


Abstract
Cannabinoids, the active components of marijuana and their endogenous counterparts, exert many of their actions in brain through the seven-transmembrane receptor CB1. This receptor is coupled to the activation of the extracellular signal-regulated kinase (ERK) cascade. However, the precise molecular mechanism for CB1-mediated ERK activation is still unknown. Here, we show that in U373 MG human astrocytoma cells, CB1 receptor activation with the cannabinoid agonist Δ8-tetrahydrocannabinol dimethyl heptyl (HU-210) was coupled to ERK activation and protection from ceramide-induced apoptosis. HU-210-induced ERK activation was inhibited by tyrphostin AG1478 and PP2, widely employed inhibitors of the epidermal growth factor receptor (EGFR) and the Src family of cytosolic tyrosine kinases, respectively. However, HU-210 stimulation resulted in neither EGFR phosphorylation, Src tyrosine phosphorylation, nor increased Src activity. In addition, dominant-negative forms of both proteins were unable to prevent cannabinoid-induced ERK activation, thus excluding the existence of CB1-mediated EGFR transactivation or Src activation. Wortmannin and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294,002), inhibitors of the phosphatidylinositol 3-kinase (PI3K) signaling pathway, blocked cannabinoid-induced ERK activation. Likewise, HU-210 stimulated the PI3K downstream targets protein kinase B (PKB), as shown by its phosphorylation in Thr 308 and Ser 473 residues, and Raf-1. Moreover, βγ subunit release mimicked ERK and PI3K/PKB activation, suggesting that activation of class IB PI3K mediates cannabinoid action. Pro-survival HU-210 action also required activation of both PI3K and ERK signaling pathways. In conclusion, CB1-induced ERK activation was mediated by PI3KIB and this effect may have important consequences in the control of cell death/survival decision.
Cannabinoids, the active components of marijuana and their endogenous counterparts exert most of their actions through two well characterized seven-transmembrane receptors designated CB1 and CB2 (Matsuda et al., 1990; Munro et al., 1993). The CB1 receptor is highly expressed in the central nervous system and is also present in peripheral and extraneural sites (Piomelli et al., 2000; Porter and Felder, 2001). In contrast, the CB2 receptor is almost restricted to the immune system. The CB1 receptor is known to be coupled to inhibition of adenylyl cyclase, inhibition of voltage-dependent Ca2+ channels, and activation of G-protein-regulated inwardly rectifying K+channels (Howlett, 1995; Porter and Felder, 2001). Besides these well established signal transduction events, novel possibilities have arisen to explain cannabinoid regulation of cell death/survival decision (Guzmán et al., 2001a,b). Thus the CB1receptor has been shown to regulate different members of the mitogen-activated protein kinase family, such as extracellular signal-regulated kinase (ERK) (Bouaboula et al., 1995; 1997;Sánchez et al., 1998), c-Jun N-terminal kinase (Liu et al., 2000;Rueda et al., 2000), and p38 (Galve-Roperh et al., 2000; Rueda et al., 2000). The CB1 receptor can also activate the phosphatidylinositol 3-kinase/protein kinase B (PI3K/PKB) signaling pathway (Gómez del Pulgar et al., 2000) and focal adhesion kinase (Derkinderen et al., 1996; 2001). In addition, the CB1 receptor modulates sphingolipid metabolism, leading to increased ceramide levels by either activating sphingomyelin hydrolysis (Sánchez et al., 1998; 2001) or enhancing ceramide synthesis de novo (Gómez del Pulgar et al., 2002).
A complex variety of mechanisms has been described to explain ERK regulation by seven-transmembrane receptors (reviewed in Gschwind et al., 2001; Marinissen and Gutkind, 2001; Pierce et al., 2001). In the case of Gi-coupled receptors such as CB1, several mechanisms could be hypothesized to mediate ERK activation, including: 1) transactivation of growth factor receptors with intrinsic tyrosine kinase activity (Gschwind et al., 2001); 2) β-arrestin-mediated receptor internalization and recruiting of cytosolic tyrosine kinases of the Src family (Pierce et al., 2001;Marinissen and Gutkind, 2001); 3) G protein-dependent class IB PI3K activation (Lopez-Ilasaca et al., 1997; Marinissen and Gutkind, 2001). In the present work, we investigated how the CB1receptor is coupled to ERK activation and its implications in cannabinoid regulation of glial cell death/survival decision.
