNAs specific for HNF-4α abolish HNF-4α protein expression, whereas protein expression of the housekeeping gene Ku-70 remains unchanged.
Bile Acids Decrease Endogenous Expression of hOAT2 in Huh7 Cells. Bile acids are known to modulate the transcription of many genes encoding hepatic drug and bile acid transporters (for review, see Eloranta and Kullak-Ublick, 2005). This is likely to be particularly important to adjust the uptake and subsequent metabolism of drugs and bile acids to tolerable levels in cholestatic situations when bile acid levels within hepatocytes are elevated. One mechanism by which bile acids can repress gene transcription is through negative interaction with the transcriptional activator HNF-4α (Zhang and Chiang, 2001; Yang et al., 2002; Chen and Chiang, 2003). Because the hOAT2 gene is also activated by HNF-4α, we investigated whether treatment of hepatoma cells with the bile acid CDCA has an influence on the endogenous mRNA levels of hOAT2. Treatment of Huh7 cells with 50 μM CDCA for 24 h led to a clear decrease in the endogenous hOAT2 mRNA, compared with control cells treated with the vehicle DMSO (Fig. 6A). Bile acids such as CDCA activate several different signaling cascades and are ligands of the nuclear bile acid receptor FXR (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999). To investigate whether an FXR-mediated mechanism is involved in the down-regulation of hOAT2 expression, we used a synthetic agonist for FXR, GW4064 (Willson et al., 2001), which would not be expected to activate the FXR-independent pathways of bile acid signaling such as the c-Jun NH2-terminal kinase pathway (Gupta et al., 2001). Incubation of Huh7 cells with 200 nM GW4064 for 24 h also decreased the endogenous hOAT2 mRNA (Fig. 6A), suggesting that the CDCA-mediated suppression of hOAT2 transcription is indeed dependent on the activation of FXR.