Materials and Methods
Reagents.
The following materials were kindly donated: pcDNA3-CD533 encoding truncated EGFR by Dr. A. Ullrich (Max-Planck Institute, Martinsried, Germany), pcDNA3-Src dominant-negative K295R by Dr. F. Mayor Jr. (Autónoma University, Madrid, Spain), the fusion construct of glutathioneS-transferase and the Ras binding domain of Raf (GST-Raf-RBD) by Dr. J.L. Bos (Utrecht University, Utrecht, The Netherlands), SR141716 by Sanofi Synthelabo (Montpellier, France), HU-210 by Dr. R. Mechoulam (Hebrew University, Jerusalem, Israel), Src substrate peptide by Dr. F. Barahona (Autónoma University, Madrid, Spain), and BB94 by Dr. C. López Otı́n (Oviedo University, Oviedo, Spain). Tyrphostins AG1296 and AG1478 were from Calbiochem (San Diego, CA); PD98059, wortmannin and LY294,002 from Alexis Biochemicals (San Diego, CA); polyclonal anti-Raf-1, Src, and Fyn antibodies as well as monoclonal anti-phospho-ERK antibody from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal anti-EGFR antibody and monoclonal anti-phosphoTyr1173-EGFR (pY1173- EGFR) and anti-α-tubulin antibodies from Upstate Biotechnology (Lake Placid, NY); anti-phosphotyrosine monoclonal antibody clone PY20 from BD Transduction Laboratories (Lexington, KY); polyclonal anti-phosphoThr308-PKB (pT308-PKB) and phosphoSer473-PKB (pS473-PKB) from Cell Signaling Technology (Beverly, MA); and anti-phosphoTyr 416-Src (pY416-Src) from Calbiochem.
Cell Culture and Transfection.
Human astrocytoma U373 MG cells were cultured as described previously (Rueda et al., 2000). Cells were transferred to serum-free medium 12 h before the experiments. Stock solutions of cannabinoids were prepared in dimethyl sulfoxide and control incubations had the corresponding dimethyl sulfoxide content. No significant influence of the vehicle was observed at the final concentration used (0.1%, v/v) in any of the parameters determined. Cells were stably transfected with LipofectAMINE 2000 (Invitrogen, Carlsbad, CA) with the aforementioned constructs, and transfected cells were selected and maintained in culture in the presence of 0.5 mg/ml G418. At least three different clones of stably transfected cells were assessed for each dominant negative construct, obtaining similar results.
Apoptosis and Cell Viability.
U373 MG cells were incubated in the presence of 10 μM C2-ceramide for 1 h, medium was removed, and cells were subsequently incubated for 15 h in the presence of cannabinoids with different pharmacological inhibitors. Apoptosis was quantified by determination of the content of oligonucleosomal DNA fragments, a hallmark of apoptotic cell death using an enzyme-linked immunosorbent assay according to manufacturer instructions (Roche Applied Science, Mannheim, Germany). Cell lysates were incubated for 90 min in a 96-well microtiter plate previously coated with an anti-histone antibody clone H11–4. After washing, the plate was incubated with a mouse anti-DNA-peroxidase–labeled antibody and peroxidase activity was determined by measuring absorbance at 405 nm after substrate incubation. In addition, for some experiments, cell viability was determined by Trypan blue exclusion.
Western Blot Analysis.
Western blots were performed as described previously (Galve-Roperh et al., 2000). After stimulation, cells were washed with ice-cold phosphate-buffered saline (phosphate-buffered saline; 10 mM NaPi, 150 mM NaCl, pH 7.4) and scraped in lysis buffer consisting of 50 mM Tris-HCl, pH 7.5, 1% (v/v) Triton X-100, 1% (w/v) sodium deoxycholate, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 10 mM sodium β-glycerophosphate, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 0.1% (v/v) 2-mercaptoethanol, 0.5 μM microcystin-LR, 17.5 μg/ml phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 20 μg/ml soybean trypsin inhibitor, and 5 μg/ml benzamidine. Cell lysates cleared by 15 min of centrifugation at 12,000g were employed for the different experimental procedures. After protein normalization samples were subjected to 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Galve-Roperh et al., 2000). EGFRblotting was performed using 8% SDS-PAGE and high-voltage transfer conditions to allow high-molecular weight proteins to be efficiently transferred. After incubation with primary antibodies (1:1000), blots were developed with appropriate horseradish peroxidase-coupled secondary antibodies (1:20,000) and enhanced chemiluminescence detection kit. Loading controls were performed with an anti-α-tubulin antibody. Densitometric analysis of the luminograms was performed using a GS-700 Imaging Densitometer (Bio-Rad, Hercules, CA) and MultiAnalyst software. Relative values of optical density of the representative experiment shown are included in the figures.
Immunoprecipitation and Src Activity Assay.
Cleared cell lysates (500 μg protein) were incubated with 2 μg of anti-EGFR, Src, or Fyn antibodies precoupled to protein G-Sepharose. After washing with lysis buffer, EGFR and Src phosphorylation status was determined in the immunoprecipitate by Western blot using the PY20 antibody. In addition, Western blot analyses of total cell extracts were performed with an anti-pY1173-EGFR or anti-pY416-Src antibody. For Src activity assays, immunoprecipitated proteins were transferred to Src activity buffer consisting of 100 mM Tris-HCl, pH 7.2, 50 mM magnesium acetate, 10 mM MnCl2, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol, and 0.5 μM microcystin-LR, 17.5 μg/ml phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 20 μg/ml soybean trypsin inhibitor, and 5 μg/ml benzamidine. Kinase activity was determined in the presence of 200 μM ATP, 5 μCi of [γ−32P]ATP, and 500 μM substrate peptide corresponding to amino acids 6 to 20 of p34cdc2. Endogenous phosphorylation was also determined in parallel. After 10 min, reactions were stopped with 75 mM orthophosphoric acid, samples were centrifugated, and supernatants spotted on Whatman P81 filter paper (Whatman, Maidstone, UK). Filters were washed and radioactivity was determined by liquid scintillation counting.
Ras Activation.
The fusion construct GST-Raf-RBD was overexpressed in E. coli DH5α following standard procedures. Cleared cell lysates (250 μg of protein) were incubated in the presence of GST-Raf-RBD precoupled to agarose-glutathione complexes (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) to detect activated RasGTP. Beads were extensively washed with phosphate-buffered saline and, after SDS-PAGE, immunoblotting was performed using monoclonal anti-pan-Ras antibody (Oncogene, Boston, MA).
Raf-1 Activity.
Raf-1 activity assays were performed as described previously (Galve-Roperh et al., 2000). In summary, immunoprecipitated Raf-1 was incubated in kinase buffer in the presence of 1 μg of myelin basic protein and [γ−32P]ATP, reactions were stopped with SDS sample buffer and substrate phosphorylation was determined after SDS-PAGE and autoradiography.
Statistical Analysis.
Results shown represent the means ± S.D. of the number of experiments indicated in every case. Statistical analysis was performed by analysis of variance. A post hoc analysis was made by the Student-Neuman-Keuls test.
Results
CB1-Induced ERK Activation Protects Astrocytoma Cells from Ceramide-Induced Apoptosis.
To elucidate the mechanism of cannabinoid activation of the ERK pathway, we first examined cannabinoid-induced ERK activation in the presence of different pharmacological inhibitors using a monoclonal anti-phospho-Tyr 204 antibody that recognizes phosphorylated ERK. CB1stimulation with the cannabinoid agonist HU-210 resulted in a time-dependent ERK activation (data not shown) that peaked at 10 min (Fig. 1A). This effect was completely prevented by the selective CB1 antagonist SR141716, thus evidencing the involvement of this receptor. To determine whether transactivation of receptors with intrinsic tyrosine kinase activity such as EGFR or platelet-derived growth factor receptor could be responsible for CB1-induced ERK activation, we employed inhibitors of tyrosine kinase activity with different selectivity. Tyrphostin AG1478, a selective EGFR inhibitor, but not tyrphostin AG1296, a selective platelet-derived growth factor receptor inhibitor, abrogated HU-210–induced ERK activation (Fig. 1A). In addition, the pyrazolopyrimidine PP2 and herbimycin A, two inhibitors of the Src family of cytosolic tyrosine kinases, blocked cannabinoid-induced ERK activation (Fig. 1A). The concentration-dependent action of tyrphostin AG1478 and PP2 on HU-210-induced ERK activation was determined (data not shown), allowing us to calculate IC50 values of 0.1 and 0.8 μM, respectively.