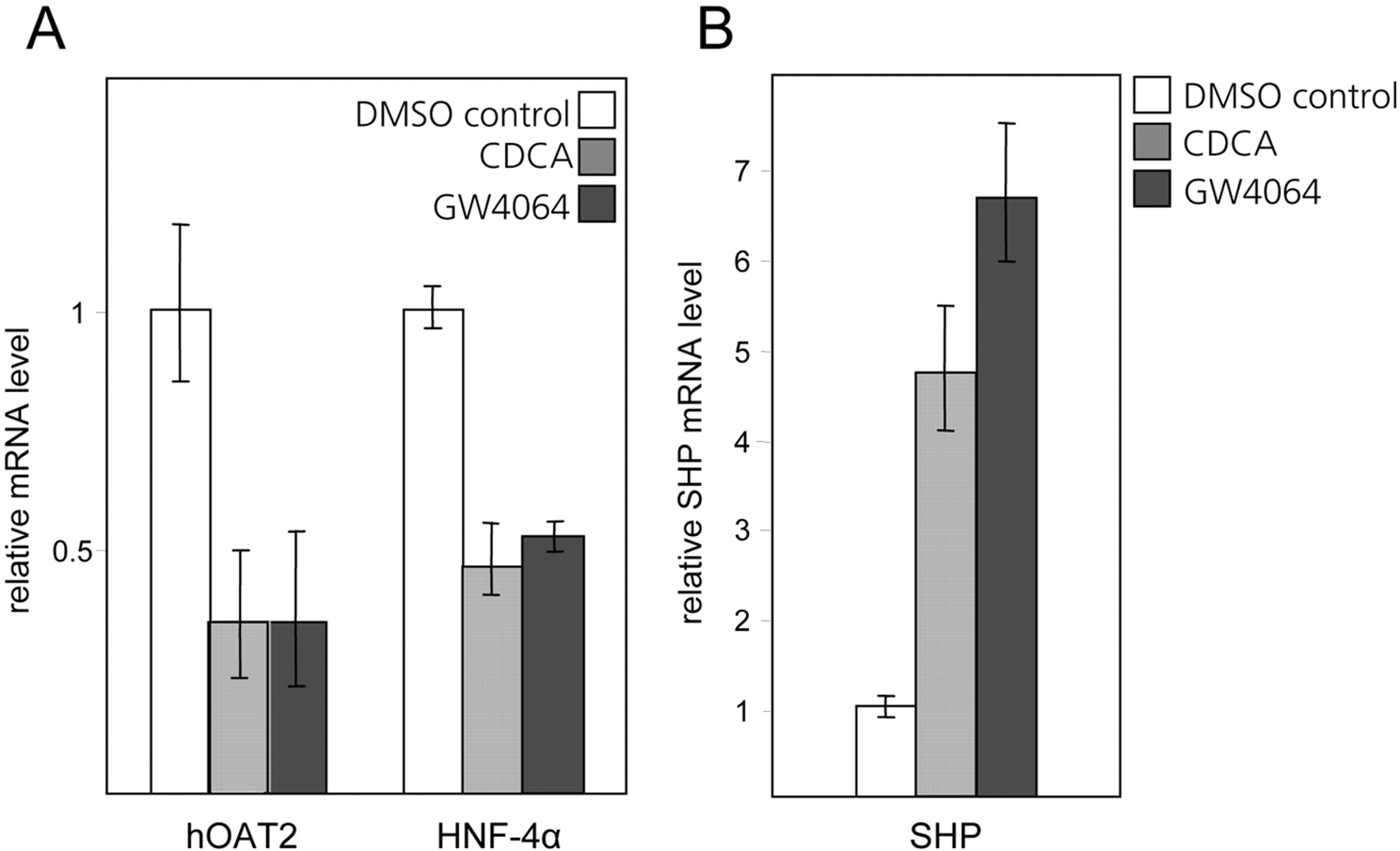
Treatment of Huh7 cells with either CDCA or the synthetic FXR agonist GW4064 decreases the endogenous mRNA levels of both hOAT2 and its transactivator HNF-4α, and induces SHP. A, Huh7 cells were treated with 50 μM CDCA, 200 nM GW4064, or the vehicle DMSO for 24 h, before isolating total cellular RNA. After reverse transcription, the cDNAs were analyzed by quantitative PCR using primers specific for hOAT2 and HNF-4α. The signals obtained for the DMSO-treated cells were set to 1, and values for the samples from CDCA- and GW4064-treated cells are shown relative to this. B, the same RNA preparations that were analyzed for hOAT2 and HNF-4α expression (A) were used to quantify changes in endogenous SHP mRNA levels by quantitative real-time PCR.
Bile Acid Treatment Decreases Cellular HNF-4α mRNA and Protein Levels. In parallel with assessing the changes in hOAT2 expression, we also measured the levels of endogenous HNF-4α mRNA after incubating Huh7 cells with either CDCA, GW4064, or the vehicle DMSO. It is interesting that, similar to hOAT2, HNF-4α mRNA levels are also suppressed by treatment with both the bile acid and the synthetic FXR agonist (Fig. 6A). This implies that FXR-dependent pathways are involved in down-regulating the endogenous mRNA level of both HNF-4α as well as its target gene hOAT2. To confirm the suppressive effect of the bile acid CDCA on HNF-4α protein expression, we performed immunoblot analysis with an antibody recognizing an epitope at the C terminus of HNF-4α. As shown in Fig. 7A, treatment of cells with 50 μM CDCA for 24 h decreased HNF-4α protein in both nuclear and whole-cell extracts compared with cells treated with the vehicle DMSO. HNF-4α was normalized for protein levels of the Ku-70 antigen that is constitutively expressed (Fig. 7A).