CB1-induced ERK activation protects astrocytoma cells from ceramide-induced apoptosis. A, U373 MG cells were incubated with 50 nM HU-210 for 10 min either alone or after 30-min preincubation with 1 μM SR141716, 1 μM tyrphostin AG1478, 30 μM tyrphostin AG1296, 10 μM herbimycin A (HerbA), or 10 μM pyrazolopyrimidine PP2. Cell lysates were subjected to Western blot analysis with an anti-phospho-ERK antibody. Loading control was carried out with an anti–α-tubulin antibody. O.D., optical density. B, U373 MG cells were incubated in the presence of 10 μM C2-ceramide (C2-CER) for 1 h, the medium was removed, and cells were incubated for 15 h in the presence of vehicle or 50 nM HU-210. Apoptosis was quantified by determination of oligonucleosomal DNA fragmentation. C, after C2-ceramide–induced apoptotic stimulation, U373 MG cells were incubated in the presence of HU-210, CP-55940, or WIN-55,212-2 (50 nM). WIN-55,212-2 incubations were also performed in the presence of 1 μM SR141716, 25 μM PD98059, or 1 μM tyrphostin AG1478. Cell viability was determined 15 h later by Trypan blue exclusion. Results correspond in A to a representative experiment of six independent experiments and in B and C to three and four different experiments, respectively. *, P < 0.01, significantly different from vehicle incubations.
The important role of the mitogenic ERK cascade in the control of cell death/survival decision (Grewal et al., 1999; Kolch, 2000) prompted us to investigate the functional consequences of CB1-induced ERK activation in astrocytoma cells. Cells incubated with the permeable ceramide analog C2-ceramide underwent apoptosis (Fig. 1B), and HU-210 inhibited this process. Moreover, exposure to other synthetic cannabinoids, such as CP-55940 and WIN-55,212-2 mimicked the antiapoptotic action of HU-210 (Fig. 1C). Cannabinoid pro-survival action relied on CB1 receptor activation, as evidenced by SR141716 antagonism (Fig. 1C). To investigate the potential involvement of the ERK cascade in CB1-mediated protection from apoptosis, we employed the specific mitogen-activated protein kinase kinase inhibitor PD98059. This compound abrogated cannabinoid prosurvival action, thus indicating the involvement of the ERK signaling pathway. Moreover, like cannabinoid-induced ERK activation, cannabinoid prosurvival effect was also inhibited by tyrphostin AG1478. These data show that CB1-induced ERK activation is functionally relevant in our experimental model and results in cell protection from apoptosis.
CB1-Induced ERK Activation Does Not Involve EGFR Transactivation.
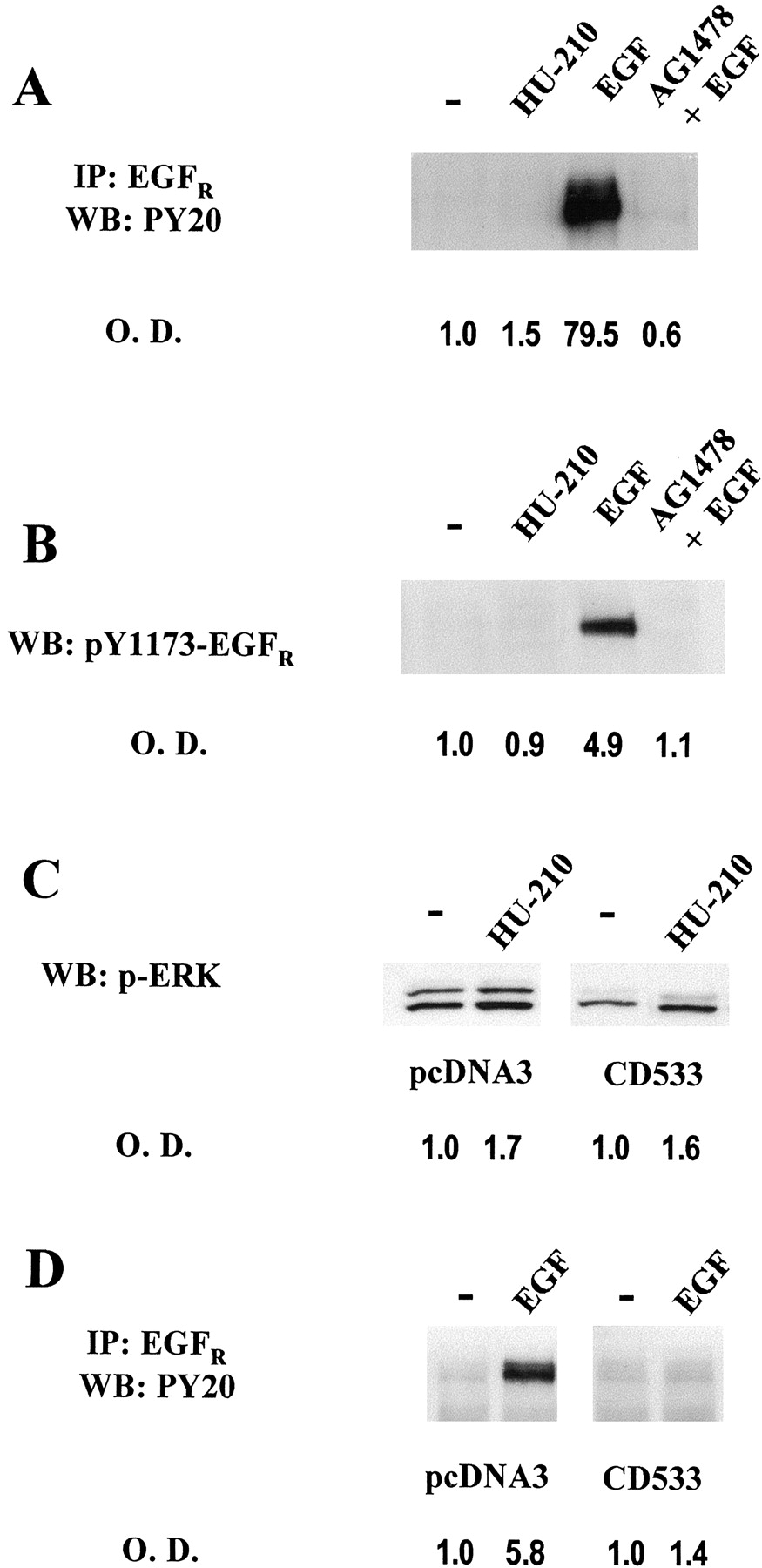
Next, we examined whether CB1-mediated transactivation of EGFR could be responsible for ERK activation. Although tyrphostin AG1478 prevented HU-210-induced ERK activation (Fig. 1A), HU-210 was not able to induce EGFRphosphorylation (Fig. 2A). Under the same experimental conditions, EGF potently increased tyrosine phosphorylation of immunoprecipitated EGFR, and this effect was indeed blocked by tyrphostin AG1478. Similar results were obtained with an antibody against activated EGFR that specifically recognizes phosphorylated Tyr 1173. HU-210 was ineffective in eliciting EGFR phosphorylation, whereas EGF induced EGFR phosphorylation (Fig. 2B). The apparent contradiction of the results obtained with tyrphostin AG1478 in ERK activation assays and EGFR phosphorylation experiments required further analysis. Therefore, we stably transfected U373 MG cells with CD533, a truncated form of EGFR that acts as a dominant negative form (Redemann et al., 1992). CD533-U373 MG cells did not show reduced cannabinoid-induced ERK activation (Fig. 2C), in line with the lack of involvement of EGFR in HU-210-induced ERK activation. In contrast, EGF-induced EGFRphosphorylation was effectively abrogated (Fig. 2D), evidencing the effectiveness of the stably generated CD533-U373 MG cells to block EGF signaling. In addition, BB94, an inhibitor of zinc-dependent metalloproteinases responsible for proheparin binding-EGF shedding required for G-protein-coupled receptor transactivation of EGFR (Prenzel et al., 1999), did not prevent HU-210-induced ERK activation (data not shown), supporting the absence of CB1-mediated EGFRtransactivation.

CB1-induced ERK activation does not involve EGFR transactivation. O.D., optical density. A, after stimulation with 50 nM HU-210 or 100 ng/ml EGF in the absence or presence of 1 μM tyrphostin AG1478, U373 MG cell extracts were immunoprecipitated (IP) with an anti-EGFR antibody and Western blot (WB) was performed using anti-phosphotyrosine PY20 antibody. B, cell lysates from stimulated cells were subjected to Western blot analysis using an anti-phospho-Tyr-1173-EGFRantibody. C, Western blot analysis of phospho-ERK in vehicle or HU-210–stimulated cell lysates from vector- and CD533-stably transfected U373 MG cells. D, tyrosine phosphorylation status of immunoprecipitated EGFR in vector- and CD533-U373 MG cells after EGF stimulation. Representative luminograms are shown. Similar results were obtained in at least two other experiments for each.