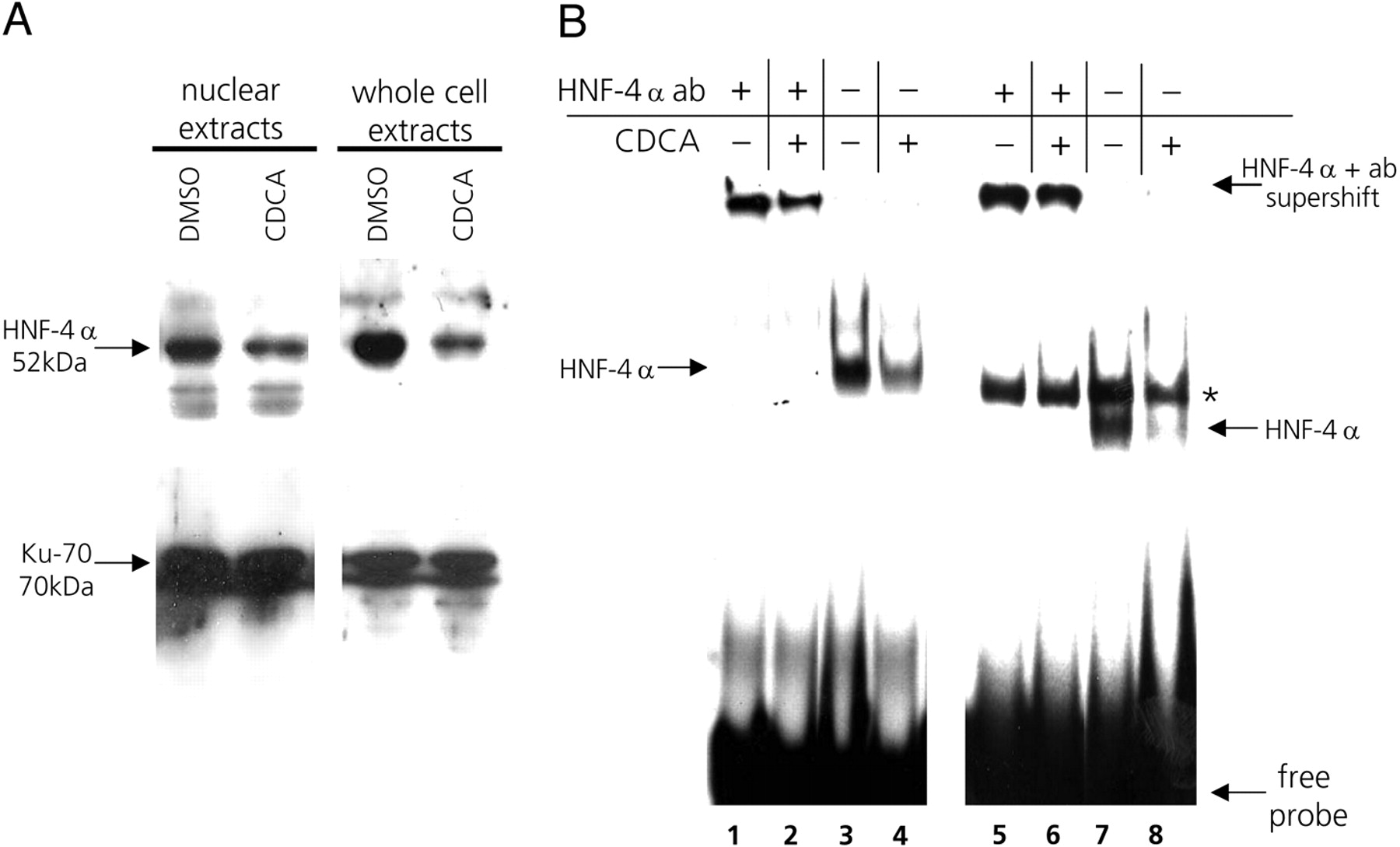
Treatment of Huh7 cells with CDCA decreases endogenous HNF-4α protein levels and leads to decreased DNA binding by HNF-4α. A, Huh7 cells were treated with 50 μM CDCA or with the vehicle DMSO for 24 h, after which proteins from either nuclei or whole cells were extracted. Samples of the protein extracts were subjected to immunoblot analysis with the HNF-4α-specific antibody. To verify equal loading of total protein in all samples, the blots were stripped and reprobed with an antibody against the constitutively expressed Ku-70 antigen. B, electrophoretic mobility shift assays using whole cell extracts prepared from cells treated with either 50 μM CDCA (lanes 2, 4, 6, and 8) or the vehicle DMSO (lanes 1, 3, 5, and 7) for 24 h. The radiolabeled probes contained either a consensus binding motif for HNF-4α (lanes 1–4) or the intact DR-1 element from the hOAT2 promoter (lanes 5–8). To identify the protein-DNA complexes specifically formed between HNF-4α and the radiolabeled probes, samples preincubated with an HNF-4α-specific antibody were included (supershift in lanes 1, 2, 5, and 6). The asterisk indicates an unidentified protein-DNA complex forming on the hOAT2 probe (lanes 5–8); however, this complex does not contain HNF-4α because it is not supershifted by the HNF-4α-specific antibody (lanes 5 and 6).