Lack of Involvement of Src Tyrosine Kinases in CB1-Induced ERK Activation.
To investigate the potential involvement of the Src family of tyrosine kinases in CB1 signaling, kinase activity assays were performed. Immunoprecipitation from U373 MG cell extracts of Src, Fyn, and Yes using an anti-pan Src antibody did not reveal any increased activity of those kinases by HU-210 stimulation, whereas under the same conditions, PDGF induced a robust Src activation(Fig.3A). Moreover, time course analysis from 5 to 60 min did not evidence stimulation of pan-Src activity by HU-210 (data not shown). As cannabinoid-induced focal adhesion kinase regulation in hippocampal neurons has been proposed to be specifically mediated by Fyn (Derkinderen et al., 2001), we examined Fyn kinase activity using a specific antibody against this protein. However, no changes in Fyn activity could be detected (Fig. 3A).

Lack of involvement of Src tyrosine kinases in CB1-induced ERK activation. O.D., optical density. A, Src family (Src, Fyn, and Yes, ■) or Fyn activity (▪) as determined by immunoprecipitation (IP) with an anti-pan-Src antibody or anti-Fyn antibody from lysates of cells stimulated with 50 ng/ml PDGF for 5 min or 50 nM HU-210 for 10 min. B, tyrosine phosphorylation status of Src after cell stimulation. Top, Src Western blot of PY20 immunoprecipitates; bottom, phosphorylated Tyr 416 of Src in total cell extracts using an specific antibody. C, Western blot analysis with anti-phospho-ERK antibody in vector and dominant-negative Src-K295R stably transfected U373 MG cells after vehicle or HU-210 stimulation. D, overexpression of Src in stable Src-K295R-transfected cells as evidenced by Western blot with an anti-Src antibody. Results correspond to six different experiments (A), and representative luminograms of three (B), six (C), and three (D) independent experiments.
To fully address the question of cannabinoid regulation of Src, we have also analyzed its phosphorylation status. Immunoprecipitation with anti-PY20 antibody and subsequent detection with the Src antibody revealed an increased global tyrosine phosphorylation of Src induced by PDGF but not HU-210 (Fig. 3B). Because tyrosine phosphorylation can reflect either increased or blockade of Src activity (Tyr 416 versus Tyr 527; Martin, 2001) we have also employed a monoclonal antibody against phosphorylated Tyr 416 of Src that reflects the activation status of the protein. Again, Western blot results show the lack of phosphorylation/activation of Src by HU-210 stimulation (Fig.3B). Moreover, enforced expression of dominant negative Src-K295R did not affect HU-210–induced ERK activation (Fig. 3C), whereas it efficiently blocked PDGF stimulation of Src activity (data not shown) because of its overexpression in transfected cells (Fig. 3D).
Involvement of the PI3K/PKB Pathway in CB1-Induced ERK Activation and Cell Survival.
Because G protein βγ subunit release may activate class IB PI3K leading to ERK activation (Lopez-Ilasaca et al., 1997; Marinissen and Gutkind, 2001), we examined whether this process occurred upon CB1 activation by employing wortmannin and LY294,002, two PI3K inhibitors. As shown in Fig. 4A, HU-210-induced ERK activation was completely abrogated by PI3K inhibition. Moreover, mastoparan-induced G protein βγ subunit release led to ERK activation, and such effect was not additive to HU-210 action (Fig.4A). Support for the involvement of the PI3K signaling pathway was obtained by showing that its primary downstream target PKB was regulated in a similar manner. Phosphorylation of PKB occurs in different residues that represent progressive steps in its activation mechanism (Brazil and Hemmings, 2001). Thus, HU-210 challenge induced PKB Thr 308 phosphorylation (Fig. 4B), which reflects PI3K/PDK1-mediated phosphorylation. HU-210 also induced PKB Ser 473 phosphorylation (Fig. 4B), indicative of the last activation step upon PI3K stimulation. Phosphorylation of both residues was antagonized by SR141716, pointing to the involvement of CB1receptor. In addition, PI3K inhibition by wortmannin and LY294,002 abrogated HU-210–induced phosphorylation of both PKB residues and therefore validated ERK data obtained with these inhibitors. Moreover, mastoparan-induced βγ subunit release increased PKB phosphorylation in both residues, and this effect was not additive to that of HU-210 (Fig. 4B). We next determined whether Ras was involved in ERK activation given that PI3K-dependent ERK activation might be mediated by this monomeric G protein (Lopez-Ilasaca et al., 1997). However, HU-210 stimulation did not induce Ras activation in U373 MG cells (Fig.4C), pointing to a PI3K-dependent mechanism of ERK activation that does not involve Ras activation. A similar mechanism has been proposed for PI3K activation of the ERK cascade by direct PAK-mediated Raf-1 phosphorylation (King et al., 1998; Chaudhary et al., 2000; Zang et al., 2001). Likewise, CB1 receptor activation enhances Raf-1 activity in U373 MG cells (Fig. 4D), similarly to what occurs in primary astrocytes (Sánchez et al., 1998) and C6 glioma cells (Galve-Roperh et al., 2000).

Involvement of the PI3K/PKB pathway in cannabinoid-induced ERK activation and cell survival. O.D., optical density. A, Western blot analysis of phospho-ERK in cells stimulated with vehicle or 50 nM HU-210 alone or in the presence of 200 nM wortmannin (WOR), 25 μM LY294,002 (LY), or 15 μM mastoparan (MAS). B, phosphorylation status of PKB as determined with antibodies against Thr 308 (top) or Ser 473 (bottom). Loading controls were carried out with an anti–α-tubulin antibody. C, Ras activation was determined after 50 nM HU-210 stimulation in cell extracts by affinity-precipitation of active Ras with GST-Raf-RBD precoupled to GSH-agarose and subsequent Western blot with an anti-Ras antibody. D, Raf-1 activity was determined by immunoprecipitation of Raf-1 from HU-210–stimulated cells. E, cell viability of U373 MG cells after 10 μM C2-ceramide (C2-CER) challenge and subsequent exposure to 50 nM HU-210 alone or in the presence of 200 nM wortmannin (WOR) or 25 μM LY294,002 (LY). Results correspond to four different experiments for each. *, P < 0.01, significantly different from vehicle incubations.
Confirmation of the functional relevance of PI3K-dependent ERK activation was investigated in the U373 MG cell model of cannabinoid anti-apoptotic action. Cannabinoid-mediated protection from ceramide-induced cell death was abrogated by wortmannin and LY294,002 (Fig. 4E). Thus, cannabinoids exert an effective pro-survival action because of their ability to activate in a concerted manner the classical survival signaling pathways PI3K/PKB and ERK via the CB1 receptor.
Discussion
Results presented herein show that CB1receptor coupling to ERK activation depends on Gi protein dissociation and subsequent PI3KIB activation. Although many G protein-coupled receptors activate different members of the mitogen-activated protein kinase family by transactivation of tyrosine kinase receptors (Gschwind et al., 2001), CB1-mediated activation of ERK occurs by an EGFR-independent mechanism. In addition, seven-transmembrane receptors have been shown to recruit Src family members during the β-arrestin–mediated internalization process, which in certain cases is coupled to ERK activation (Marinissen and Gutkind, 2001; Pierce et al., 2001). Regarding the CB1 receptor, our results exclude the involvement of the Src family of cytosolic tyrosine kinases in ERK activation, in line with data showing that internalization-defective CB1 receptors exhibit correct ERK activation capacity (Roche et al., 1999).
As previously highlighted by others (e.g., Davies et al., 2000), the study of cannabinoid-induced ERK activation has taught us the necessity for caution when interpreting data obtained using pharmacological inhibitors claimed in the literature to be highly selective. Thus, whereas experiments conducted with pharmacological inhibitors suggested the involvement of certain mechanism for CB1-mediated ERK activation, detailed study of such pathways (EGFR transactivation or Src involvement) was fruitless and pointed in different directions. It should be noted that the IC50 values for AG1478 and PP2 action obtained in this study (0.1 and 0.8 μM, respectively) indicate that the concentrations employed (1 and 10 μM) are high enough to inhibit the desired target (EGFR and Src) but not so massive as to explain the nonspecific actions observed. The existence of potential targets for tyrphostins different from the paradigmatic ones can be exemplified by tyrphostin AG213, which, although claimed to be specific for the EGFR, also exerts complex actions on the EGF-ERK signaling pathway (Levitzki, 1999). Paradoxical stimulation of the ERK pathway by tyrphostins has been described and attributed to a potential inhibitory effect on tyrosine phosphatase activity (Nowak et al., 1997). In addition, different tyrphostins (AG213, AG34, and AG82) have been shown to alter Src-initiated signaling and cellular transformation, as well as to inhibit Bcr-Abl tyrosine kinase (Agbotounou et al., 1994; Levitzki, 1999). Moreover, in astrocytes, tyrphostin AG1478 was able to prevent thrombin-induced ERK activation, but EGFRtransactivation could not be evidenced (Wang et al., 2002). Like quinoline-derived tyrphostins, pyrazolopirimidine-derived inhibitors of the Src family, such as PP2, act as ATP binding competitors. In this regard, interference of both types of inhibitors toward unrelated postulated targets has been described. Thus, in certain cases, tyrphostins may inhibit Src family-mediated actions (Agbotounou et al., 1994; Levitzki, 1999) and pyrazolopirimidines may inhibit tyrosine kinase receptors (Susa et al., 2000).