To further confirm that CDCA diminishes binding of HNF-4α to its response element in the hOAT2 promoter, we used the same Huh7 whole cell extracts in electromobility shift assays that were used for immunoblotting. When extracts from CDCA-treated cells were incubated with either the consensus HNF-4α oligonucleotide (Fig. 7B, lane 4) or the hOAT2 DR-1 oligonucleotide (Fig. 7B, lane 8), HNF-4α-DNA complex formation was decreased compared with extracts from DMSO-treated cells (Fig. 7B, lanes 3 and 7). The hOAT2 DR-1 element also forms a second complex with a protein of unknown identity (Fig. 7B, asterisk). However, this second complex of slower mobility is unrelated to HNF-4α, because it is not recognized by the HNF-4α-specific antibody (Fig. 7B, lanes 5 and 6).
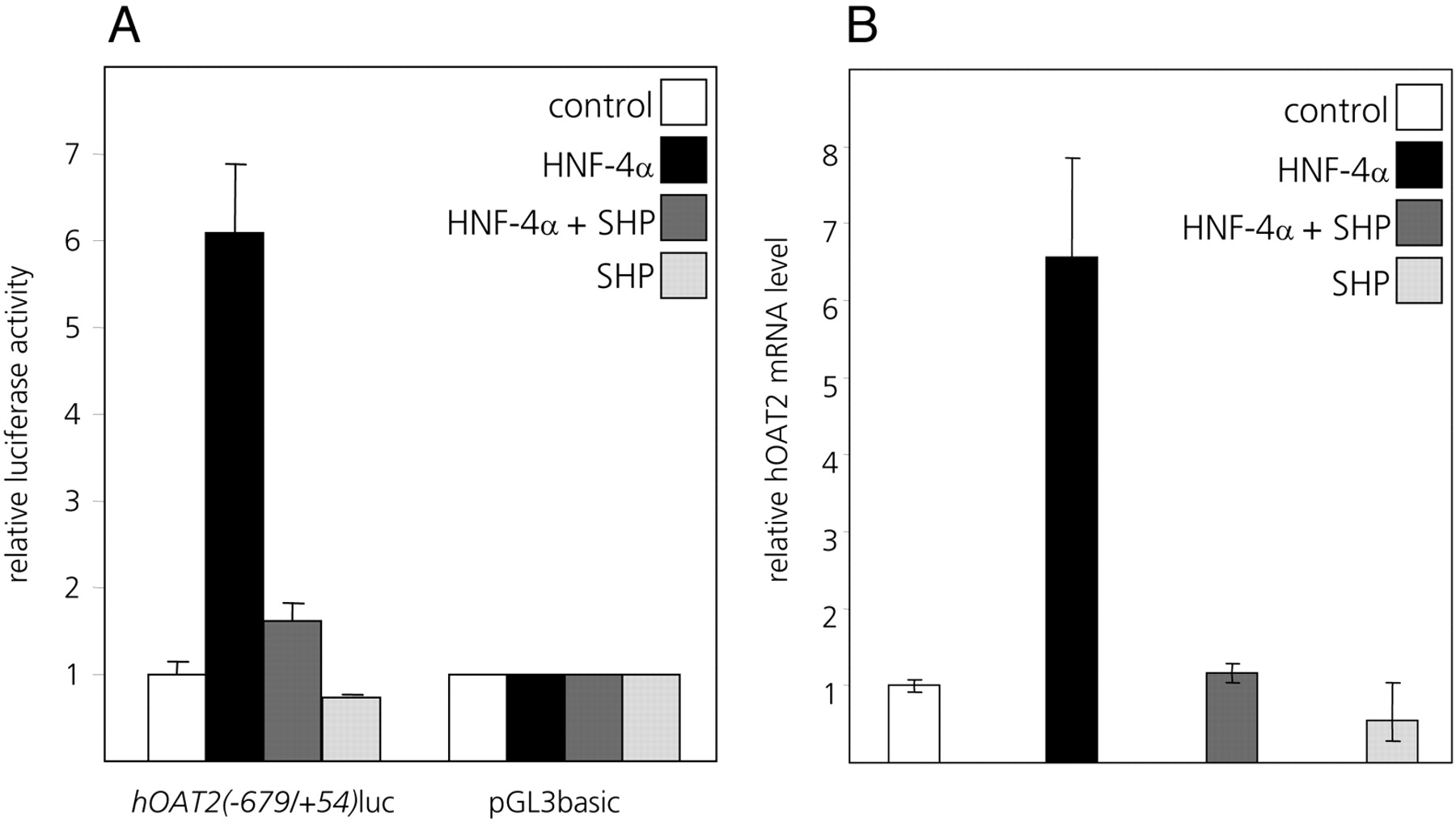
SHP Interferes with the HNF-4α-Mediated Transactivation of thehOAT2Promoter. FXR-mediated transcriptional repression often involves the FXR-induced transcriptional repressor SHP (Goodwin et al., 2000; Lu et al., 2000). As shown in Fig. 6B, endogenous levels of SHP mRNA were increased in cells treated with CDCA and GW4064. Because SHP has previously been shown to negatively interfere with HNF-4α-mediated transactivation (Lee et al., 2000; Zhang and Chiang, 2001; Yang et al., 2002; Chen and Chiang, 2003; Jung and Kullak-Ublick, 2003; Hirokane et al., 2004), we next investigated whether SHP is involved in the bile acid-mediated suppression of the hOAT2 promoter and whether it negatively targets activation of the hOAT2 promoter by HNF-4α. In Huh7 cells cotransfected with SHP and HNF-4α expression plasmids, the HNF-4α-mediated transactivation of the hOAT2(–679/+54)luc promoter construct was strongly suppressed (Fig. 8A). Cotransfection of SHP alone had a mild suppressing effect on hOAT2(–679/+54)-driven luciferase expression, possibly because of negative interference of SHP with endogenously expressed HNF-4α in Huh7 cells (Fig. 8A). Endogenous hOAT2 mRNA levels are also markedly induced in Huh7 cells upon cotransfection of HNF-4α expression plasmid, and this HNF-4α-mediated activation of the endogenous hOAT2 gene is abolished when SHP is cotransfected together with HNF-4α (Fig. 8B). These results further confirm that SHP exerts a repressive effect on the HNF-4α-mediated transactivation of the hOAT2 gene.
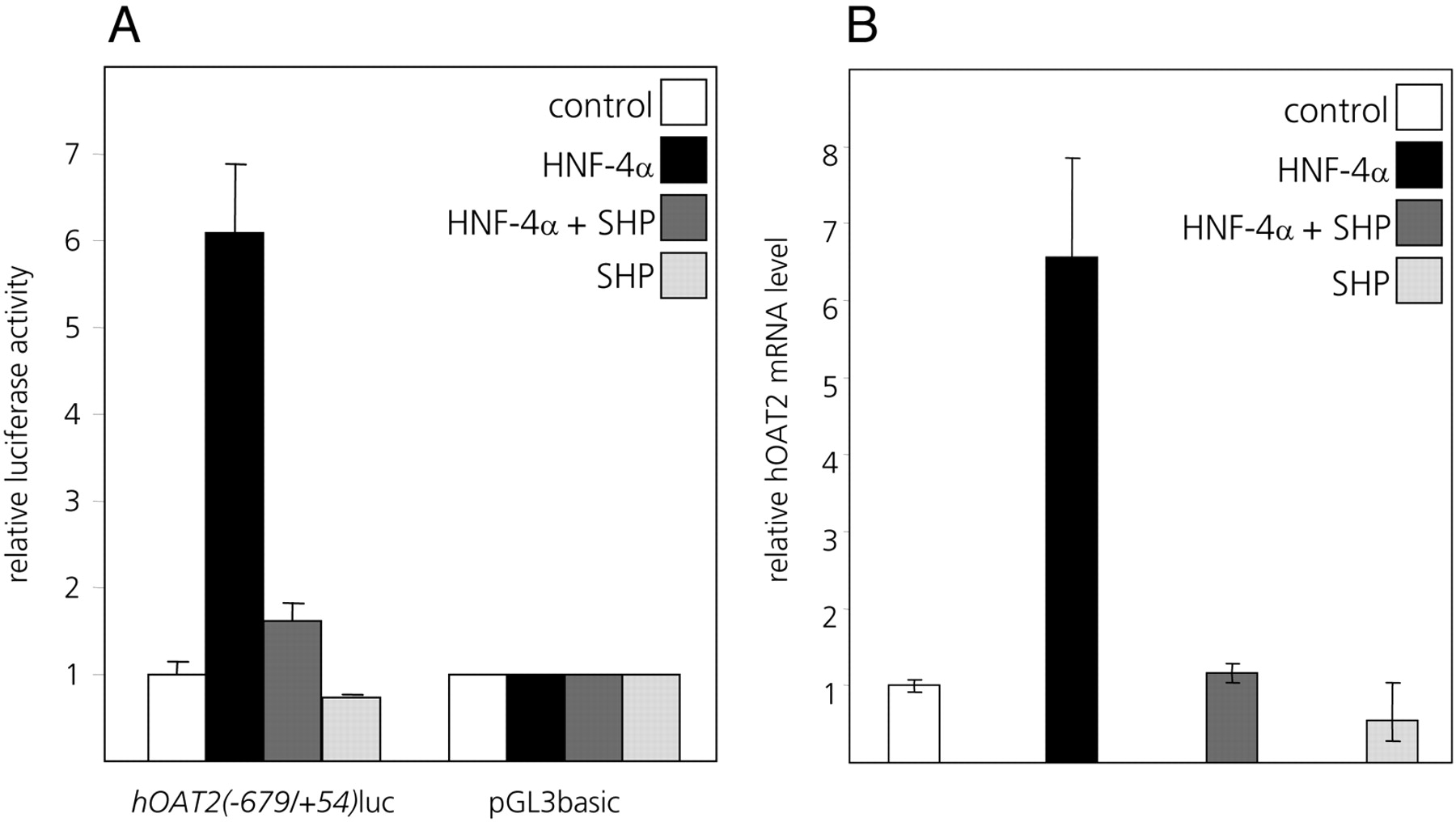
SHP down-regulates endogenous hOAT2 expression in Huh7 cells. A, HNF-4α-mediated transactivation of the human OAT2 promoter is inhibited by cotransfection of SHP in Huh7 cells. Cells were transfected with the hOAT2(–679/+54)luc construct or the promoterless vector pGL3basic. Together with the reporter constructs, cytomegalovirus promoter-driven expression vectors for HNF-4α or SHP or both were cotransfected. For all combinations of the cotransfected effector vectors, the values obtained for pGL3basic were set to 1, and the values obtained for the hOAT2(–679/+54)luc construct are shown relative to this. B, HNF-4α activated transcription of the endogenous hOAT2 gene can be