Our results highlight the existence of cannabinoid-induced activation of survival signaling pathways in a coordinated manner. Thus, CB1 activation results in PI3K/PKB stimulation (Gómez del Pulgar et al., 2000; this study) in concert with the ERK pathway (Guzmán et al., 2001b; this study). The important role of the mitogenic ERK cascade (Grewal et al., 1999; Kolch, 2000) as well as of the anti-apoptotic PI3K/PKB pathway (Brazil and Hemmings, 2001) in the control of the cell death/survival decision is well known. In general, activation of these signaling pathways leads to cell protection from death stimuli by acting either independently or coordinately. Thus, CB1-mediated acute ERK and PI3K/PKB activation protects glial cells from ceramide induction of the apoptotic program. On the other hand, CB1-activation can also promote apoptosis, particularly in transformed cells, owing to its ability to induce sustained ceramide generation and ERK activation (Galve-Roperh et al., 2000; Guzmán et al., 2001b). In fact, recent research has evidenced growth arrest and deleterious actions of the ERK pathway when activated in a sustained manner. For example, sustained ERK activation is in part responsible for neural cell death after brain ischemia or glutamate excitotoxicity (Alessandrini et al., 1999). Similarly, in PC12 neuronal-like cells, whereas short-term ERK activation promotes proliferation, sustained ERK activation results in cell cycle arrest and neuronal differentiation (Grewal et al., 1999). Regarding glial cells, different magnitude and kinetics of CB1-induced ERK activation produces opposite cellular outcomes (Guzmán et al., 2001a). Thus, whereas short-term ERK activation protects glial cells from ceramide-induced apoptosis (this study), sustained ERK activation promotes apoptosis or growth arrest (Galve-Roperh et al., 2000; Fanton et al., 2001). Current research in our laboratory focuses on the identification of the downstream targets of these signaling pathways that would be ultimately responsible for CB1-mediated cell protection or death.
Acknowledgments
We are indebted to Dr. H. Rosenfeldt (National Institutes of Health) and Dr. D. L. Altschuller (University of Pittsburgh) for advice in Src activity assay and Ras activation assay, respectively, to Dr. G. Reiser (Otto-von-Guericke University, Magdeburg, Germany) for sharing unpublished data, and to T. Aguado for technical assistance.
Footnotes
- Received May 2, 2002.
- Accepted September 3, 2002.
-
This study was supported by grants from Comisión Interministerial de Ciencia y Tecnologı́a (PM 98/0079), Comunidad Autónoma de Madrid (CAM 08.1/0079/2000), Complutense University (PR48/01–9846), and Fundación Ramón Areces.
Abbreviations
- ERK
- extracellular signal-regulated kinase
- PI3K
- phosphatidylinositol 3-kinase
- PKB
- protein kinase B
- PDGF
- platelet derived growth factor
- GST-Raf-RBD
- glutathioneS-transferase-Ras binding domain of Raf
- SR141716
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride
- AG1296
- 6,7-dimethoxy-3-phenylquinoxaline
- AG1478
- 4-(3-chloroanilino)-6,7-dimethoxyquinazoline
- PD98059
- 2′-amino-3′-methoxyflavone
- LY294,002
- 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one
- PAGE
- polyacrylamide gel electrophoresis
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- CP-55940
- (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol
- WIN-55,212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone
- BB94
- [4-(N-hydroxyamino)-2R-isobutyl-3S-(thiopen-2-ylthiomethyl)-succinyl]-l-phenylalanine-N-methylamide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}