suppressed by SHP. Cells were cotransfected with either HNF-4α or SHP expression construct alone, or both expression constructs together. After 24 h, total RNA from each transfectant was extracted and reverse-transcribed, and levels of endogenous hOAT2 expression were analyzed using quantitative real-time PCR with hOAT2-specific PCR primers.
Discussion
This study identifies HNF-4α as a transcriptional activator of the human SLC22A7 gene that codes for the liver-enriched drug transporter hOAT2. We show that both endogenous hOAT2 mRNA expression and reporter-linked hOAT2 promoter function are transactivated by exogenously expressed HNF-4α in the hepatoma cell line Huh7 (Figs. 3 and 8). When HNF-4α expression was reduced to 10% in Huh7 cells by siRNA targeted against HNF-4α, expression of the endogenous hOAT2 gene was suppressed to a corresponding degree (Fig. 5). A near-consensus HNF-4α binding site within the proximal hOAT2 promoter, arranged as a DR-1 element (nt –329/–317 relative to the hOAT2 transcription start site), is a functional mediator of HNF-4α transactivation of the hOAT2 gene (Figs. 2A and 3). It is of note that this element is conserved in a corresponding location in the rat Oat2 promoter, even if the overall conservation of the proximal promoters between species is low (Fig. 2B). Given this evolutionary conservation of the HNF-4α binding element, it is likely to serve a physiologically important role in the regulation of one of the major drug and xenobiotic transporters in mammalian hepatocytes.
During the completion of our study Odom et al. (2004), using chromatin immunoprecipitation assays combined with promoter microarrays, demonstrated that the hOAT2 promoter is one of more than 1500 promoters bound by HNF-4α in human primary hepatocytes. HNF-4α is a liver-enriched transcription factor of the nuclear receptor family known to have indispensable roles in hepatocyte differentiation and in the maintenance of hepatic gene expression. Target genes encode proteins involved in a wide range of physiological processes, notably cholesterol and glucose metabolism (Sladek et al., 1990; Cereghini, 1996; Hayhurst et al., 2001). HNF-4α is also a positive transcriptional regulator of the gene encoding HNF-1α (Tian and Schibler, 1991; Kuo et al., 1992), which similarly has a prominent role in the regulation of liver-specific genes. Most nuclear receptors require binding of an agonistic ligand for their transactivation ability; however, for HNF-4α, no ligands have been identified to date.
In this study, expression of the endogenous hOAT2 mRNA in cultured Huh7 cells was suppressed by treatment with the bile acid CDCA (Fig. 6A). Similarly to hOAT2, expression levels of several other hepatic transporters have previously been shown to be suppressed by bile acids. The basolateral OCT1 is down-regulated in obstructive cholestasis in rats (Denk et al., 2004), presumably because of a repressive effect of bile acids on gene transcription. In rodent models of cholestasis, both the mRNA and protein levels of the major basolateral sodium-dependent bile acid uptake system Na+-taurocholate cotransporting polypeptide are suppressed (Gartung et al., 1996; Dumont et al., 1997; Fickert et al., 2001; Wolters et al., 2002). We hypothesize that the decrease in the expression of hOAT2 and other basolateral drug or bile acid transporters serves to reduce the hepatic uptake of the respective transport substrates from sinusoidal blood when the liver is already burdened with a higher than desired bile acid load. Our finding also implies that the efficiency of hOAT2-mediated drug absorption may be impaired in cholestatic situations.
Bile acids affect transcription of their target genes chiefly by acting as ligands for the farnesoid X receptor (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999). Bile acids potentiate the transactivation ability of FXR, thus leading to induction of FXR target genes. One of the FXR target genes encodes the transcriptional repressor SHP (Seol et al., 1996). SHP is another orphan member of the nuclear receptor family, without a known ligand. SHP lacks a DNA binding domain, but it suppresses other DNA-binding transcription factors such as HNF-4α (Lee et al., 2000), through protein-protein interactions (for review, see Eloranta and Kullak-Ublick, 2005).
In this study, we show that the specific FXR ligand GW4064 is as effective as the bile acid CDCA in decreasing the endogenous expression of the hOAT2 mRNA in cultured hepatoma cells (Fig. 6A), indicating that the suppressive effect on hOAT2 expression is at least largely dependent on FXR. We further show that overexpression of the FXR-inducible transcriptional repressor SHP can counteract HNF-4α-mediated activation of both the endogenous and reporter-linked hOAT2 promoter (Fig. 8). The hOAT2 gene is not the first shown to be both transactivated by HNF-4α and negatively regulated by bile acids. The most notable examples of other such genes are those encoding the bile acid synthesizing enzymes CYP7A1 (De Fabiani et al., 2001, 2003), CYP8B1 (Zhang and Chiang, 2001; Yang et al., 2002), and CYP27A1 (Chen and Chiang, 2003). For the CYP8B1 gene, it has been shown that HNF-4α-mediated transactivation is suppressed by bile acids through both FXR- and SHP-dependent and -independent mechanisms (Sinal et al., 2000; Zhang and Chiang, 2001; Yang et al., 2002). It remains to be seen whether alternative or parallel FXR- and SHP-independent pathways are operative in the context of the hOAT2 promoter.
In addition to bile acids negatively affecting the transactivation potential of HNF-4α bound to the DR-1 element on the hOAT2 promoter, they also decrease HNF-4α mRNA and protein levels in Huh7 cells and consequently reduce nuclear HNF-4α binding (Fig. 7). Consistent with our results, bile acid feeding has previously been shown to decrease HNF-4α mRNA expression in rat liver (Yang et al., 2002). We further show that the down-regulation of the HNF-4α gene seems to be FXR-dependent, because the specific FXR agonist GW4064 is as effective as the bile acid CDCA in suppressing HNF-4α mRNA levels in Huh7 cells (Fig. 6A).
It is possible that FXR-induced SHP is responsible for the suppression of the HNF-4α gene, although the responsible transcription factor and its binding element within the HNF-4α promoter are still under investigation. The human HNF-4α promoter has been shown to be regulated by the coordinated action of the transcription factors HNF-1α, HNF-1β, HNF-6, and GATA-6 (Hatzis and Talianidis, 2001), but none of these factors have so far been reported to be direct targets for negative interference by SHP. It is worth noting that the HNF-1α promoter is regulated by HNF-4α, the latter being a frequent target for SHP. Indeed, bile acids are known to suppress HNF-1α-mediated activation of the gene encoding another transporter on the basolateral membrane of human hepatocytes, namely, OATP1B1 (SLCO1B1 gene), through a cascade where SHP negatively targets HNF-4α bound to the HNF-1α promoter (Jung and Kullak-Ublick, 2003). Apart from acting via a SHP-dependent mechanism, FXR may directly bind to a negative response element located in the HNF-4α promoter, a mechanism shown to be operative in the context of two human apolipoprotein genes (Claudel et al., 2002; Claudel et al., 2003).
In summary, we have shown that HNF-4α transactivates the hOAT2 promoter and that the bile acid CDCA decreases the transactivation potential of HNF-4α. Figure 9 shows a hypothetical scheme according to which bile acids negatively target the HNF-4α-mediated activation of hOAT2 by two parallel FXR-dependent mechanisms, resulting in decreased expression of hOAT2 in conditions associated with elevated intrahepatic concentrations of bile acids.

Potential pathways through which bile acids may target HNF-4α, resulting in suppression of the hOAT2 promoter. 1, bile acids bind to and activate FXR, which subsequently induces the expression of the transcriptional repressor SHP. SHP directly interferes with the transcriptional activator HNF-4α, which binds to the DR-1 element on the hOAT2 promoter. 2a, SHP may interfere with an as yet unidentified transactivator protein, which putatively binds to the HNF-4α promoter. This would result in decreased levels of the HNF-4α protein and consequently reduced transactivation of the hOAT2 promoter. 2b, bile acids may decrease the expression of the HNF-4α gene in a SHP-independent manner through direct binding of FXR to a negative response element within the HNF-4α promoter. This would also lead to a reduced degree of transcription from the hOAT2 promoter. act, activating nuclear factor; rep, repressing nuclear factor.
Acknowledgments
We thank Drs. David Moore, David Mangelsdorf, and Daniel Berger for generously donating the HNF-4α expression plasmid, SHP expression plasmid, and GW4064, respectively. The expert technical assistance of Claudia Seitz is gratefully acknowledged.
Footnotes
-
This work was supported by grant 632-062773 from the Swiss National Science Foundation.
-
Parts of this work were presented at the 55th Annual Meeting of the American Association for the Study of Liver Diseases and have been published in abstract form: Popowski K, Eloranta JJ, Fried M, Meier PJ, and Kullak-Ublick GA (2004) The human liver organic anion transporter 2 gene (hOAT2, SLC22A7) is transactivated by the hepatocyte nuclear factor HNF4α and is suppressed by bile acids through decreased nuclear HNF4α levels. Hepatology 40 (Suppl. 1):211A.
-
K.P. and J.J.E. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.010223.
-
ABBREVIATIONS: OAT, organic anion transport; OATP, organic anion-transporting polypeptide; hOAT2, human organic anion transporter 2; HNF, hepatocyte nuclear factor; nt, nucleotide(s); FXR, farnesoid X receptor; PCR, polymerase chain reaction; DR, direct repeat; FBS, fetal bovine serum; CDCA, chenodeoxycholic acid; DMSO, dimethyl sulfoxide; SHP, small heterodimer partner; siRNA, short interfering RNA; PBS, phosphate-buffered saline; DTT, dithiothreitol; PBS-T, phosphate-buffered saline/Tween 20; EMSA, electrophoretic mobility shift assay.
- Received December 13, 2004.
- Accepted February 2, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}